

Abstract
Arthrogryposis multiplex congenita (AMC) has been described and defined in thousands of articles, but the terminology used has been inconsistent in clinical and research communities. A definition of AMC was recently developed using a modified Delphi consensus method involving 25 experts in the field of AMC from 8 countries. Participants included health care professionals, researchers, and individuals with AMC. An annotation of the definition provides more in‐depth explanations of the different sentences of the AMC definition and is useful to complement the proposed definition. The aim of this study was to provide an annotation of the proposed consensus‐based AMC definition. For the annotation process, 17 experts in AMC representing 10 disciplines across 7 countries participated. A paragraph was developed for each sentence of the definition using an iterative process involving multiple authors with varied and complementary expertise, ensuring all points of view were taken into consideration. The annotated definition provides an overview of the different topics related to AMC and is intended for all stakeholders, including youth and adults with AMC, their families, and clinicians and researchers, with the hopes of unifying the understanding of AMC in the international community.
Keywords: annotation, arthrogryposis multiplex congenita, collaboration, definition, expert opinion
1. INTRODUCTION
An updated definition of arthrogryposis multiplex congenita (AMC) was developed following an extensive literature review and a modified Delphi approach for consensus building (Cachecho & Dahan‐Oliel, 2019). A panel of 25 international experts in the field of AMC consisting of health care professionals, researchers, and adults with AMC participated in this iterative process that leads to agreement on a definition of AMC. This consensus‐based definition of AMC includes eight concise sentences each targeting a different element of AMC in order to standardize the terminology and concepts related to AMC. A common understanding of AMC across disciplines and borders, including a unified language and common definition of AMC, is important to help harmonize research and clinical endeavors, to facilitate knowledge building in AMC, and to improve communication between families, clinicians, and researchers.
In order for the AMC definition to be well understood by all stakeholders (i.e., clinicians, youth and adults with AMC and their families, and researchers) and easily applicable in different settings and contexts, additional information to expand concepts and clarify the content of the definition is useful. An annotation of the AMC definition is a way to complement this definition and provides more in‐depth information about its different sentences. This method of annotation was previously used in the definition of cerebral palsy by Rosenbaum et al. (2007). The aim of this study was to provide an annotation of the proposed consensus‐based AMC definition. For purposes of consistency, the abbreviated term AMC will be used throughout this manuscript to signify AMC, arthrogryposis, or multiple congenital contractures.
2. METHODS
The process for the development of the consensus‐based definition for AMC was comprised of five phases, as depicted in Figure 1. The first four phases focused on the development of the definition, consisting of a literature review, preliminary survey, draft definition, and modified Delphi process, as detailed in Cachecho and Dahan‐Oliel (2019). Phase 5 consisted of the expansion of each sentence of the definition by annotating it to provide additional explanations on the different aspects of the definition and is the focus of this paper.
Figure 1.
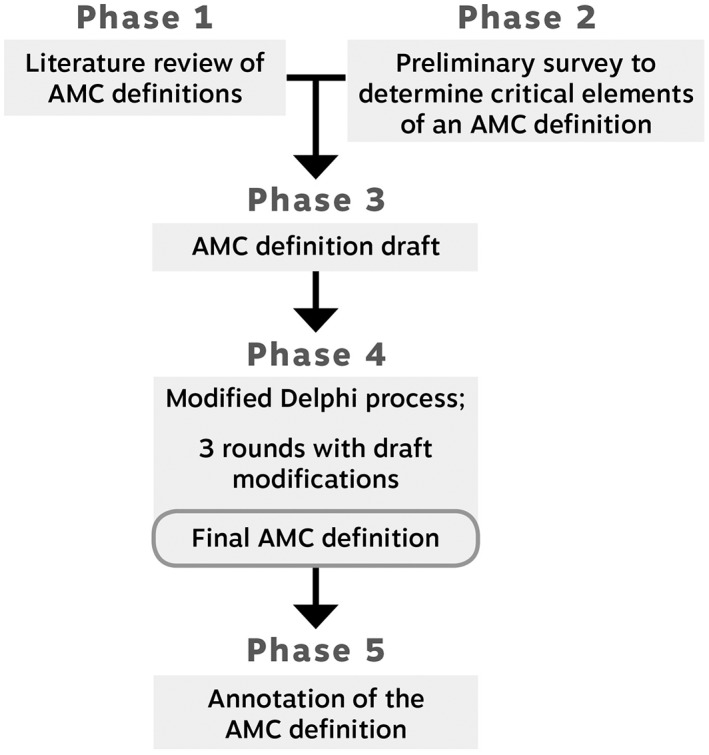
Five phases of the AMC definition study. AMC, arthrogryposis multiplex congenita
Participants in the modified Delphi process (Phase 4) were invited to collaborate on the annotation of the definition. Out of the 25 participants in Phase 4, 14 expressed an interest in participating in the annotation step of the study. Seven additional authors with specific expertise, who had not participated in the initial consensus process, were also invited to the annotation process. In total, 21 experts were invited to participate in this study.
The definition agreed to by the consensus methodology of the Delphi process is presented in Table 1 (Cachecho & Dahan‐Oliel, 2019). The definition was comprised of eight sentences to be annotated. The corresponding sentences with the elements of AMC being addressed are depicted in Table 2. Participants were asked to annotate one of the eight sentences in the form of a paragraph of up to one page. As well, participants were instructed to elaborate on the topics that emanated from the modified Delphi (Phase 4) but had not been directly included in the definition in order to keep it concise. A 4–6‐week window was provided for the development of the initial draft of each of the eight sentences. Drafts were reviewed by the first two authors (N.D.‐O. and S.C.) and sent back for editing to ensure the key concepts identified in Phase 4 were addressed and explained. The following draft versions were then reassigned to another participant with complementary expertise in the field. This process was used to ensure that various disciplines and points of view were taken into account. For example, authors who contributed to Sentence 1 had expertise in epidemiology, orthopedics, and genetics. Table 2 shows the expertise consulted for each sentence. Drafts were reviewed until no new content or additional comments were provided. This iterative process resulted in eight sections describing each of the eight sentences of the definition (i.e., one section per sentence).
Table 1.
Consensus‐based arthrogryposis multiplex congenita definition
| Arthrogryposis multiplex congenita (AMC) is a term used to describe a group of congenital conditions characterized by joint contractures in two or more body areas. While the precise cause may be unknown for some individuals, causes are variable and may include genetic, parental, and environmental factors, as well as abnormalities during fetal development. Individuals with AMC have limited joint movement, with or without muscle weakness, in the involved body areas. Contractures vary in distribution and severity, do not progress to previously unaffected joints, but may change over time due to growth and treatment. Spinal deformities may be present at birth or develop throughout childhood and adolescence. Depending on the underlying diagnosis, other body systems such as the central nervous system (CNS), respiratory, gastrointestinal, and genitourinary systems may be affected. Cognition may be affected if the CNS is involved; sensation is usually intact. The impact on mobility, activities of daily living, and participation is variable. |
Table 2.
Sentences of the arthrogryposis multiplex congenita definition and expertise consulted
| Overall sentence | Complete sentence | Expertise consulted for each sentence |
|---|---|---|
| Sentence 1: general description of arthrogryposis multiplex congenita (AMC) | AMC is a term used to describe a group of congenital conditions characterized by joint contractures in two or more body areas | Epidemiology, genetics, and orthopedics |
| Sentence 2: causes | While the precise cause may be unknown for some individuals, causes are variable and may include genetic, parental, and environmental factors, as well as abnormalities during fetal development | Genetics and neurology |
| Sentence 3: impact on joints and muscles | Individuals with AMC have limited joint movement, with or without muscle weakness, in the involved body areas | Plastic surgery and orthopedics |
| Sentence 4: evolution of the clinical presentation | Contractures vary in distribution and severity, do not progress to previously unaffected joints, but may change over time due to growth and treatment | Physiotherapy, plastic surgery, and dentistry |
| Sentence 5: impact on the spine | Spinal deformities may be present at birth or develop throughout childhood and adolescence | Orthopedics |
| Sentence 6: impact on other body systems | Depending on the underlying diagnosis, other body systems such as the central nervous system (CNS), respiratory, gastrointestinal, and genitourinary systems may be affected | Neurology, genetics, epidemiology, and dentistry |
| Sentence 7: impact on cognition and sensation | Cognition may be affected if the CNS is involved; sensation is usually intact | Neurology and orthopedics |
| Sentence 8: impact on function | The impact on mobility, activities of daily living and participation is variable | Lived experience, genetics, occupational therapy, and dentistry |
3. RESULTS
From the 21 individuals who were invited to participate, 15 contributed to the annotation of the definition, in addition to the two first authors.
The annotation for each sentence of the AMC definition is provided below. Table 3 describes the medical terms used in the different paragraphs.
Table 3.
Glossary
| Term | Definition | Examples |
|---|---|---|
| Antagonist muscle | Muscle opposite to the one contracting | When the biceps contracts, the triceps relaxes. In this case, the triceps is the antagonist muscle |
| Chiari malformation | Condition in which brain tissue or cerebellum extends into the spinal canal. It occurs when part of the skull is abnormally small or misshapen, pressing on the brain and forcing it downward | – |
| Chromosomal aberrations | Abnormality in the structure or number of chromosomes | Trisomy 21, trisomy 13, trisomy 18, microdeletions, and microduplications |
| Coronal plane | Any vertical plane that divides the body into ventral and dorsal (belly and back) sections | – |
| Cryptorchidism | A condition in which one or both of the testes fail to descend from the abdomen into the scrotum | – |
| Deformative | Altered morphogenesis secondary to mechanical forces | – |
| Diastematomyelia | Congenital disorder in which a part of the spinal cord is split | – |
| Disruptive (cause) | Pathological arrest in development of a normal tissue | Amniotic band sequence, vascular or infectious causes |
| Dysplasia | The abnormal growth or development of a tissue or organ | Hip dysplasia |
| Embryogenesis | The formation and development of an embryo | – |
| Genotype | Genetic constitution of an organism that participates in determining its observable characteristics | – |
| Hydrocephalus | Excess cerebrospinal fluid which builds up within the ventricles (fluid‐containing cavities) of the brain and may increase pressure within the head | – |
| Iatrogenic (cause) | Relating to illness caused by medical examination or treatment | Failed abortion |
| ICD 10 coding system | International classification of diseases, tenth revision, clinical modification is a system used by physicians and other healthcare providers to classify and code all diagnoses, symptoms, and procedures | – |
| Ischemic lesion | Lack of blood flow to the brain that leads to limited oxygen supply | – |
| Lissencephaly | Brain malformation characterized by the absence of normal convolutions (folds) in the cerebral cortex and an abnormally small head | – |
| Intrauterine growth retardation | A condition in which a baby does not grow to normal weight during pregnancy | – |
| Microcephaly | A condition in which the brain does not develop properly resulting in a smaller than normal head | – |
| Microdeletions | Loss of a tiny piece of a chromosome, not seen on a karyotype | – |
| Microduplications | Tiny extra piece of a chromosome, not seen on a karyotype | – |
| Myopathy | Disease of the muscle in which the muscle fibers do not function properly | – |
| Neural axis | The axis of the CNS, that is, brain and spinal cord | – |
| Neurography | Neurography or magnetic resonance neurography is the direct imaging of nerves in the body. It is a modification of magnetic resonance imaging | |
| Neuropathy | A condition that affects the normal activity of the nerves of the peripheral nervous system. It causes weakness, numbness, and pain | – |
| Oligohydramnios (or oligoamnios) | An abnormal condition occurring during pregnancy resulting from lack of amniotic fluid (fluid surrounding the baby in the uterus) | – |
| Phenotype | Observable characteristics of an organism that result from the interaction of its genotype with the environment | Behaviour, biochemical properties, color, shape, and size |
| Polydactyly | A condition where someone is born with one or more extra fingers or toes | – |
| Polymicrogyria | A condition characterized by abnormal development of the brain before birth. The brain develops too many folds, and the folds are unusually small | – |
| Pulmonary hypoplasia | Incomplete development of the lungs, resulting in an abnormally low number or size of bronchopulmonary segments or alveoli | – |
| Retrognathia | A condition in which the lower jaw is set further back than the upper jaw | |
| Sagittal plane | Parallel to the longitudinal axis of the organism, or from the mouth to the tail | – |
| Short gut syndrome | Malabsorption disorder caused by a lack of functional small intestine | – |
| Syndactyly | Term used to describe webbed or conjoined digits (fingers or toes) | – |
| Synostosis | Union or fusion between adjacent bones | Radioulnar synosthosis |
| Teratogenic (cause) | A drug or other substance capable of interfering with the development of a fetus, causing birth defects | Alcohol, chemicals, and radiation exposure |
| Tethered cords | A condition in which the spinal cord is “stuck” to a structure within the spine, limiting the movement of the spinal cord within the spinal column | |
| Whole exome sequence | Genomic technique for sequencing all of the protein‐coding region of genes in a genome | |
| Whole genome sequence | The process of determining the complete DNA sequence of an organism's genome |
3.1. Sentence 1: AMC is a term used to describe a group of congenital conditions characterized by joint contractures in two or more body areas
The term arthrogryposis is Greek, from arthron (joint) + gryp (curved) + osis (condition). Multiplex is Latin for multiple, and congenita, also Latin, is from the word congenitus meaning present at birth. Thus, AMC is a term for multiple curved joints that are present at birth. AMC, arthrogryposis, and multiple congenital contractures are all terms that have been used interchangeably. In the past, the term AMC was used as a diagnosis before it was clear that although cases had similar features of multiple congenital contractures, they represented different specific entities (Hall, 1985). AMC is now considered a descriptive term rather than a specific diagnosis, representing a heterogeneous group of over 400 specific diagnoses (Hall, Kimber, & van Bosse, 2017). The term may seem confusing as it is still often used incorrectly as a distinct diagnosis rather than phenotypically observed characteristics.
The contractures in AMC are present at birth; however, they develop throughout pregnancy and can be detected in utero. We define a contracted joint as one lacking a full passive range of motion, be it restrictions in flexion, extension, or both. The limbs, spine, and jaw can be affected. A flexion contracture is when a joint lacks full anatomical extension, so that the two bony segments on either side of the joint cannot be maximally separated. Alternatively, an extension contracture is when a joint lacks full expected flexion, so that the two bony segments on either side of their connecting joint cannot be brought together. Hyperextension is when there is excessive extension at the joint and the bone segments on either side of the joint move past the anatomic limit in the direction opposite to flexion. AMC contractures do not include an isolated contracture, such as an isolated clubfoot or a hip dislocation, but affect two or more joints in different body areas (Staheli, Hall, Jaffe, & Paholke, 1998), hence the alternative description of multiple congenital contractures.
Over 50% of individuals with AMC are diagnosed with either Amyoplasia or distal arthrogryposis (DA) (Hall et al., 2017). Amyoplasia is the most common form of AMC and DA the second most common. A retrospective study on 560 individuals with Amyoplasia showed that there is involvement of all four limbs in 63% of the cases (i.e., contractures in the upper limbs and lower limbs). There is three‐limb involvement in 5% of the cases, upper limb involvement only in 17% of cases and lower limb involvement only in 15% of cases in Amyoplasia (Hall, Aldinger, & Tanaka, 2014). DA is comprised of a group of syndromes characterized by specific genetic mutations (Bamshad, Van Heest, & Pleasure, 2009). Individuals with DA present mainly with joint contractures in the hands and feet as well as other physical abnormalities specific to each syndrome. Prevalence of DA syndromes is not yet known (Kimber, 2015). Distribution and severity of joint contractures are detailed further under Sentence 4.
3.2. Sentence 2: While the precise cause may be unknown for some individuals, causes are variable and may include genetic, parental, and environmental factors, as well as abnormalities during fetal development
Regardless of the underlying cause, the common denominator is fetal hypo‐ or akinesia, ultimately leading to multiple congenital contracture or AMC. Though the exact distribution of the possible causes of AMC has not been established, genetic causes seem to represent the largest part (Hall, 2014a). To date, more than 400 gene mutations have been associated with AMC and this number is increasing (Hall & Kiefer, 2016). Point mutations, chromosomal aberrations such as trisomies (especially 21, 13, and 18), as well as microdeletions or microduplications are all well described. Some genetic causes always lead to multiple congenital contractures, while in others, multiple joint contractures are present only in the more severe end of the phenotypic spectrum. The most commonly identified genetic causes of AMC affect the motor unit, that is, the anterior horn cell, its axon, the neuromuscular junction, or the striated skeletal muscle cell (Darin, Kimber, Kroksmark, & Tulinius, 2002; Ravenscroft et al., 2011). As well, genes involved in the functioning of the central nervous system (CNS) have also been identified in association with AMC (Kiefer & Hall, 2019). The emergence of new technologies for sequencing of the human genome has drastically changed the diagnostic possibilities, allowing, among other things, for the discovery of more genetic causes.
Among parental causes, transient neonatal myasthenia with maternal antibodies directed against the fetal acetylcholine receptor at the neuromuscular junction is a well‐known and a rare cause of AMC (Dalton et al., 2006; Gilhus & Hong, 2018; Vincent et al., 1995). Another rare maternal cause is prolonged maternal hypotension during pregnancy (Farrell & McGillivray, 1983; Hall, 2012).
Bacterial or viral infections (e.g., Zika virus) (van der Linden et al., 2016), iatrogenic (e.g., failed abortion) (Hall, 2012), or teratogenic (e.g., certain drugs such as skeletal muscle relaxants and antiepileptics) (Hall, 2014a) causes of AMC are rare, but have been reported. Since 2015, Zika virus has become the most frequent infectious and therefore environmental cause of AMC (C. Lage et al., 2019; van der Linden et al., 2016). Previously, only congenital infections with cytomegalovirus (Perlman, Burns, Twickler, & Weinberg, 1995), varicella zoster virus (Huang, Lin, Chiu, & Hung, 2001), and rubella virus (Hall & Reed, 1982) had been known to cause AMC in humans.
Extrinsic causes such as mechanical limitations to fetal movement from a multiple pregnancy, oligohydramnios, amniotic bands, or anatomical abnormalities of the uterus (e.g., bicornuate) may also cause AMC (Hall, 1981; Hall, 2014b).
Regardless of whether the cause is intrinsic or extrinsic, the common denominator is fetal hypo‐ or akinesia, ultimately leading to multiple congenital contracture or AMC.
3.3. Sentence 3: Individuals with AMC have limited joint movement, with or without muscle weakness, in the involved body areas
Whatever the underlying condition causing the appearance of a baby with multiple joint contractures, they all share the phenomena of decreased fetal movement (fetal akinesia) (Hall, 1997). The body sites involved and the severity of the contracture are directly related to the timing and the duration of the event causing the fetal akinesia such that the earlier and longer the duration of decreased fetal movements, the more severe the contractures will be at birth (Hall, 1997). Early and short events will primarily affect the upper extremities; later events will primarily affect the lower extremities. Longer lasting events will affect more body parts and more severely.
Decreased fetal movement can be associated with an increase of connective tissue around the joints, as well as joint fibrosis and fibrotic replacement of muscle, which further limits the joint movement and increases the degree of contracture (Drachman & Coulombre, 1962; Fuller, 1975; Gordon Robertson, Williamson, & Blattner, 1955; Hall, 1997; Swinyard & Bleck, 1985). Muscles may be atrophied due to misuse, or replaced by fibrous or fatty tissue. The lack of functional muscle in the fetus may lead to fetal akinesia and secondarily to joint contractures. Alternatively, restricted joint motion caused by soft tissue abnormalities may lead to disuse atrophy of muscles, thus increasing the likelihood of contractures and of muscle weakness. A joint contracture is almost always associated with muscle atrophy (Kimber, 2015; Swinyard & Bleck, 1985). The degree of muscle atrophy found in AMC will depend on the pathology leading to fetal akinesia and the timing of its onset (Kimber, 2015). The known pathologies can involve nerves, muscles, motor endplates, connective tissue, space limitation in utero, maternal illness, intrauterine vascular compromise, and metabolic disturbances (Bamshad et al., 2009; Hall, 1997; Swinyard & Bleck, 1985). In some conditions, such as Escobar syndrome, if a joint is in flexion and not moving, a skin web (i.e., pterygium) may form across the joint (Escobar, Bixler, Gleiser, Weaver, & Gibbs, 1978). A web implies a longstanding lack of movement of that joint.
In most forms of AMC, joint development is normal during embryogenesis (Hall, 2014a). Lack of movement of the joint can lead to both articular deformities and soft tissue dysplasia. Therefore, most of the changes seen in AMC are deformational.
3.4. Sentence 4: Contractures vary in distribution and severity, do not progress to previously unaffected joints, but may change over time due to growth and treatment
Joint contractures or stiffness in AMC are the result of complex developmental changes in the joint surfaces, and surrounding soft tissues including muscle, which prevent the limb from moving into its full range of motion (Bamshad et al., 2009; Hall, 1997). The severity of the condition varies depending on how many joints are affected and the extent of joint stiffness. The heterogeneity of AMC means that there is wide variation in clinical presentation from a few contractures in the hands or feet to contractures in all limbs, the spine, and the jaw (Hall, 2014a). Restricted jaw opening, weakness of the masticatory musculature, and other jaw anomalies (e.g., high‐arched palate, clefting, and retrognathia) may affect oral hygiene and feeding (Emmanouil, Roumani, & Petsi, 2010; Steinberg, Nelson, Feinberg, & Calhoun, 1996). Individuals with AMC do not develop new contractures in unaffected joints, but the more joints involved, the greater the impact on independence in mobility and activities of daily living (ADLs) (Amor, Spaeth, Chafey, & Gogola, 2011; Steen, Wekre, & Vollestad, 2018).
Following birth, the contractures will typically show improvement, especially with early physiotherapy and occupational therapy involving stretching and maintaining positions with night splints (Bevan et al., 2007; Mennen, van Heest, Ezaki, Tonkin, & Gericke, 2005). In association with therapy, surgical intervention can enable an improvement in the range of joint movement and functional position of the joints (Bevan et al., 2007; Kowalczyk & Feluś, 2016; Mennen et al., 2005). The passive range of motion of joints sometimes exceeds the active range of motion because of weakness or absence of a functioning muscle (Steen, Christensen, & Samargian, 2017).
As the child grows older, contractures may get tighter due to muscle imbalance (Hahn, 1985) coupled with the natural process of decreasing flexibility in all humans. Individuals with AMC have disproportions in muscle strength and elasticity—the stronger muscle will pull the joint into a more contracted position as the weaker antagonist muscle is unable to overpower it. This is particularly evident during the growth spurts. It is therefore important to enhance muscle strength and length through exercising, stretching, and maintenance of optimum joint position in order to prevent joint contractures from worsening and to maintain the maximum range of movement possible throughout growth (Bevan et al., 2007; Hahn, 1985; Steen et al., 2018). Improving one's function and independence allows them to maintain joint range of motion through everyday activities.
3.5. Sentence 5: Spinal deformities may be present at birth or develop throughout childhood and adolescence
There are different types of spinal deformities in AMC. Scoliosis is a curvature of the spine in the coronal plane, and may be an unbalanced “C”‐type curve, or a more compensated “S” curve. The “S” curve will have smaller curves proximal and/or distal to the main curve to attain a more balanced posture. These deformities can occur at any level in the spine, from cervical, thoracic, and lumbar. In the sagittal plane, hyperkyphosis is an exaggeration of the normal posterior convexity of the thoracic spine, and hyperlordosis is a pathological increase in the normal lumbar posterior postural concavity. In severe deformities, kyphosis can occur in the lumbar spine, and/or lordosis in the thoracic spine.
In children with Amyoplasia or syndromic arthrogryposes, spinal deformities may be present at birth and truly are a contracture of the spine due to akinesia, reflecting the position of the fetus in utero (Drummond & Mackenzie, 1978; Komolkin, Ulrich, Agranovich, & van Bosse, 2017). Although these deformities are present at birth, they are not labeled “congenital scoliosis” as that term is used to describe abnormal spine alignment at birth associated with vertebral malformations. Like most joint abnormalities in AMC, the spine abnormalities are deformations—the underlying structures were normally formed, then misshapen during intrauterine development (Hall, 2014a). These “at‐birth” or essential spinal deformities may include “C”‐shaped thoracolumbar scoliosis with or without associated hyperlordosis (Greggi et al., 2010; Herron, Westin, & Dawson, 1978; Soultanis et al., 2007). Babies presenting with essential spine deformities generally have severe upper and lower limbs contractures, fibro‐fatty degeneration of the muscles and a more guarded potential for independent daily living and ambulation (Komolkin et al., 2017). Treatment may include early physiotherapy for stretching, bracing, and seat/bed modifications, with the goal of preventing rapid progression of the spine deformity, providing for stable seating, and positioning for comfortable sleep. Curves can either progress rapidly during the infantile period, or conversely remain stable until late‐childhood (Komolkin et al., 2017). It is not unusual for these curves to undergo spontaneous fusion, making surgical correction more complicated (Herron et al., 1978).
Spine deformities may also develop de novo during childhood in Amyoplasia, some forms of DA (e.g., Freeman–Sheldon and Beals syndrome) or syndromic arthrogryposis (Herron et al., 1978). Treatment should focus on limiting progression while maximizing the child's function and independence. The curves associated with the distal arthrogryposes tend to be thoracolumbar and are often the most aggressively progressive curves (Komolkin et al., 2017). Treatment options for developmental curves are spinal casting for infantile curves and bracing for curves between 20° and 50° to prevent rapid progression (Sanders et al., 2009). In curves measuring 50° or greater with rapid progression (more than 5° per year), surgery is indicated, either an expandable implant for those with significant remaining growth or a spinal fusion for nearly mature patients (Astur et al., 2014; Greggi et al., 2010). Magnetic resonance imaging (MRI) scans of the entire neural axis should be obtained prior to surgical intervention, or if pathologic neurologic signs develop, to rule out spinal cord abnormalities, such as Chiari malformations, tethered cords, or diastematomyelia.
3.6. Sentence 6: Depending on the underlying diagnosis, other body systems such as the CNS, respiratory, gastrointestinal, and genitourinary systems may be affected
Having an underlying diagnosis in AMC is very useful in preventing complications related to that diagnosis, understanding the cause of the clinical findings, thereby allowing planning adequate support and treatment, and providing a prognosis of functional development.
Over the last two decades, Hall's clinical classification of AMC has been widely used (Hall, 1997). It divides individuals with AMC into three groups. Group 1 includes individuals with primarily limb involvement, Group 2 includes individuals with musculoskeletal involvement plus other system anomalies, and Group 3 encompasses individuals with limb and CNS involvement. In AMC, organ system involvement may be present depending on the underlying cause, including one or more of the CNS, cardiovascular, respiratory, gastrointestinal, genitourinary, ophthalmological, and otorhinolaryngeal systems (Staheli et al., 1998). More recently, Zika virus syndrome, with microcephaly and AMC, has become prevalent in endemic parts of the world (C. Lage et al., 2019; van der Linden et al., 2016). An example of CNS and other organ system involvement is trisomy 18 with involvement of heart, brain, kidneys and gastrointestinal system. Another example is Pena Shokeir syndrome (also known as fetal akinesia deformation sequence), which involves intrauterine growth retardation, pulmonary hypoplasia, short gut syndrome, cryptorchidism, and brain malformations (Hall, 2009). These syndromes are rare and may be lethal due to complications of pulmonary hypoplasia (i.e., lungs are underdeveloped), such as in lethal multiple ptergyium and Pena Shokeir syndromes (Hall, 1984; Hall, Opitz, & Reynolds, 1986; Staheli et al., 1998). Limb anomalies may also be present in AMC and can include loss of fingers or toes, loss of a limb, polydactyly, syndactyly, and synostoses. Anomalies of the jaw and teeth (e.g., irregular tooth development and position, delayed eruption of teeth, and tooth crowding) may also occur in individuals with AMC (Rodrigues, Botelho, Vaz, Mesquita, & Ponces, 2015).
A careful clinical investigation is necessary to direct the genetic investigation and to perform a comprehensive evaluation of organ involvement. If CNS involvement is suspected, MRI of the brain and spinal cord should be performed. In all cases of suspected delay of cognitive development, evaluation of cognition, vision, and hearing is important, and neuro‐ophthalmological examination should also be carried out. Echocardiogram (or echo, is a diagnostic test that utilizes sound waves to generate images of the heart), evaluation of the respiratory, gastrointestinal, and genitourinary systems may be necessary depending on the clinical symptoms. Electroencephalogram (or EEG, is a test used to evaluate the electrical activity of the brain) may need to be done, as epilepsy may also be an associated problem in AMC with CNS involvement. If there is a suspicion of a myopathic involvement, muscle enzymes, electromyogram (or EMG, is a test to detect abnormal muscle activity), and muscle biopsy may be necessary for diagnostic purposes in addition to targeted genetic testing. In suspicion of neuropathic involvement, a neurography should be performed. Metabolic investigation and testing may be necessary. These workups are necessary if there is an unusual presentation of AMC and do not need to be performed on all individuals presenting with multiple congenital contractures.
A multidisciplinary approach is needed to ensure a comprehensive investigation and appropriate follow‐up. The interdisciplinary team should include orthopedic surgeons, neurologist, geneticist, rehabilitation specialist, and social worker. Throughout the lifespan, other specialties such as cardiology, urology, pulmonology, ophthalmology, plastic surgery, dentistry, and orthodontics may be consulted depending on the clinical presentation and underlying diagnosis. The neonatologist and/or pediatrician may be involved early in life. A physiatrist may also be involved throughout the lifespan to oversee musculoskeletal issues and pain management. Challenges in transitioning from pediatric to adult services may arise, particularly if many different specialists are involved. Care coordination involving the youth, family, and multidisciplinary team should start early to plan for a seamless transition. Genetic counseling is recommended for purposes of family planning, especially in inherited conditions.
3.7. Sentence 7: Cognition may be affected if the CNS is involved; sensation is usually intact
Multiple syndromes and chromosomal anomalies have phenotypic expressions of multiple contractures and have concomitant cognitive impairment (Hoff, Loane, Gilhus, Rasmussen, & Daltveit, 2011). Cognition includes aspects of perception, thinking, memory, attention, and is linked with language development. A Dutch study concerning the diagnostic management of newborns with AMC confirmed various cerebral developmental defects in 21 out of 75 (28%) (Hageman et al., 1988), which were theorized to have intrauterine fetal weakness leading to reduced motion and, hence, congenital contractures. A Swedish study found cerebral involvement to be present in 23 out of 68 cases (34%) (Darin et al., 2002) and a Canadian study on the prevalence of AMC found that 33 out of 103 cases (32%) belonged to Group 3 of Hall's classification (i.e., limb and CNS involvement) (Lowry, Sibbald, Bedard, & Hall, 2010). In the Swedish and Canadian studies, individuals corresponding to Group 2 of Hall's classification (i.e., musculoskeletal involvement plus other system anomalies) presented with conditions and syndromes in which cognitive impairment may be a part of the clinical picture (Darin et al., 2002; Lowry et al., 2010). In a French retrospective study, 8 out of 42 children (19%) belonged to Group 1 (i.e., primarily limb involvement), 14 to Group 2 (33%), and 20 to Group 3 (48%) of Hall's classification (Wallach et al., 2018). The single most common etiology, present in 19% of cases, was a neurological process, predominantly involving problems with brain gyration formation of a polymicrogyria type. The predominance of Groups 2 and 3 in this study is high, probably due to a recruitment bias.
Signs and symptoms of CNS dysfunction include delayed intellectual development, seizures, microcephaly, ischemic lesions on brain MRI, as well as various structural anomalies, such as polymicrogyria, lissencephaly, hydrocephalus, and absence of the corpus callosum (Hageman, Willemse, Van Ketel, & Verdonck, 1987; Hall, 2014a; Saito, Hayashi, Miyazono, Shimogama, & Ohno, 2006; Wallach et al., 2018). The clinical signs and symptoms may also be found in cases where the primary cause of AMC is presumed not to be of cerebral origin (Hageman et al., 1987). Given the fact that a significant proportion of AMC is linked to CNS anomalies and syndromes, which may affect cognitive functions, it is prudent to assess cognition in individuals in Groups 2 and 3 of Hall's classification.
In AMC, it has generally been considered that sensation is intact. It is known that different types of congenital neuropathies may contribute to the genesis of joint contractures (Bamshad et al., 2009; Evangelista et al., 2015; Ravenscroft et al., 2011; Scoto et al., 2015). Most are motor neuropathies, but some do affect proprioception, at least in animal studies (Haliloglu et al., 2017; Hengel et al., 2017; Masingue, Fauré, Solé, Stojkovic, & Léonard‐Louis, 2019). Auditory sensory abnormalities have been identified in some children with DA. A Swedish report found almost a 10% incidence of hearing impairments (Kimber, Tajsharghi, Kroksmark, Oldfors, & Tulinius, 2012). Deafness interferes with language development and must be investigated. An interdisciplinary approach to individuals with AMC with cognition dysfunction is highly recommended to assign an appropriate diagnosis and to develop a comprehensive management plan. Important team members already mentioned earlier include geneticists, neurologists, and orthopedic surgeons, in addition to developmental pediatricians and the allied health therapy disciplines (e.g., speech therapist, occupational and physical therapists.
3.8. Sentence 8: The impact on mobility, ADLs, and participation is variable
In individuals with AMC, limitations in joint movements and muscle strength are the main factors that impact mobility, ADLs, and participation (Steen et al., 2018). Children and adults with AMC may use different modes of mobility to get around in their environments (Ho & Karol, 2008). On one end, individuals with little or no lower extremity involvement may walk independently. On the other end, those with more severe involvement of the limbs may need to use powered wheelchairs. In between, some individuals walk unaided but with an altered gait, and others use walking devices (e.g., crutches and walkers) and/or leg braces. Some may be able to walk at home but use a wheelchair or a scooter in the community to compensate for their limited walking endurance and balance. The level of independence for ambulation is related to muscle strength, severity and type of joint contractures, cognitive functioning, and response to therapy. The ability to use walking aids and/or a manual wheelchair is also affected by the degree of involvement of the upper extremities (Ho & Karol, 2008). With age, getting around may require increased effort and energy; therefore, individuals with AMC may use additional assistive devices for mobility (Ho & Karol, 2008).
There is also great variability in the ability of individuals with AMC to perform ADLs independently (Amor et al., 2011; Dai et al., 2018). Many live and function completely independently while others require significant attendant care (Dai et al., 2018; Steen et al., 2018). Independence in ADLs is related to antigravity muscle strength and/or a high degree of passive range of motion mostly of the upper extremities (Steen et al., 2018). People with AMC use many compensatory strategies such as taking advantage of their passive range of motion or using other body parts to increase their independence (Steen et al., 2017). For example, leaning a forearm on a table to flex the elbow to bring food closer to the mouth when one cannot actively bend his/her elbow is a frequently used adaptation. Another example is using the feet or mouth for grooming and hygiene. As individuals with AMC may use their mouth for everyday activities, tooth wear may occur over time and therefore preventative care is recommended (Hanif, Rashid, & Nasim, 2015). Assistive devices may sometimes be required to facilitate certain tasks (e.g., dressing stick and universal cuff for feeding) (Steen et al., 2017). Children and adults with AMC may choose to get assistance with tasks that they could perform on their own, but with difficulty, in order to save their time and energy for more meaningful pursuits such as education and employment (Steen et al., 2017). Physical and occupational therapists can provide treatment and home exercise programs, recommend appropriate assistive devices and compensatory strategies to promote independence in daily activities (Steen et al., 2018). Speech therapists can assist for speech and feeding difficulties. The ways that this population adapts to their physical limitations is impressive, and their solutions are often clever and unique.
Participation in meaningful activities includes leisure, social roles and relationships, education, and paid or unpaid work. In both children and adults with AMC, participation in physical activities may be decreased compared to able‐bodied peers (Amor et al., 2011; Nouraei, Sawatzky, MacGillivray, & Hall, 2017). This may be overcome by finding different ways to participate in leisure activities using compensatory strategies and through adapted sports (e.g., wheelchair sports and painting by mouth). When cognition is not affected, individuals with AMC can attain high education levels and lead productive and social lives (Nouraei et al., 2017; Steen et al., 2018). When psychosocial support is needed, a consultation with a psychologist is recommended. Individuals with AMC generally report having a positive mental attitude and a good quality of life (Nouraei et al., 2017).
4. DISCUSSION
The aim of this study was to propose a consensus‐based AMC definition, annotated for clarity and completeness. The definition provides a global overview of AMC, and the annotations, developed by individuals with lived experience and by international experts from various disciplines, afford a more in‐depth understanding of the different topics of the AMC definition. The different backgrounds ensure that all points of view were considered, so that the final annotation was meaningful to all stakeholders, including patient representatives (Domecq et al., 2014).
AMC is not an actual diagnosis, but an umbrella term for the many conditions associated with multiple joint contractures. Therefore, the proposed definition for AMC is not diagnosis specific, but attempts to describe and encapsulate the varied presentations of the spectrum of possible underlying causes of multiple congenital contractures, along with the associated impairments and limitations. The annotated definition is designed to provide an overview on different topics related to AMC and may guide the diagnostic process and/or interventions. The annotation gives a general description of AMC, expands on the causes, and explains the evolution of the clinical presentation, using the most recent epidemiological data. It also strives to deepen the understanding of joint contractures and muscle weakness, presents the range of different body systems that can be affected, and the overall impact on the individual's level of function. The annotation's content was based on both clinical experience and the latest research evidence. The provided glossary is meant to make the included terms accessible to all stakeholders regardless of their background.
A unified definition of AMC may be a step toward facilitating more accurate prevalence rates for AMC. In fact, these are challenging to ascertain due to the heterogeneity of conditions that are associated with AMC and the lack of a standard case definition. The reported prevalence of AMC ranges between one per 3,300 and 12,000 infants (Lowry et al., 2010). This wide range in prevalence is perhaps due to differences in case definition, case ascertainment, and disease coding practices among institutions and between studies. The International Classification of Diseases‐ 10th revision (ICD‐10) coding system, which is often used by population‐based studies (Bedard & Lowry, 2019), is difficult to use for AMC and diseases associated with AMC. The most cited prevalence of AMC is 1 per 3,000 infants, likely following the report by Laitinen and Hirvensalo (1966) on a small hospital‐based study in Finland (11 cases from 1961 to 1965). A more recent Finnish population‐based study by Pakkasjärvi et al. (2006) reported an extrapolated prevalence of 1 in 4,600 (214 cases from 1987 to 2002) which is comparable to the population‐based study by Lowry et al. (2010) of 1 in 4,300 (103 cases from 1997 to 2007).
Inconsistencies of inclusion and exclusion criteria for AMC across studies may also affect prevalence rates. In fact, certain conditions with congenital contractures require special consideration. Examples are spina bifida and spinal muscular atrophy (SMA). Spina bifida is caused by the incomplete closure of the neural tube of the spine (Williams et al., 2015), representing a malformation of the fetus, and therefore has a distinct etiology from AMC. Although spina bifida may present with orthopedic deformities of the spine and limbs, including clubfoot, lower extremity contractures, and hip dislocations, (Thompson et al., 2019; Wynne‐Davies, Williams, & O'Connor, 1981) due to partial or complete paralysis of the lower limbs and/or trunk, it does not adhere to our definition of AMC. The term SMA describes a group of heritable neurodegenerative diseases with a spectrum of severities, age of onset, and underlying causes. Bürglen et al. (1996) noted at the International SMA Consortium that AMC is an exclusion criterion for SMA, but they themselves found an association of AMC with certain types of SMA (Bürglen et al., 1996). These types of SMA include Type 2 or intermediate SMA (Yoshioka et al., 2018) and Type 0 or severe‐fatal SMA (Falsaperla et al., 2001; Grotto et al., 2016; Storbeck et al., 2017), as these babies present with reduced fetal movement, congenital contractures, and fractures at birth, but no malformations. Genetic work‐up is essential for any baby or child with an atypical presentation of AMC to ensure essential and timely treatment, as well as parental counseling. Advances in genetic technologies, such as whole exome or whole genome sequencing, used to identify specific diagnoses, may also help develop more accurate epidemiological data and coding systems.
5. CONCLUSIONS
This annotated definition is expected to provide a unified understanding of AMC, ensuring the use of a coherent language for improved communication and collaborations. It is meant to be accessible and meaningful not only to the international community of clinicians and researchers, but also to all stakeholders, including youth and adults with AMC, and their families. As research on diagnostic methods, classification systems and treatments of AMC continue to evolve, the definition and its annotation will need to be periodically reviewed, revised, and updated in the future.
ACKNOWLEDGMENTS
We acknowledge the methodological contribution provided by Dr Peter Rosenbaum, Canada Research Chair in Childhood Disability, Professor of Pediatrics at McMaster University, and co‐founder of CanChild Centre for Childhood Disability Research. We thank Carla Evangeliste from Shriners Hospital for Children—Canada for formatting assistance. This project was supported by a clinical research fund (79150‐CAN‐18 and 79150‐CAN‐19) awarded by the Shriners Hospitals for Children.
Dahan‐Oliel N, Cachecho S, Barnes D, et al. International multidisciplinary collaboration toward an annotated definition of arthrogryposis multiplex congenita. Am J Med Genet Part C. 2019;181C:288–299. 10.1002/ajmg.c.31721
Corrections added on 29 July, 2019, after first online publication: author has updated the funding acknowledgment and reference.
REFERENCES
- Amor, C. J. , Spaeth, M. C. , Chafey, D. H. , & Gogola, G. R. (2011). Use of the pediatric outcomes data collection instrument to evaluate functional outcomes in arthrogryposis. Journal of Pediatric Orthopaedics, 31(3), 293–296. [DOI] [PubMed] [Google Scholar]
- Astur, N. , Flynn, J. M. , Ramirez, N. , Glotzbecker, M. , van Bosse, H. J. , Hoashi, J. S. , … Sawyer, J. R. (2014). The efficacy of rib‐based distraction with VEPTR in the treatment of early‐onset scoliosis in patients with arthrogryposis. Journal of Pediatric Orthopaedics, 34(1), 8–13. [DOI] [PubMed] [Google Scholar]
- Bamshad, M. , Van Heest, A. E. , & Pleasure, D. (2009). Arthrogryposis: A review and update. Journal of Bone and Joint Surgery. American Volume, 91(Suppl 4), 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard, T. , & Lowry, R. B. (2019). Disease coding systems for arthrogryposis multiplex congenita. American Journal of Medical Genetics. Part C (in press). [DOI] [PubMed] [Google Scholar]
- Bevan, W. P. , Hall, J. G. , Bamshad, M. , Staheli, L. T. , Jaffe, K. M. , & Song, K. (2007). Arthrogryposis multiplex congenita (amyoplasia): An orthopaedic perspective. Journal of Pediatric Orthopaedics, 27(5), 594–600. [DOI] [PubMed] [Google Scholar]
- Bürglen, L. , Amiel, J. , Viollet, L. , Lefebvre, S. , Burlet, P. , Clermont, O. , … Melki, J. (1996). Survival motor neuron gene deletion in the arthrogryposis multiplex congenita‐spinal muscular atrophy association. The Journal of Clinical Investigation, 98(5), 1130–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. Lage, M. L. , Carvalho, A. L. , Ventura, P. A. , Taguchi, T. B. , Fernandes, A. S. , Pinho, S. F. , … Nascimento‐Carvalho, C. M. (2019). Clinical, neuroimaging, and neurophysiological findings in children with microcephaly related to congenital Zika virus infection. International Journal of Environmental Research and Public Health, 16(3), pii: E309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachecho, S., Elfassy, C., Hamdy, R., Rosenbaum, P., Dahan‐Oliel, N. (2019). Arthrogryposis Multiplex Congenita definition: Update using an international consensus‐based approach. Am J Med Genet C Semin Med Genet (In press). [DOI] [PubMed] [Google Scholar]
- Dai, S. , Dieterich, K. , Jaeger, M. , Wuyam, B. , Jouk, P. S. , & Pérennou, D. (2018). Disability in adults with arthrogryposis is severe, partly invisible, and varies by genotype. Neurology, 90(18), e1596–e1604. [DOI] [PubMed] [Google Scholar]
- Dalton, P. , Clover, L. , Wallerstein, R. , Stewart, H. , Genzel‐Boroviczeny, O. , Dean, A. , & Vincent, A. (2006). Fetal arthrogryposis and maternal serum antibodies. Neuromuscular Disorders, 16(8), 481–491. [DOI] [PubMed] [Google Scholar]
- Darin, N. , Kimber, E. , Kroksmark, A. K. , & Tulinius, M. (2002). Multiple congenital contractures: Birth prevalence, etiology, and outcome. The Journal of Pediatrics, 140(1), 61–67. [DOI] [PubMed] [Google Scholar]
- Domecq, J. P. , Prutsky, G. , Elraiyah, T. , Wang, Z. , Nabhan, M. , Shippee, N. , & Erwin, P. (2014). Patient engagement in research: A systematic review. BMC Health Services Research, 14(1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman, D. , & Coulombre, A. (1962). Experimental clubfoot and arthrogryposis multiplex congenita. The Lancet, 280(7255), 523–526. [DOI] [PubMed] [Google Scholar]
- Drummond, D. S. , & Mackenzie, D. A. (1978). Scoliosis in arthrogryposis multiplex congenita. Spine, 3(2), 146–151. [DOI] [PubMed] [Google Scholar]
- Emmanouil, D. E. , Roumani, T. , & Petsi, G. (2010). Arthrogryposis multiplex congenita: Dental findings and treatment of an 8‐year‐old child. Journal of Disability and Oral Health, 11(1), 32–36. [Google Scholar]
- Escobar, V. , Bixler, D. , Gleiser, S. , Weaver, D. D. , & Gibbs, T. (1978). Multiple pterygium syndrome. American Journal of Diseases of Children, 132(6), 609–611. [DOI] [PubMed] [Google Scholar]
- Evangelista, T. , Bansagi, B. , Pyle, A. , Griffin, H. , Douroudis, K. , Polvikoski, T. , … Lochmüller, H. (2015). Phenotypic variability of TRPV4 related neuropathies. Neuromuscular Disorders, 25(6), 516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsaperla, R. , Romeo, G. , Di Giorgio, A. , Pavone, P. , Parano, E. , & Connolly, A. M. (2001). Long‐term survival in a child with arthrogryposis multiplex congenita and spinal muscular atrophy. Journal of Child Neurology, 16(12), 934–936. [DOI] [PubMed] [Google Scholar]
- Farrell, K. , & McGillivray, B. C. (1983). Arthrogryposis following maternal hypotension. Developmental Medicine & Child Neurology, 25(5), 648–650. [DOI] [PubMed] [Google Scholar]
- Fuller, D. J. (1975). Immobilization of fetal joints: A cause of progressive prenatal deformity. Journal of Bone and Joint Surgery: British & European Volume, 57, 115. [Google Scholar]
- Gilhus, N. E. , & Hong, Y. (2018). Maternal myasthenia gravis represents a risk for the child through autoantibody transfer, immunosuppressive therapy and genetic influence. European Journal of Neurology, 25(12), 1402–1409. [DOI] [PubMed] [Google Scholar]
- Gordon Robertson, G. , Williamson, A. P. , & Blattner, R. J. (1955). A study of abnormalities in early chick embryos inoculated with Newcastle disease virus. Journal of Experimental Zoology, 129(1), 5–43. [Google Scholar]
- Greggi, T. , Martikos, K. , Pipitone, E. , Lolli, F. , Vommaro, F. , Maredi, E. , … Di Silvestre, M. (2010). Surgical treatment of scoliosis in a rare disease: Arthrogryposis. Scoliosis, 5(1), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotto, S. , Cuisset, J. M. , Marret, S. , Drunat, S. , Faure, P. , Audebert‐Bellanger, S. , … Journel, H. (2016). Type 0 spinal muscular atrophy: Further delineation of prenatal and postnatal features in 16 patients. Journal of Neuromuscular Diseases, 3(4), 487–495. [DOI] [PubMed] [Google Scholar]
- Hageman, G. , Ippel, E. P. , Beemer, F. A. , de Pater, J. M. , Lindhout, D. , & Willemse, J. (1988). The diagnostic management of newborns with congenital contractures: A nosologic study of 75 cases. American Journal of Medical Genetics, 30(4), 883–904. [DOI] [PubMed] [Google Scholar]
- Hageman, G. , Willemse, J. , Van Ketel, B. A. , & Verdonck, A. F. M. M. (1987). The pathogenesis of fetal hypokinesia. Neuropediatrics, 18(01), 22–33. [DOI] [PubMed] [Google Scholar]
- Hahn, G. (1985). Arthrogryposis. Pediatric review and habilitative aspects. Clinical Orthopaedics and Related Research, (194), 104–114. [PubMed] [Google Scholar]
- Haliloglu, G. , Becker, K. , Temucin, C. , Talim, B. , Küçükşahin, N. , Pergande, M. , … Cirak, S. (2017). Recessive PIEZO2 stop mutation causes distal arthrogryposis with distal muscle weakness, scoliosis and proprioception defects. Journal of Human Genetics, 62(4), 497. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (1981). An approach to congenital contractures (arthrogryposis). Pediatric Annals, 10(7), 15–26. [PubMed] [Google Scholar]
- Hall, J. G. (1984). The lethal multiple pterygium syndromes. American Journal of Medical Genetics, 17(4), 803–807. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (1985). Genetic aspects of arthrogryposis. Clinical Orthopaedics and Related Research, 194, 44–53. [PubMed] [Google Scholar]
- Hall, J. G. (1997). Arthrogryposis multiplex congenita: Etiology, genetics, classification, diagnostic approach, and general aspects. Journal of Pediatric Orthopaedics. Part B, 6(3), 159–166. [PubMed] [Google Scholar]
- Hall, J. G. (2009). Pena‐Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Research. Part A, Clinical and Molecular Teratology, 85(8), 677–694. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (2012). Arthrogryposis (multiple congenital contractures) associated with failed termination of pregnancy. American Journal of Medical Genetics. Part A, 158(9), 2214–2220. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (2014a). Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. European Journal of Medical Genetics, 57(8), 464–472. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (2014b). Oligohydramnios sequence revisited in relationship to arthrogryposis, with distinctive skin changes. American Journal of Medical Genetics. Part A, 164(11), 2775–2792. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. , Aldinger, K. A. , & Tanaka, K. I. (2014). Amyoplasia revisited. American Journal of Medical Genetics Part A, 164(3), 700–730. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. , & Kiefer, J. (2016). Arthrogryposis as a syndrome: Gene ontology analysis. Molecular Syndromology, 7(3), 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. G. , Kimber, E. , & van Bosse, H. J. (2017). Genetics and classifications. Journal of Pediatric Orthopaedics, 37, S4–S8. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. , Opitz, J. M. , & Reynolds, J. F. (1986). Analysis of Pena Shokeir phenotype. American Journal of Medical Genetics, 25(1), 99–117. [DOI] [PubMed] [Google Scholar]
- Hall, J. G. , & Reed, S. D. (1982). Teratogens associated with congenital contractures in humans and in animals. Teratology, 25(2), 173–191. [DOI] [PubMed] [Google Scholar]
- Hanif, A. , Rashid, H. , & Nasim, M. (2015). Tooth surface loss revisited: Classification, etiology, and management. Journal of Restorative Dentistry, 3(2), 37. [Google Scholar]
- Hengel, H. , Magee, A. , Mahanjah, M. , Vallat, J. M. , Ouvrier, R. , Abu‐Rashid, M. , … Bauer, P. (2017). CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurology Genetics, 3(2), e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron, L. D. , Westin, G. W. , & Dawson, E. G. (1978). Scoliosis in arthrogryposis multiplex congenita. The Journal of Bone and Joint Surgery. American Volume, 60(3), 293–299. [PubMed] [Google Scholar]
- Ho, C. A. , & Karol, L. A. (2008). The utility of knee releases in arthrogryposis. Journal of Pediatric Orthopaedics, 28(3), 307–313. [DOI] [PubMed] [Google Scholar]
- Hoff, J. M. , Loane, M. , Gilhus, N. E. , Rasmussen, S. , & Daltveit, A. K. (2011). Arthrogryposis multiplexa congenita: An epidemiologic study of nearly 9 million births in 24 EUROCAT registers. European Journal of Obstetrics & Gynecology and Reproductive Biology, 159(2), 347–350. [DOI] [PubMed] [Google Scholar]
- Huang, C. S. , Lin, S. P. , Chiu, N. C. , & Hung, H. Y. (2001). Congenital varicella syndrome as an unusual cause of congenital malformation: Report of one case. Acta Paediatrica Taiwan, 42(4), 239–242. [PubMed] [Google Scholar]
- Kiefer, J. , & Hall, J. G. (2019). Gene ontology analysis of Arthrogryposis (multiple congenital contractures). American Journal of Medical Genetics. Part C (In press). [DOI] [PubMed] [Google Scholar]
- Kimber, E. (2015). AMC: Amyoplasia and distal arthrogryposis. Journal of Children's Orthopaedics, 9(6), 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimber, E. , Tajsharghi, H. , Kroksmark, A. K. , Oldfors, A. , & Tulinius, M. (2012). Distal arthrogryposis: Clinical and genetic findings. Acta Paediatrica, 101(8), 877–887. [DOI] [PubMed] [Google Scholar]
- Komolkin, I. , Ulrich, E. V. , Agranovich, O. E. , & van Bosse, H. J. (2017). Treatment of scoliosis associated with arthrogryposis multiplex congenita. Journal of Pediatric Orthopaedics, 37, S24–S26. [DOI] [PubMed] [Google Scholar]
- Kowalczyk, B. , & Feluś, J. (2016). Arthrogryposis: An update on clinical aspects, etiology, and treatment strategies. Archives of Medical Science: AMS, 12(1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen, O. , & Hirvensalo, M. (1966). Arthrogryposis multiplex congenita. Annales Paediatriae Fenniae, 12(2), 133–138. [PubMed] [Google Scholar]
- Lowry, R. B. , Sibbald, B. , Bedard, T. , & Hall, J. G. (2010). Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(12), 1057–1061. [DOI] [PubMed] [Google Scholar]
- Masingue, M. , Fauré, J. , Solé, G. , Stojkovic, T. , & Léonard‐Louis, S. (2019). A novel nonsense PIEZO2 mutation in a family with scoliosis and proprioceptive defect. Neuromuscular Disorders, 29(1), 75–79. [DOI] [PubMed] [Google Scholar]
- Mennen, U. , van Heest, A. , Ezaki, M. B. , Tonkin, M. , & Gericke, G. (2005). Arthrogryposis multiplex congenita. Journal of Hand Surgery: British & European Volume, 30(5), 468–474. [DOI] [PubMed] [Google Scholar]
- Nouraei, H. , Sawatzky, B. , MacGillivray, M. , & Hall, J. (2017). Long‐term functional and mobility outcomes for individuals with arthrogryposis multiplex congenita. American Journal of Medical Genetics Part A, 173(5), 1270–1278. [DOI] [PubMed] [Google Scholar]
- Pakkasjärvi, N. , Ritvanen, A. , Herva, R. , Peltonen, L. , Kestilä, M. , & Ignatius, J. (2006). Lethal congenital contracture syndrome (LCCS) and other lethal arthrogryposes in Finland—An epidemiological study. American Journal of Medical Genetics Part A, 140(17), 1834–1839. [DOI] [PubMed] [Google Scholar]
- Perlman, J. M. , Burns, D. K. , Twickler, D. M. , & Weinberg, A. G. (1995). Fetal hypokinesia syndrome in the monochorionic pair of a triplet pregnancy secondary to severe disruptive cerebral injury. Pediatrics, 96(3), 521–523. [PubMed] [Google Scholar]
- Ravenscroft, G. , Sollis, E. , Charles, A. K. , North, K. N. , Baynam, G. , & Laing, N. G. (2011). Fetal akinesia: Review of the genetics of the neuromuscular causes. Journal of Medical Genetics, 48(12), 793–801. [DOI] [PubMed] [Google Scholar]
- Rodrigues, H. M. , Botelho, P. C. , Vaz, P. , Mesquita, P. , & Ponces, M. J. (2015). Arthrogryposis multiplex congenita associated with intraoral changes–multidisciplinary approach. Revista Portuguesa de Estomatologia, Medicina Dentária e Cirurgia Maxilofacial, 56(3), 182–187. [Google Scholar]
- Rosenbaum, P. , Paneth, N. , Leviton, A. , Goldstein, M. , Bax, M. , Damiano, D. , … Jacobsson, B. (2007). A report: The definition and classification of cerebral palsy April 2006. Developmental Medicine and Child Neurology, 109(Suppl 109), 8–14. [PubMed] [Google Scholar]
- Saito, Y. , Hayashi, M. , Miyazono, Y. , Shimogama, T. , & Ohno, K. (2006). Arthrogryposis multiplex congenita with callosal agenesis and dentato‐olivary dysplasia. Brain and Development, 28(4), 261–264. [DOI] [PubMed] [Google Scholar]
- Sanders, J. O. , D'Astous, J. , Fitzgerald, M. , Khoury, J. G. , Kishan, S. , & Sturm, P. F. (2009). Derotational casting for progressive infantile scoliosis. Journal of Pediatric Orthopedics, 29(6), 581–587. [DOI] [PubMed] [Google Scholar]
- Scoto, M. , Rossor, A. M. , Harms, M. B. , Cirak, S. , Calissano, M. , Robb, S. , … Fallon, P. (2015). Novel mutations expand the clinical spectrum of DYNC1H1‐associated spinal muscular atrophy. Neurology, 84(7), 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanis, K. C. , Payatakes, A. H. , Chouliaras, V. T. , Mandellos, G. C. , Pyrovolou, N. E. , Pliarchopoulou, F. M. , & Soucacos, P. N. (2007). Rare causes of scoliosis and spine deformity: Experience and particular features. Scoliosis, 2(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Staheli L. T., Hall J. G., Jaffe K. M., & Paholke D. O. (Eds.). (1998). Arthrogryposis: A text atlas. Cambridge University Press: UK. [Google Scholar]
- Steen, U. , Christensen, E. , & Samargian, A. (2017). Adults living with amyoplasia: Function, psychosocial aspects, and the benefit of AMC support groups. Journal of Pediatric Orthopaedics, 37, S31–S32. [DOI] [PubMed] [Google Scholar]
- Steen, U. , Wekre, L. L. , & Vollestad, N. K. (2018). Physical functioning and activities of daily living in adults with amyoplasia, the most common form of arthrogryposis. A cross‐sectional study. Disability and Rehabilitation, 40(23), 2767–2779. [DOI] [PubMed] [Google Scholar]
- Steinberg, B. , Nelson, V. S. , Feinberg, S. E. , & Calhoun, C. (1996). Incidence of maxillofacial involvement in arthrogryposis multiplex congenita. Journal of Oral and Maxillofacial Surgery, 54(8), 956–959. [DOI] [PubMed] [Google Scholar]
- Storbeck, M. , Eriksen, B. H. , Unger, A. , Hölker, I. , Aukrust, I. , Martínez‐Carrera, L. A. , … Houge, G. (2017). Phenotypic extremes of BICD2‐opathies: From lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. European Journal of Human Genetics, 25(9), 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinyard, C. A. , & Bleck, E. E. (1985). The etiology of arthrogryposis (multiple congenital contracture). Clinical Orthopaedics and Related Research, 194, 15–29. [PubMed] [Google Scholar]
- Thompson, R.M. , Foley, J. , Dias, L. , & Swaroop, V.T. (2019). Hip Status and Long‐term Functional Outcomes in Spina Bifida. J Pediatr Orthop. 39(3):e168–e172. [DOI] [PubMed] [Google Scholar]
- van der Linden, V. , Rolim Filho, E. L. , Lins, O. G. , van der Linden, A. , Aragão, M. D. F. V. V. , Brainer‐Lima, A. M. , … do Amaral, F. J. (2016). Congenital Zika syndrome with arthrogryposis: Retrospective case series study. BMJ, 354, i3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, A. , Newland, C. , Beeson, D. , Riemersma, S. , Newsom‐Davis, J. , Brueton, L. , & Huson, S. M. (1995). Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. The Lancet, 346(8966), 24–25. [DOI] [PubMed] [Google Scholar]
- Wallach, E. , Walther‐Louvier, U. , Espil‐Taris, C. , Rivier, F. , Baudou, E. , & Cances, C. (2018). Arthrogryposis in children: Etiological assessments and preparation of a protocol for etiological investigations. Archives de Pédiatrie, 25(5), 322–326. [DOI] [PubMed] [Google Scholar]
- Williams, J. , Mai, C. T. , Mulinare, J. , Isenburg, J. , Flood, T. J. , Ethen, M. , … Kirby, R. S. (2015). Updated estimates of neural tube defects prevented by mandatory folic acid fortification—United States, 1995–2011. MMWR. Morbidity and Mortality Weekly Report, 64(1), 1–5. [PMC free article] [PubMed] [Google Scholar]
- Wynne‐Davies, R. , Williams, P. F. , & O'Connor, J. C. (1981). The 1960s epidemic of arthrogryposis multiplex congenita: A survey from the United Kingdom, Australia and the United States of America. The Journal of Bone and Joint Surgery. British Volume, 63(1), 76–82. [DOI] [PubMed] [Google Scholar]
- Yoshioka, M. , Morisada, N. , Toyoshima, D. , Yoshimura, H. , Nishio, H. , Iijima, K. , … Kosaki, K. (2018). Novel BICD2 mutation in a Japanese family with autosomal dominant lower extremity‐predominant spinal muscular atrophy‐2. Brain and Development, 40(4), 343–347. [DOI] [PubMed] [Google Scholar]