

Abstract
The amount of plastic waste is continuously increasing. Besides conventional recycling, one solution to deal with this problem could be to use this waste as a resource for novel materials. In this study, polyesters are hydrogenated to give polyether polyols by using in situ‐generated Ru‐Triphos catalysts in combination with Lewis acids. The choice of Lewis acid and its concentration relative to the ruthenium catalyst are found to determine the selectivity of the reaction. Monitoring of the molecular weight during the reaction confirms a sequential mechanism in which the diols that are formed by hydrogenation are etherified to the polyethers. To probe the applicability of this tandem hydrogenation etherification approach, a range of polyester substrates is investigated. The oligoether products that form in these reactions have the chain lengths that are appropriate for application in the adhesives and coatings industries. This strategy makes polyether polyols accessible that are otherwise difficult to obtain from conventional fossil‐based feedstocks.
Keywords: hydrogenation, polymers, ruthenium, sustainable chemistry, waste prevention
Introduction
The world‐wide production volume of polymers is predicted to rise in the future as these materials are used in the manufacturing of an increasing number of consumer goods.1 As a result, there will be an increase in plastic waste. To counteract the pollution of the environment, as well as to conserve natural resources, several strategies have been proposed and developed for the recycling of polymeric materials.2 Depolymerization and mechanical recycling are among the most applied ones (Figure 1). In the first approach, some polymers can be converted back into their monomers by heating,3 hydrogenation,4 or hydrolysis.5 At the moment, this strategy is questionable from an economic point of view, given the low prices of conventional monomer feedstocks. In contrast, mechanical recycling is starting to be implemented on an industrial scale for PVC‐based products.6 However, there are disadvantages associated with this approach. The molecular weight will drop owing to chain scissions by thermal stress during extrusion or reaction with impurities may lead to inferior mechanical properties. The chemical conversion of one polymer into another could be a viable alternative recycling method. In addition, it could establish new routes to novel materials.
Figure 1.
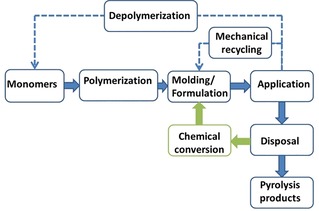
Possible polymer life cycles.
Herein we report on a tandem hydrogenation–acid‐catalyzed etherification strategy to synthesize oligoethers in suitable molecular weight ranges for use in adhesives from polyesters. Whereas in recent years many highly active and selective homogeneous catalysts for ester hydrogenation have been reported, most of these catalysts require base for their activation.7 Alternatively, they must contain an acid‐labile ligand, such as BH4 − or ‐CH2Si(Me)3,8 which also makes them useless for hydrogenation under acidic conditions. To date, the only homogeneous hydrogenation catalysts known to tolerate acidic conditions are bipyridine complexes of rhodium or iridium9 and combinations of ruthenium10 or cobalt11 with a Triphos ligand. In particular the Triphos‐based metal complexes have achieved much attention, owing to their broad applicability in reactions including the reduction of esters12 and of CO2,13 hydrogenation of lactams to cyclic amines,14 hydrogenolysis of polyesters to diols4c and direct reductive etherification of carboxylic acid esters to ethers (Scheme 1).15 This versatility and their stability towards acids and high temperatures resulted in our decision to investigate this system for the chemical recycling of polyesters to polyether oligomers.
Scheme 1.

Previous work on reductive etherification using hydrogen as reductant (met=2‐methylallyl).
Results and Discussion
To achieve realistic conditions, industrial grade poly(hexene 1,12‐dodecanate) (PHDD; Figure 2) was used as a model substrate. Initially, a solvent screening was conducted (Table 1). Toluene and cyclohexene were chosen as apolar solvents. Protic solvents were excluded as they would react with the potential intermediates. Diethyl ether, tetrahydrofuran (THF), and 1,4‐dioxane were employed as polar aprotic solvents. Although not a green solvent, chloroform was included in the screening as it is known to positively affect selectivity and rate in reductive etherification via hydrosilylation.16
Figure 2.
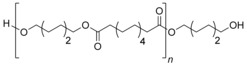
Structure of poly(hexene‐1,12‐dodecanate) (PHDD).
Table 1.
Solvent screening for the reduction of PHDD to the polyether.[a]

| Entry | Solvent | Conv.[b,c] [%] | Yield[b,d] [%] | Sel.[b] [%] |
|---|---|---|---|---|
| 1 | Et2O | 33 | 0 | 0 |
| 2 | THF | 44 | 37 | 84 |
| 3 | cyclohexane | 40 | 2 | 4 |
| 4 | CHCl3 | 24 | 2 | 9 |
| 5 | 1,4‐dioxane | 38 | 17 | 46 |
| 6 | toluene | 17 | 0 | 0 |
[a] Conditions: PHDD (200 mg, 1.28 mmol COOR groups), Ru(acac)3 (5 mg, 13 μmol, 1 mol %), Triphos ligand (12 mg, 20 μmol, 1.5 mol %), Al(OTf)3 (15 mg, 33 μmol, 2.5 mol %), solvent (2 mL), 140 °C, 24 h. [b] Calculated from NMR spectra. [c] Conversion of COOR. [d] Yield of R−CH2OCH2−R groups.
In all solvents, conversion of the ester groups of the polymer was observed. However only in 1,4‐dioxane and THF a significant formation of ether bonds was observed. In THF the selectivity was twice as high as in dioxane at nearly the same conversion. Based on this, THF was chosen as the solvent for further experiments. The effect of the different metal triflates as Lewis acid co‐catalysts was also studied (Figure 3). All of the investigated metal triflates were found to facilitate the hydrogenation of the ester moieties.
Figure 3.
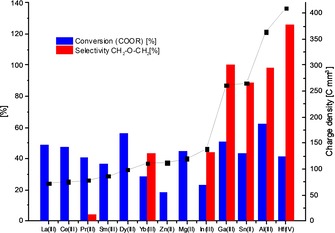
Effect of different metal triflates on the conversion and selectivity of the reductive etherification of PHDD compared to their corresponding estimated charge densities. Reaction conditions: PHDD (200 mg, 1.28 mmol COOR groups), Ru(acac)3 (1 mol %), Triphos (1.5 mol %), Lewis acid (2.5 mol %), THF (2 mL), T=140 °C, 40 bar H2, t=16 h.
The role of the Lewis acid is twofold: It provides protons from the reaction with water impurities, which are thought to be crucial for the activation of the catalytic system.15a In addition, the Lewis acid is needed to catalyze the etherification reaction (Scheme 2, pathway I; see also the Supporting Information, Table S4). The selectivity for ether bond formation seems to correlate with the charge density, which can be estimated from the charge number and the respective ionic radii.17 In addition, there seems to be a certain threshold below which no etherification is possible. The highest selectivities were obtained with GaIII, SnII, and AlIII triflates. Curiously, addition of HfIV triflate results in a selectivity for ether formation of over 100 %. This finding can be rationalized by the capability of HfIV triflate to open the THF ring at higher temperatures, which, in combination with the reducing conditions, results in the formation of n‐butyl ethers alongside n‐alkanes, as reported by Marks and co‐workers.18
Scheme 2.
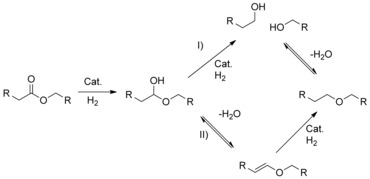
Possible reaction pathways for the formation of oligoethers from polyesters.
To exclude the possibility that THF ring‐opening by aluminum triflate is the reason behind the observed high selectivities, we performed a control experiment. Heating THF together with Al(OTf)3 to 140 °C for 24 h did not yield any oligomer of THF or other ethers (see the Supporting Information for details). Having identified a selective system with Al(OTf)3, we further attempted to increase the conversion by varying the aluminum triflate/ruthenium ratios. Interestingly, at higher ruthenium loadings (1 mol %), increasing the Al(OTf)3 content led to lower conversions (Table 2, entries 2 and 5), whereas at lower ruthenium loadings a large excess of Al(OTf)3 is beneficial and increased the conversion (Table 2, entries 3 and 4). When a 1:1 ratio of Ru(acac)3 and Al(OTf)3 was used, no ether linkages were formed, although full conversion occurred. The reaction rather seemed to yield only the free diols, since no oligomers were detected by gel permeation chromatography (GPC; Table 2, entry 8). This indicates that a certain Lewis acid concentration is necessary to facilitate etherification of the free alcohol groups formed by the hydrogenation.
Table 2.
Effect of different Al/Ru‐ratios.[a]
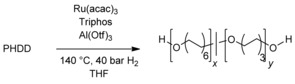
| Entry | Al/Ru | Ru[b]
[mol %] |
Conv. [%][c] |
Sel. [%] |
Mw
[d]
[g mol−1] |
M
n
[d]
[g mol−1] |
|---|---|---|---|---|---|---|
| 1 | 2.5 | 3 | 91 | 92 | 1200 | 600 |
| 2 | 2.5 | 1 | 62 | 98 | 2200 | 800 |
| 3 | 10 | 0.25 | 33 | 73 | 2250 | 1500 |
| 4 | 20 | 0.125 | 62 | 100 | 2400 | 1400 |
| 5 | 15 | 1 | 28 | 41 | 2100 | 1300 |
| 6 | 7.5 | 1 | 49 | 100 | 2400 | 1400 |
| 7 | 2.5 | 1 | 58 | 100 | 2700 | 1600 |
| 8 | 1 | 1 | >99 | 0 | n.d.[e] | n.d.[e] |
[a] Conditions: PHDD (200 mg, 1.28 mmol COOR), Ru(acac)3 (5 mg, 13 μmol, 1 mol %), Triphos ligand (12 mg, 20 μmol, 1.5 mol %), Al(OTf)3 (15 mg, 33 μmol, 2.5 mol %), solvent (2 mL), 140 °C, 24 h. [b] w.r.t. amount of COOR groups in the polyester. [c] Calculated from NMR spectra. [d] Determined by GPC. [e] No oligomers were detected inside the calibration range of the GPC method
Thus, the reaction proceeds according to a tandem hydrogenation–etherification mechanism (Scheme 2, pathway I), consistent with the observations reported by Gooßen and co‐workers (Scheme 1).15b However, a mechanism whereby etherification occurs through acid‐catalyzed elimination of water from the hemiacetal intermediate (Scheme 2, pathway II) was also deemed plausible by Beller and co‐workers.15a
To further verify which hypothesis is correct for this reaction, we monitored the molecular weight of the resulting polyester/polyether mixture during the course of the reaction. At the start of the reaction, a steep drop in the molecular weight of the substrate was detected (Figure 4), in line with the formation of the mixture of diols. Over the course of the reaction, the formed alcohols were slowly etherified to give polyether oligomers. The polycondensation of diols with Brønsted or Lewis acids is a known method to obtain polyethers that cannot be obtained by ring‐opening polymerization of cyclic ethers.19 Additional proof was obtained in a control experiment in which 1,12‐dodecanediol was treated with Al(OTf)3 at 140 °C. After 16 h, complete conversion was achieved and oligomers were obtained with a M w of 1850–2000, as indicated by GPC analysis (see the Supporting Information for details).
Figure 4.
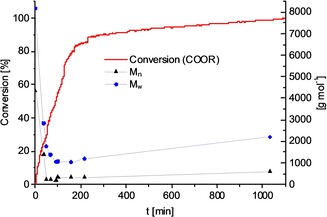
Molecular weight over time in the hydrogenation of PHDD. Conditions: Polymer (3.0 g), THF (30 mL), Ru(acac)3 (3 mol %), Triphos (4.5 mol %), Al(OTf)3 (7.5 mol %), T=140 °C, 60 bar H2.
As expected, switching the setup from simple vials to a mechanically stirred autoclave resulted in the reduction of all ester groups in a reasonable period of time. To get more insight into the generality of this approach, we subjected several other polyesters (Figure 5) to the reaction conditions.
Figure 5.

Other polyesters subjected to the hydrogenation procedure. PEGP=poly[(2‐(ethoxy)ethyl)phthalate]; PHA=poly(hexyl‐1,6‐adipate); PBA=poly(butyl‐1,4‐adipate); C36‐co‐PHA=poly(hexene‐1,6‐adipate‐co‐distearate).
Table 3 shows a comparison of the polyether molecular weights compared to the molecular weights of the starting polyesters. From the results obtained, it is clear that the catalytic protocol also works for other commercially available polyesters. The polyether polyols were obtained with molecular weights suitable for application in adhesives. It was not possible to obtain products with higher molecular weights, owing to the presence of water, which forms in the etherification reaction and cannot be removed, leading to an equilibrium with relatively low‐M w oligoethers. We tried adding drying agents to the hydrogenation reactions, but these did not have the desired effect.
Table 3.
Results of the hydrogenation/re‐etherification of the polyesters shown in Figure 5.[a]
| Entry | Polyester |
M
n(GPC)[b]
[g mol−1] |
M
w(GPC)[b]
[g mol−1] |
M
n(GPC)[c]
[g mol−1] |
M
w(GPC)[c]
[g mol−1] |
M
n(NMR)[c,d]
[g mol−1] |
Conv.[e] [%] | Yield[f] [%] |
|---|---|---|---|---|---|---|---|---|
| 1 | PHDD | 4200 | 13 000 | 600 | 2000 | 614 | >99 | 80 |
| 2[g] | PHA | 3000 | 13 000 | 900 | 2100 | 892 | 96 | 90 |
| 3 | PEGP | 3200 | 13 000 | n.d. | n.d. | n.d. | 49[i] | n.a. |
| 4[h] | PBA | 4200 | 11 000 | 700 | 1100 | 563 | >99 | >99 |
| 5 | C36‐co‐PHA | 2400 | 4000 | 1500 | 2800 | 1000 | >99 | 80 |
| 6[j] | PHA+1,4‐BDM | – | – | 1100 | 2000 | 1238 | >99 | 89 |
[a] Conditions (unless otherwise stated): Polymer (3.0 g), Ru(acac)3 (3 mol %), Triphos (4.5 mol %), Al(OTf)3 (7.5 mol %), THF (30 mL), 40 bar H2 at RT, 140 °C, 24 h. [b] Determined by GPC with THF as eluent, before hydrogenation of the polyester sample. [c] Measured after hydrogenation. [d] Calculated from NMR spectra by end group analysis. [e] Conversion of COOR groups as analyzed by NMR spectroscopy. [f] Calculated w.r.t. the theoretical conversion of all ester groups into ether groups. [g] T=180 °C; [h] Ru(acac)3 (2 mol %), Triphos (3 mol %), Al(OTf)3 (5 mol %). [i] Determined from hydrogen consumption. [j] 1,4‐benzene dimethanol (22 mol %) was added; yield based only on the ether groups obtained by the conversion of original ether groups.
Notably, industrial grade polyesters were used without purification, except drying under vacuum. Poly[(2‐(ethoxy)ethyl)phthalate] (PEGP), in contrast, was not fully reduced and yielded a complicated mixture of polyesters and polyethers (Table 3, entry 3). Polyethylene terephthalate (PET) was also subjected to the procedure but no conversion of the ester groups occurred. The most likely reason for the absence of reactivity is the insolubility of this polymer under the reaction conditions. However, a polyester polyol based on a dimer of a fatty acid could be easily reduced to a branched polyether structure (Table 3, entry 5). Integration of the peaks in the aliphatic region of the 1H NMR spectra clearly shows the absence of THF incorporation in all obtained polyether polyols. To provide further evidence for the stepwise mechanism, we performed an experiment in which an exogenous diol was added, which, according to the mechanism, should be inserted into the polyether chain. Thus, 1,4‐benzene dimethanol (1,4‐BDM) was added as co‐substrate to the hydrogenation reaction of PHA (Table 3, entry 6). 1,4‐BDM is a suitable probe for this since there should be no 1H NMR signal overlap between its free diol or ether form with the formed aliphatic polyether. PHA was chosen as polyester substrate to further simplify the NMR analysis of the product. Analysis of the 1H NMR spectrum revealed that 8–10 % of the added diol was incorporated in the oligomer, which had a slightly increased molecular weight of 1238 g mol−1 compared to the other products in Table 3. Further verification was provided by converting the free hydroxy groups into the corresponding trifluoroacetates, which did not result in any significant change with respect to the chemical shift of the signal assigned to the benzylic protons of the 1,4‐BDM ether fragment. This signal consisted of two peaks indicating the presence of bonds of the 1,4‐BDM to each other as well as to the 1,6‐hexandiol fragments (Figure 6). Thus, if different diols are present in the reaction mixture they are, as expected, randomly incorporated in the oligomer chain.
Figure 6.
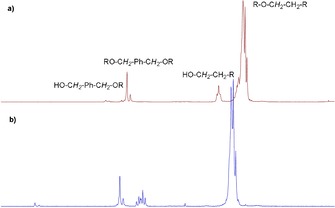
1H NMR spectra (3.0–5.7 ppm) of the oligoether described in Table 3, entry 6: a) before and b) after the addition of excess trifluoroacetic acid anhydride.
The molecular weight of these polyether polyols obtained from end‐group analysis is in agreement with the GPC‐derived M n value. This is a strong indication that the content of cyclic oligomers is low. The reduced molecular weight of the obtained polyethers is still in the range of 400–2000 g mol−1, as for other common polyols in polyurethane‐based adhesives.20 Their usefulness was further verified by adding 1.1 equivalents of 2,4‐toluene diisocyanate to the polyether obtained in entry 2 of Table 3, followed by moisture curing under ambient conditions. This procedure yielded a polyurethane film with a leather like haptic. Figure S2 shows this film which is flexible but still tightly engulfs a PTFE stirring bar. DSC measurements showed that poly[oxy(hexane‐1,6‐diyl)] obtained by the reduction of poly(hexene‐1,6‐adipate) retained its crystallinity. However the melting range was reduced from 45–50 °C (PHA) to 14–17 °C (the hydrogenated product). This can be seen as an advantage of the widely used poly‐THF polyether that melts at 28 °C, which usually requires heated distribution pipes and storage tanks.
Conclusions
In conclusion, we have shown that a catalyst combination consisting of Ru(acac)3, Triphos and Al(OTf)3 can perform the hydrogenolysis of polyesters to polyethers. It was also possible to replace aluminum triflate with gallium or tin triflate. Control experiments supported a tandem hydrogenation etherification pathway. The reaction worked well with linear aliphatic polyesters. The polyether products could be converted into polyurethanes, opening up the possibility of adding novel polyols to the polyurethane chemist's toolbox. In addition, it provides an alternative to established hydrolysis and hydrogenolysis approaches for the recycling of polyesters. Although the molecular weight is reduced compared to the starting polyesters, the MW range is right for use in polyurethane‐based adhesives.
Experimental Section
Hydrogenation and re‐etherification of polyesters (Table 3)
PHDD (3 g; n(COOR)=20 mmol) was added to a 100 mL Hastelloy autoclave equipped with a mechanical stirrer, followed by Ru(acac)3 (240 mg, 3 mol %), Triphos (581 mg, 5 mol %), and Al(OTf)3 (735 mg, 7.5 mol %). The reactor was then subjected to three vacuum–argon cycles. Next, THF (30 mL) was added to the reactor while having the latter under a flow of argon. Then the reactor was sealed, purged two times with N2 (10 bar), pressurized with H2 (40 bar), and heated to 140 °C. The temperature was controlled by a thermocouple inside the reactor. During the next 16 h, the pressure was recorded by a digital pressure read‐out. After approximately 120 min, extra pressure of H2 was added again to keep the pressure at 40–50 bar. Samples (100 μL) were taken according to the observed speed of pressure drop; these were filtered over silica, diluted with THF (ca. 1000 μL) and analyzed by GPC. Table S2 contains the GPC results of the reaction of PHDD at 140 °C. Figures S1 and S2 show the pressure, temperature, and calculated conversion over the course of the reaction. The polyether products were isolated by removing the solvent under reduced pressure followed by dissolution in toluene and filtration through silica to remove the residual catalyst and Lewis acid.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank E. Gerova‐Martin for her invaluable analytical support, as well as Dr. A. Brandt, Dr. A. Taden, and Dr. H. Beck from the AR‐Bio‐renewables at Henkel AG & Co. KGaA for the kind gift of polyester samples. Prof. Dr. M. Beller is thanked for helpful discussions. This work was funded by the European Union's Horizon 2020 research and innovation program, as part of the Bio‐Based Industries program under grant agreement No 720695.
B. M. Stadler, S. Hinze, S. Tin, J. G. de Vries, ChemSusChem 2019, 12, 4082.
References
- 1. Brems A., Baeyens J., Dewil R., Therm. Sci. 2012, 16, 669–685. [Google Scholar]
- 2. Ignatyev I. A., Thielemans W., Vander Beke B., ChemSusChem 2014, 7, 1579–1593. [DOI] [PubMed] [Google Scholar]
- 3. Yamaguchi T., Watanabe S., Shimada Y., Chemosphere 1973, 2, 7–10. [Google Scholar]
- 4.
- 4a. Braun D., W. von Gentzkow , Rudolf A. P., Polym. Degrad. Stab. 2001, 74, 25–32; [Google Scholar]
- 4b. Fuentes J. A., Smith S. M., Scharbert M. T., Carpenter I., Cordes D. B., Slawin A. M., Clarke M. L., Chem. Eur. J. 2015, 21, 10851–10860; [DOI] [PubMed] [Google Scholar]
- 4c. Westhues S., Idel J., Klankermayer J., Sci Adv 2018, 4, eaat9669; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Farrar-Tobar R. A., Wozniak B., Savini A., Hinze S., Tin S., J. G. de Vries , Angew. Chem. Int. Ed. 2019, 58, 1129–1133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1141–1145. [Google Scholar]
- 5. Tokiwa Y., Suzuki T., Nature 1977, 270, 76. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Janajreh I., Alshrah M., Zamzam S., Sustainable Cities Soc. 2015, 18, 13–20; [Google Scholar]
- 6b. Ciacci L., Passarini F., Vassura I., Resour. Conserv. Recycl. 2017, 123, 108–116. [Google Scholar]
- 7.
- 7a. Zhang J., Leitus G., Ben-David Y., Milstein D., Angew. Chem. Int. Ed. 2006, 45, 1113–1115; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1131–1133; [Google Scholar]
- 7b. Kuriyama W., Matsumoto T., Ogata O., Ino Y., Aoki K., Tanaka S., Ishida K., Kobayashi T., Sayo N., Saito T., Org. Process Res. Dev. 2012, 16, 166–171; [Google Scholar]
- 7c. O W. N., Morris R. H., ACS Catal. 2013, 3, 32–40; [Google Scholar]
- 7d. Spasyuk D., Smith S., Gusev D. G., Angew. Chem. Int. Ed. 2013, 52, 2538–2542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2598–2602; [Google Scholar]
- 7e. Werkmeister S., Junge K., Wendt B., Alberico E., Jiao H., Baumann W., Junge H., Gallou F., Beller M., Angew. Chem. Int. Ed. 2014, 53, 8722–8726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8867–8871; [Google Scholar]
- 7f. Tan X., Wang Y., Liu Y., Wang F., Shi L., Lee K.-H., Lin Z., Lv H., Zhang X., Org. Lett. 2015, 17, 454–457; [DOI] [PubMed] [Google Scholar]
- 7g. Dub P. A., Scott B. L., Gordon J. C., Organometallics 2015, 34, 4464–4479; [Google Scholar]
- 7h. Stadler B. M., Puylaert P., Diekamp J., R. van Heck , Fan Y., Spannenberg A., Hinze S., J. G. de Vries , Adv. Synth. Catal. 2018, 360, 1151–1158; [Google Scholar]
- 7i. Srimani D., Mukherjee A., Goldberg A. F. G., Leitus G., Diskin-Posner Y., Shimon L. J. W., Ben David Y., Milstein D., Angew. Chem. Int. Ed. 2015, 54, 12357–12360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12534–12537. [Google Scholar]
- 8. Yuwen J., Chakraborty S., Brennessel W. W., Jones W. D., ACS Catal. 2017, 7, 3735–3740. [Google Scholar]
- 9. Brewster T. P., Miller A. J., Heinekey D. M., Goldberg K. I., J. Am. Chem. Soc. 2013, 135, 16022–16025. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. T. vom Stein , Meuresch M., Limper D., Schmitz M., Holscher M., Coetzee J., Cole-Hamilton D. J., Klankermayer J., Leitner W., J. Am. Chem. Soc. 2014, 136, 13217–13225; [DOI] [PubMed] [Google Scholar]
- 10b. Cui X., Li Y., Topf C., Junge K., Beller M., Angew. Chem. Int. Ed. 2015, 54, 10596–10599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10742–10745; [Google Scholar]
- 10c. Latifi E., Marchese A. D., Hulls M. C. W., Soldatov D. V., Schlaf M., Green Chem. 2017, 19, 4666–4679. [Google Scholar]
- 11. Korstanje T. J., J. I. van der Vlugt , Elsevier C. J., B. de Bruin , Science 2015, 350, 298–302. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Teunissen H. T., Elsevier C. J., Chem. Commun. 1998, 1367–1368; [Google Scholar]
- 12b. M. C. van Engelen , Teunissen H. T., J. G. de Vries , Elsevier C. J., J. Mol. Catal. A 2003, 206, 185–192. [Google Scholar]
- 13. Wesselbaum S., T. vom Stein , Klankermayer J., Leitner W., Angew. Chem. Int. Ed. 2012, 51, 7499–7502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7617–7620. [Google Scholar]
- 14. Meuresch M., Westhues S., Leitner W., Klankermayer J., Angew. Chem. Int. Ed. 2016, 55, 1392–1395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1414–1417. [Google Scholar]
- 15.
- 15a. Li Y., Topf C., Cui X., Junge K., Beller M., Angew. Chem. Int. Ed. 2015, 54, 5196–5200; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5285–5289; [Google Scholar]
- 15b. Erb B., Risto E., Wendling T., Goossen L. J., ChemSusChem 2016, 9, 1442–1448. [DOI] [PubMed] [Google Scholar]
- 16. Biermann U., Metzger J. O., ChemSusChem 2014, 7, 644–649. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Shannon R. D., Acta Crystallogr. Sect. A 1976, 32, 751–767; [Google Scholar]
- 17b. Rayner-Canham G., Overton T., Descriptive Inorganic Chemistry, 5th ed., W. H. Freeman, New York, 2010. [Google Scholar]
- 18. Li Z., Assary R. S., Atesin A. C., Curtiss L. A., Marks T. J., J. Am. Chem. Soc. 2014, 136, 104–107. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Rhoad M. J., Flory P. J., J. Am. Chem. Soc. 1950, 72, 2216–2219; [Google Scholar]
- 19b. Kobayashi S., Tadokoro H., Chatani Y., Macromol. Chem. Phys. 1968, 112, 225–241; [Google Scholar]
- 19c. Zhang S., Feret A., Lefebvre H., Tessier M., Fradet A., Chem. Commun. 2011, 47, 11092–11094. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Somani K. P., Kansara S. S., Patel N. K., Rakshit A. K., Int. J. Adhes. Adhes. 2003, 23, 269–275; [Google Scholar]
- 20b. Rahman M. M., Kim H.-D., J. Appl. Polym. Sci. 2006, 102, 5684–5691; [Google Scholar]
- 20c. Šebenik U., Krajnc M., Int. J. Adhes. Adhes. 2007, 27, 527–535. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary