

Abstract
Despite its anticipated clinical potential, anti‐PD‐1 immunotherapy has only yielded poor outcomes in recent clinical trials for glioblastoma patients. Strategies combining anti‐PD‐1 antibody with other treatment modalities are being explored to alter the immunosuppressive microenvironment that appears to characterize these anti‐PD‐1‐insensitive tumors. Here, we evaluated whether introducing wild‐type p53 gene via a tumor‐targeting nanomedicine (termed SGT‐53) could provide immune stimulation and augment anti‐PD‐1 therapy in mouse syngeneic GL261 tumor models (either subcutaneous or intracranial). In both models, anti‐PD‐1 monotherapy had no demonstrable therapeutic effect. However, combining anti‐PD‐1 with our investigational nanomedicine SGT‐53 was very effective in inhibiting tumor growth, inducing tumor cell apoptosis and increasing intratumoral T‐cell infiltration. A significant survival benefit was observed in mice bearing intracranial glioblastoma receiving combination treatment. Importantly, SGT‐53 upregulated PD‐L1 expression both in vitro and in vivo. Transcriptome analysis revealed modulation of genes linked to either cancer progression or immune activation after combination treatment. Our data suggest that SGT‐53 can boost antitumor immunity and sensitize glioblastoma to anti‐PD‐1 therapy by converting immunologically “cold” tumors into “hot” tumors. Combining SGT‐53 with anti‐PD‐1 might benefit more patients from anti‐PD‐1 immunotherapy and our data support evaluation of this combination in patients with glioblastoma.
Keywords: anti‐PD‐1, glioblastoma, nanomedicine, p53, tumor‐targeted delivery
Short abstract
What's new?
Antibodies targeting the programmed cell death protein 1 (anti‐PD‐1) pathway represent a promising immunotherapeutic strategy against glioblastoma. Nonetheless, in recent trials, significant numbers of glioblastoma patients failed to respond to anti‐PD‐1. Here, the authors explored the possibility of overcoming this insensitivity by combining anti‐PD‐1 with SGT‐53, a tumor‐targeting nanomedicine that restores p53 function. In glioblastoma mouse models, the combined immunotherapy approach effectively inhibited tumor growth and increased intratumoral infiltration of immune effector cells. Increased immunogenicity was associated with significant survival benefits. The data provide a strong mechanistic rationale for combining SGT‐53 nanomedicine and anti‐PD‐1 immunotherapy in the treatment of glioblastoma.
Abbreviations
- ARG1
arginase 1
- BBB
blood–brain barrier
- Casp3
active caspase 3
- CRT
calreticulin
- CTL
cytotoxic T lymphocyte
- CTLA‐4
cytotoxic T lymphocyte‐associated antigen 4
- FoxP3
forkhead box P3
- GzmB
granzyme B
- ICD
immunogenic cell death
- ICI
immune checkpoint inhibitor
- NOS2
nitric oxide synthase 2
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand 1
- scFv
single chain antibody fragment
- scL
single chain liposome
- TAM
tumor‐associated macrophage
- TfR
transferrin receptor
- Treg
regulatory T‐cell
- wtp53
wild‐type TP53
Introduction
Glioblastoma is the most aggressive and lethal brain tumors in adults.1 Conventional therapies, including surgery, radiotherapy and chemotherapy, have not resulted in major improvements in the survival outcomes of patients with glioblastoma.2 Intrinsic unresponsiveness and acquired drug resistance coupled with restricted accessibility of brain tumors to drugs tied to the impermeability of the blood–brain barrier (BBB) may all contribute to the treatment failure. In addition, glioblastoma has developed an array of strategies to evade/suppress the antitumor immune responses, which also likely contribute to the failure.3 Therefore, novel therapeutic approaches are greatly needed to improve the outcomes currently seen with conventional therapies.
In recent years, immune checkpoint blockade has come to the forefront of cancer immunotherapies as a powerful and promising strategy to stimulate antitumor T‐cell responses.4, 5 Multiple clinical trials have demonstrated significant response rates with immune checkpoint antibodies that target the cytotoxic T lymphocyte‐associated antigen 4 (anti‐CTLA‐4) or programmed cell death protein 1 pathway (anti‐PD‐1/PD‐L1) in patients with late‐stage melanoma and lung cancer.6, 7 However, a significant number of patients either did not respond or developed resistance to immune checkpoint inhibitors (ICIs).8 Several ICIs are currently undergoing clinical development for treatment of primary and recurrent brain tumors, but results to date have disappointed.2, 5 A large number of trials are underway that combine checkpoint blockade with a wide range of therapeutic agents (i.e., chemotherapy and/or radiation therapy) seeking synergy that might lead to enhancements in inhibition of tumor growth and overall survival.9, 10
Our approach to improve ICIs focuses on the restoration of p53 function via tumor‐targeting nanomedicine delivering the tumor suppressor gene TP53 (termed SGT‐53, a.k.a. scL‐p53). The p53 protein has a diverse range of functions including regulation of cell cycle arrest and apoptosis in response to genotoxic and oncogenic stresses. Inactivation of p53 is a critical and early event in tumorigenesis and p53‐null mice are highly predisposed to cancer development.11 Loss of p53 function is present in the majority of human cancers12 and is often associated with therapeutic unresponsiveness and poor clinical outcomes.13 A dysfunctional p53 pathway is particularly prevalent in CNS malignancies and confers a poor prognosis. Thus, restoring p53 function has long been recognized as an attractive cancer therapeutic strategy.14 Importantly, recent studies suggest that p53 participates in immune regulation and p53 pathway can be exploited to alter the immunological landscape of tumors.15, 16 Accordingly, we hypothesized that introducing functional p53 via SGT‐53 treatment would augment antitumor immunity and improve the efficacy of anti‐PD‐1 therapy for glioblastoma.
Here, we have investigated our hypothesis using the investigational agent SGT‐53 in combination with anti‐PD‐1 antibody in syngeneic mouse models of glioblastoma. SGT‐53 is a novel cationic liposome encapsulating a plasmid for human wild‐type TP53 (wtp53). The surface of liposome is decorated with a single chain antibody fragment recognizing the transferrin receptor (TfRscFv) that targets cancer cells overexpressing TfR with exquisite specificity.17 Moreover, this delivery system termed scL (for single chain liposome) can actively ferry payloads across the BBB by virtue of the elevated expression of TfRs on cerebral endothelium and receptor‐mediated transcytosis.17 This approach, that is, systemic administration of SGT‐53 nanocomplex as an anticancer nanomedicine, has been translated into completed Phase I clinical trials18, 19 and is currently in multiple Phase II studies including a trial for glioblastoma, a disease wherein a therapeutic agent that actively traverses the BBB would clearly be advantageous. Here, we show that the combination of SGT‐53 and anti‐PD‐1 resulted in a significant inhibition of tumor growth with a survival benefit conferred compared to either agent individually. The synergy between SGT‐53 and anti‐PD‐1 was observed in both subcutaneous and intracranial GL261 syngeneic tumor models. SGT‐53 treatment induced immunogenic changes in tumor cells and increased infiltration of the tumors by immune‐relevant cells leading to an enhancement of both innate and adaptive immune responses when used in combination with anti‐PD‐1.
Materials and Methods
Cell lines
Mouse glioblastoma cell line GL261 (RRID:CVCL_Y003) was obtained from the Tumor Repository at NCI (Bethesda, MD). Mouse macrophage cell line Raw 264.7 (RRID:CVCL_0493) was obtained from ATCC (Manassas, VA). Cells were maintained at 37°C in a 5% CO2 in DMEM (Mediatech, Manassas, VA) supplemented with 10% FBS (Sigma, St. Louis, MO). All experiments were performed with mycoplasma‐free cells.
SGT‐53 nanocomplex preparation
Cationic liposome consisting of 1,2‐dioleoyl‐3‐trimethylammonium propane and dioleolylphosphatidyl ethanolamine (Avanti Polar Lipids, Alabama, AL), referred to as Lip, was prepared as described previously.20 TfRscFv/Lip/p53 (scL immunoliposome nanocomplex encapsulating p53 plasmid DNA, a.k.a. SGT‐53) was prepared as previously described.20 For in vitro transfection, SGT‐53 was diluted in serum‐free media and added to culture (10 pg DNA/cell). For animal injections, 5% dextrose was added to SGT‐53.
In vivo studies
All animal experiments were performed in accordance with Georgetown University GUACUC protocols. For the subcutaneous tumor model, 6 week old female C57BL/6NHsd mice (Envigo, Frederick, MD) were inoculated on their flanks with GL261 cells (1.0 × 106 cells/mouse). For the intracranial tumor model, 6‐week‐old female albino C57BL/6 mice (B6N‐Tyr c‐Brd/BrdCrCrl, Charles River Laboratories, Frederick, MD) were stereotactically inoculated with GL261 cells (2.0 × 105 cells/mouse) as described previously.21 Mice with established tumors were injected twice weekly with either SGT‐53 (30 μg DNA/mouse, intravenously administered), anti‐PD‐1 antibody (RMP1‐14, BioXCell, West Lebanon, NH; 200 μg/mouse, intraperitoneally administered) or the combination of both agents.
Immunohistochemistry
Harvested tumors were fixed in formalin and embedded in paraffin. Tumor sections were stained for active caspase‐3 (Casp3, Cell Signaling Technology, Danvers, MA), granzyme B (GzmB, Abcam, Cambridge, MA), forkhead box P3 (FoxP3, eBioscience, San Diego, CA), nitric oxide synthase 2 (NOS2, ThermoFisher, Waltham, MA), or Arginase 1 (ARG1, ThermoFisher) antibodies. Captured images were analyzed using IHC Profiler plugin in ImageJ.
Flow cytometry
Tumor cells were dissociated using Tumor Dissociation Kit and gentleMACS Octo Dissociator (Miltenyi Biotec, San Diego, CA), labeled with Zombie‐NIR viability dye (BioLegend, San Diego, CA), stained with antibodies and analyzed on LSRFortessa flow cytometer (BD Biosciences, Boston, MA).
RT‐qPCR
Total RNAs were extracted using PureLink RNA Mini Kit (Ambion, Foster City, CA) and reverse transcribed using Superscript IV (Life Technologies, San Francisco, CA). PCR was performed in triplicate using TaqMan assays (Life Technologies). Relative mRNA expression was analyzed using StepOne Software v2.3 via the ΔΔCt method with normalization to Gapdh.
NanoString analysis
Two commercially available gene panels (PanCancer Pathways and Immune Profiler) containing total 1,330 unique genes were used. RNA was hybridized with probes and counted on nCounter analyzer (NanoString Technologies, Seattle, WA). Raw data were analyzed using nSolver 3.0 software (NanoString Technologies).
Statistical analysis
Presented data represent mean ± the standard error of the mean (SEM). Statistical significance was determined by one‐way ANOVA or by t‐tests. Values of p < 0.05 were considered significant. All graphs and statistical analysis were prepared using SigmaPlot 11.2 (Systat Software, San Jose, CA).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Combination of anti‐PD‐1 and SGT‐53 effectively inhibits GL261 tumor growth
Mice bearing subcutaneously established syngeneic GL261 tumors were treated with anti‐PD‐1, SGT‐53 or combination of both agents. No significant inhibition of tumor growth was seen with anti‐PD‐1 monotherapy indicating that GL261 tumors are intrinsically unresponsive to anti‐PD‐1 therapy (Fig. 1 a). SGT‐53 treatment alone also resulted in no significant inhibition of GL261 tumor growth. However, the combination treatment resulted in a significantly retarded tumor growth leading to markedly smaller tumors compared to those in mice given either agent individually. The average tumor weight from animals that received combination treatment was significantly lower (225.5 ± 43.0 mg) than untreated control (1,132.9 ± 212.5 mg) or treated with either single agents (1,064.3 ± 128.9 mg and 855.7 ± 168.2 mg for anti‐PD‐1 alone and SGT‐53 alone, respectively) at harvest on day 17 (Fig. 1 b). Transcriptome analysis of GL261 tumors using the NanoString nCounter revealed modulation of genes linked to either cancer progression (e.g., proliferation and invasion) or immune activation (e.g., antigen presentation and T‐cell activation) by the combination treatment of SGT‐53 plus anti‐PD‐1 (Fig. 1 c). In tumors treated with this combination, immunohistochemical analysis revealed increased Casp3 staining indicative of enhanced tumor cell apoptosis compared to tumors in untreated mice or those treated with the individual agents (Figs. 1 d and 1e). We also observed a significant increase of GzmB‐positive staining in tumors (Figs. 1 d and 1f) indicating increased cytotoxic T‐cell (CTL) infiltration of the tumors after the combination treatment. These data indicate that the combination of anti‐PD‐1 and SGT‐53 is more effective in this model than either agent individually in inhibiting tumor growth, inducing tumor cell apoptosis and recruiting T cells to the tumors.
Figure 1.
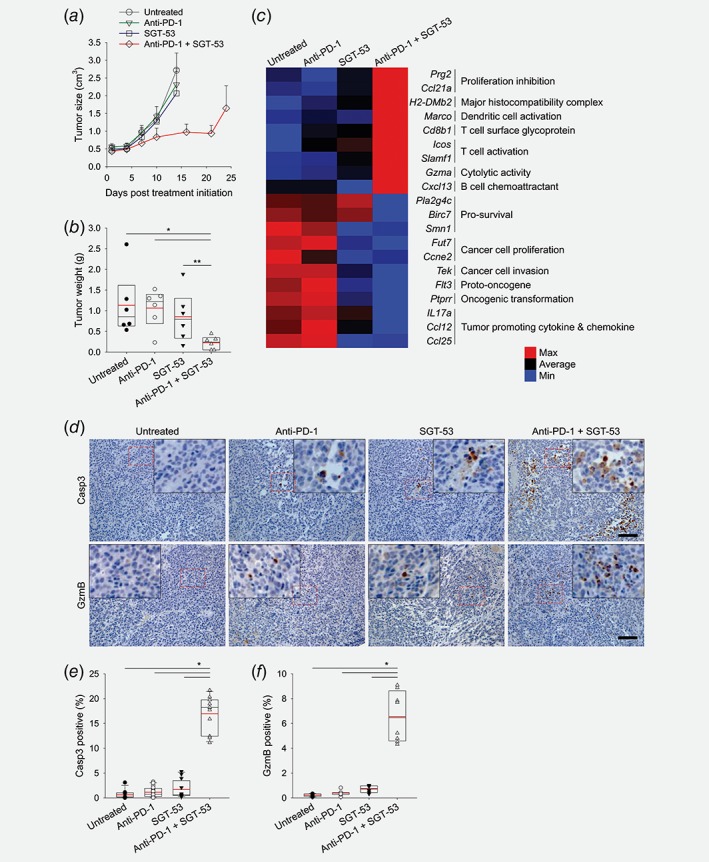
The combination of anti‐PD‐1 and SGT‐53 inhibits tumor growth. C57BL/6 mice with subcutaneous GL261 tumors were randomized to therapy with anti‐PD‐1 (200 μg antibody), SGT‐53 (30 μg DNA), or the combination of both agents twice weekly for 2.5 weeks. (a) Changes in tumor sizes were plotted. (b) Quantification of tumor weight at harvest on day 17. *p < 0.001, **p < 0.05, t‐test with ANOVA. n = 6. Red lines indicate average tumor weight. (c) Changes in gene expression of tumors using NanoString analysis. (d) Representative immunohistochemical staining of Casp3 and GzmB. Scale bars, 100 μm. Red dashed boxes indicate the magnified areas. Quantifications of positive stain of (e) Casp3 and (f) GzmB. *p < 0.001, t‐test with ANOVA (n = 8–12/group).
Combination of anti‐PD‐1 and SGT‐53 enhances immune responses in vivo
To investigate whether the observed antitumor response is associated with enhanced host immunity, we assessed tumor‐infiltrating immune cells using flow cytometry analysis. In subcutaneous tumors treated with anti‐PD‐1 plus SGT‐53, there was a significant increase of both CD45+CD11b− lymphocytes (Fig. 2 a) and CD45+CD11b+ myeloid cells among isolated tumor cells (Fig. 2 b). Further analysis of regulatory T cells (Treg, CD3+CD4+FoxP3+) and CTLs (CD3+CD8+GzmB+) showed the substantially increased number of CTLs present in tumors with the combination treatment, while the number of Tregs remained similar among the groups (Figs. 2 c–2 e). Importantly, an increase in the ratio of CTLs and Tregs, which is associated with improved prognoses in many cancers,22 was observed only in tumors with the combination treatment (Fig. 2 f) with neither of monotherapies resulting in a significant increase of the CTL/Treg ratio. These data are consistent with indication that SGT‐53 in combination with anti‐PD‐1 can increase immune cell infiltration of the tumors to alter the tumor microenvironment to produce a tumor that is more immunologically “hot.”
Figure 2.
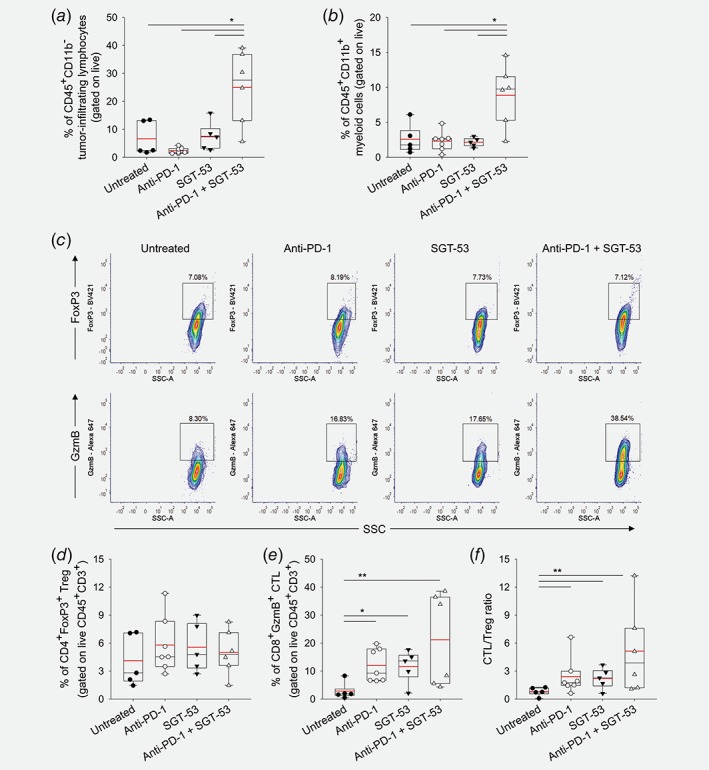
The combination of anti‐PD‐1 and SGT‐53 enhances immune responses in vivo. Tumor‐infiltrating immune cells were assessed via flow cytometry. (a) Tumor‐infiltrating lymphocytes and (b) myeloid cells were identified by gating CD45+CD11b− live cells and CD45+CD11b+ live cells, respectively. *p < 0.001, **p < 0.05, t‐test with ANOVA. n = 5–7/group. (c) Representative FACS plots of Treg (CD3+CD4+FoxP3+, upper panels) and CTL (CD3+CD8+GzmB+, lower panels). Quantifications of (d) Treg and (e) CTL were plotted. (f) The ratio of CTL/Treg. *p < 0.001, **p < 0.05, t‐test with ANOVA. n = 5–7/group. Red lines indicate average percentage of cells. [Color figure can be viewed at wileyonlinelibrary.com]
SGT‐53 increases immunogenicity of tumor cells
After exposure of GL261 cells in culture to SGT‐53 or a tumor‐targeting nanocomplex loaded with an empty vector (scL‐vec), RT‐qPCR was performed to assess expression of genes associated with immune responses (Fig. 3 a). After SGT‐53 treatment, increased mRNA levels of type I interferon (Ifna2 and Ifnb1) and several components of antigen presenting machinery (MHC I (H‐2K), Tap1 and Tap2) were evident at both 24 and 48 hr after treatment. Notably, we also observed a significant increase in the level of Pd‐l1 mRNA after SGT‐53 treatment (Fig. 3 a). We have further investigated the impact of SGT‐53 treatment to alter immunogenicity of tumor cells in vivo. Flow cytometric analysis of subcutaneous GL261 tumors revealed a significantly increased surface expression of immune cell recognition molecules including calreticulin (CRT), FAS, MHC class I (H‐2Kb/H‐2Db), PD‐L1 and ICAM1 after SGT‐53 treatment (Figs. 3 b and 3c). Notably, we observed an increased surface expression of endoplasmic reticulum protein CRT, an indicative of immunogenic cell death (ICD), which promotes immunogenic phagocytosis for antigen presentation.23 PD‐L1 has been suggested as a biomarker that might be used to predict response to anti‐PD‐1 antibody in the cancer patients.24 Our data indicate that expression of functional p53 is responsible for both induction of ICD and alterations in the immunogenicity of GL261 cells. In short, the tumors treated with SGT‐53 appear to be more immunologically “hot,” and this change would be expected to enhance the impact of checkpoint inhibitors on the body's immune response to previously “cold” glioblastoma tumors.
Figure 3.
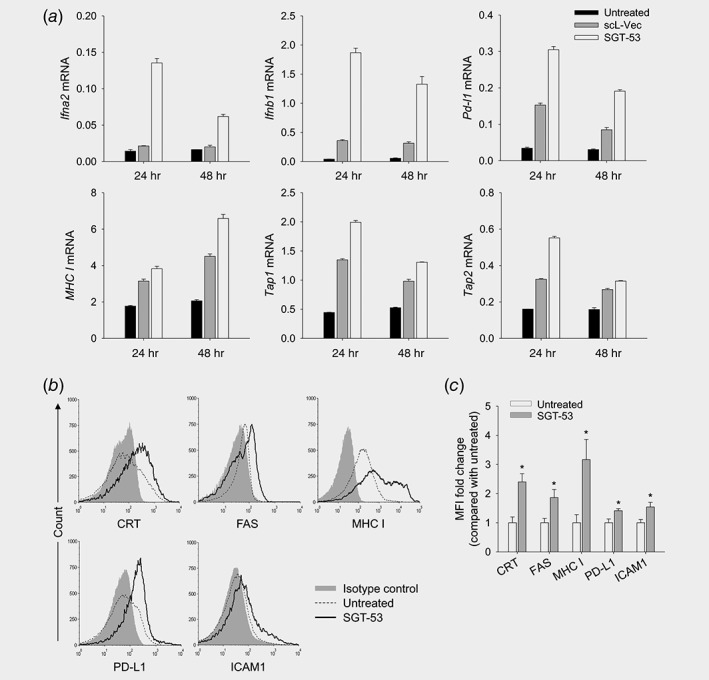
SGT‐53 increases immunogenicity of tumor. (a) GL261 cells were transfected with SGT‐53 and expression of Ifna2, Ifnb1, Pd‐l1, MHC I (H‐2K), Tap1 and Tap2 was assessed by RT‐qPCR. (b) Mice bearing GL261 tumor were treated with SGT‐53 and expression of cell surface components of immunogenicity was examined via flow cytometry. Tumor cells were identified by gating CD45−CD31− live cells. (c) The fold changes of mean fluorescence intensity (MFI) were plotted in comparison with those in tumor from untreated mice. *p < 0.05 compared to untreated group, Student t‐test (n = 3).
Combined SGT‐53 and anti‐PD‐1 treatment leads to improved survival in an intracranial glioblastoma model
To determine in vivo efficacy of combining SGT‐53 and anti‐PD‐1 therapies, we performed a survival study using an intracranial GL261 tumor model. The presence of tumor was confirmed by MRI and treatment was initiated on Day 6 (Fig. 4 a). As was seen in the subcutaneous tumor model, an enhanced antitumor effect was evident with anti‐PD‐1 plus SGT‐53 (Fig. 4 b). We observed a significant increase of Casp3 (Fig. 4 c) and GzmB (Fig. 4 d) positive staining together with a significant decrease of FoxP3 (Fig. 4 e) positive cells in tumors indicating increased tumor cell apoptosis and CTL immunity. MRI‐based measurement of tumor volume revealed a significant inhibition of tumor growth with the combination treatment (Figs. 4 f and 4g). Note that anti‐PD‐1 alone showed no antitumor activity, and SGT‐53 alone showed only a moderate antitumor effect compared to untreated controls. All untreated mice succumbed to their disease prior to Day 21, and no animals survived beyond Day 24 in the groups receiving either monotherapy indicating no significant survival benefit was conferred (Fig. 4 h). However, mice receiving SGT‐53 plus anti‐PD‐1 therapy demonstrated significantly increased survival times. Collectively, these observations support the contention that the combination of SGT‐53 and anti‐PD‐1 antibody is more efficacious as an immunotherapy regimen than checkpoint blockade alone. If our observations in the syngeneic murine glioblastoma models were to be replicated in humans, this new therapeutic strategy might markedly improve outcomes for patients with glioblastoma.
Figure 4.
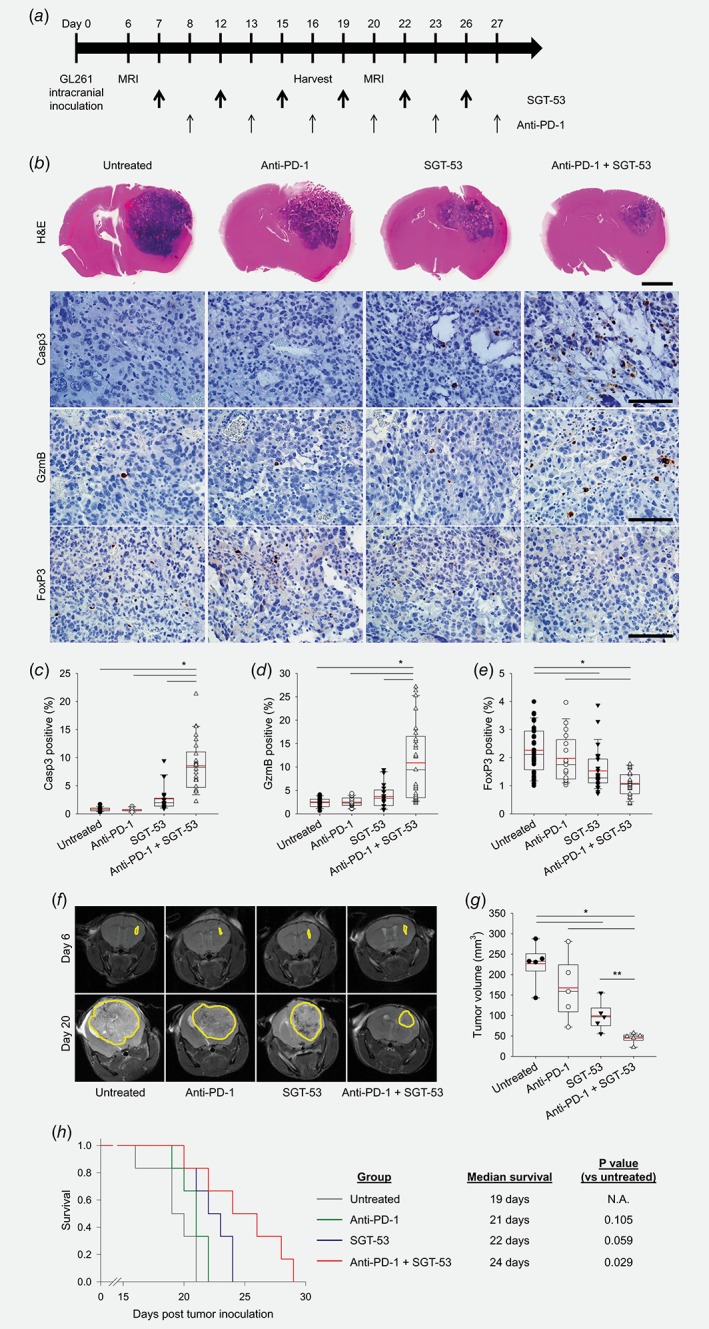
Combination treatment enhances tumor growth inhibition and survival in intracranial GL261 tumors. (a) Treatment schedule. Based on the MRI on Day 6, tumor‐bearing mice were randomized to therapy with anti‐PD‐1 (200 μg antibody), SGT‐53 (30 μg DNA) or combination of both agents (n = 10). (b) Histological examination of mouse brains on Day 16. Representative H&E (Scale bar, 2 mm) and immunohistochemical staining of Casp3, GzmB, and FoxP3 (Scale bars, 100 μm). Quantifications of positive stain of (c) Casp3, (d) GzmB, and (e) FoxP3. *p < 0.001, **p < 0.05, t‐test with ANOVA (n = 20). (f) Representative MR images acquired on Days 6 and 20. Yellow lines surround tumor area. (g) A volumetric assessment of brain tumors. *p < 0.001, **p < 0.05, t‐test with ANOVA (n = 5). Red lines indicate average tumor volume. (h) Kaplan–Meier survival curves (left panel). Median survival and statistical significance were determined by log‐rank test (right panel, n = 6). [Color figure can be viewed at wileyonlinelibrary.com]
SGT‐53 modulates polarization of macrophages
Macrophages, as highly plastic cells, play an important role in tumor immunity and regulate the growth or regression of tumors. Specifically, anti‐inflammatory M2 macrophages predominate in human cancers and actively stimulate tumor growth, while proinflammatory M1 macrophages can slow or stop cancer growth.25 Here, we examined the effects of SGT‐53 on tumor‐associated macrophage (TAM) phenotypes both in vitro and in vivo. As proof‐of‐concept, we examined mouse macrophage Raw 264.7 cells reprogramed toward the M2 phenotype by Th2 cytokines (IL4 and IL13), which substantially increased M2 phenotype (CD45+CD11b+F4/80+CD206+, from 1.56% to 25.25%) while SGT‐53 treatment clearly suppressed their reprogramming toward M2 phenotype (11.34% rather than 25.25%; Fig. 5 a). As a result, SGT‐53 significantly increased the M1/M2 ratio, while the control nanocomplex scL‐vec did not have a significant effect (Fig. 5 b). Consistent with this observation, RT‐qPCR analysis showed a significant upregulation of Tnf (an M1 marker) and downregulation of Cd206 (an M2 marker) after treatment with SGT‐53 plus Th2 cytokine (Fig. 5 c). These data suggest that introduction of wtp53 under M2‐polarizing conditions thwarts the establishment of the M2 phenotype. Modulation of macrophage polarization by SGT‐53 was further investigated in vivo using the GL261 tumor model and employing NanoString to assess expression of M1 and M2 genes (Fig. 5 d). Consistent with the results in cultured Raw 264.7 cells, SGT‐53 treatment significantly increased the average counts of M1‐related genes (Nos2, Tnf, Csf2, Il12 and Irf5) and significantly decreased the average counts of M2‐related genes (Cd206, Myc and Il4) in tumors. RT‐qPCR analysis of tumors further revealed that SGT‐53 treatment can significantly downmodulate the genes involved in M2 macrophage activation such as Snail, Myc and Hif1a (Fig. 5 e). In addition, immunohistochemical analysis with NOS2 (an M1 marker) and ARG1 (an M2 marker) confirmed the significant increase of M1 and decrease of M2 macrophages in GL261 tumors after SGT‐53 treatment compared to untreated tumors (Figs. 5 f and 5g). Thus, our results indicate that SGT‐53 treatment alters the phenotype of TAMs, and this effect may contribute to the inhibition of tumor progression by SGT‐53 in the context of immunotherapy.
Figure 5.
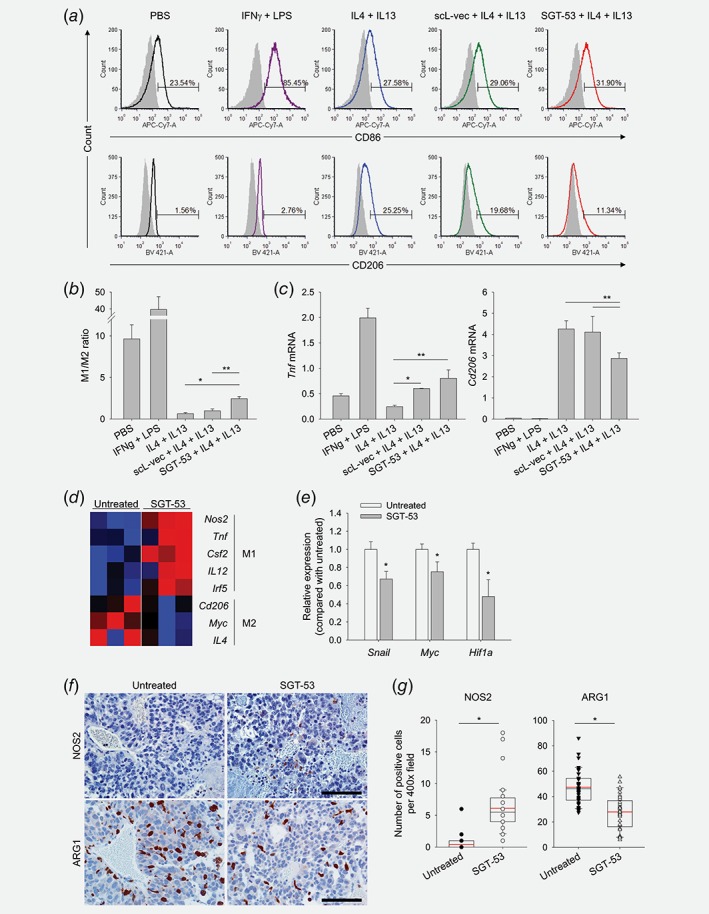
SGT‐53 regulates polarization of macrophages. M1 or M2 polarization of Raw 264.7 cells were induced using IFNγ (10 ng/ml) + LPS (100 ng/ml) for M1 or IL4 (20 ng/ml) + IL13 (20 ng/ml) for M2. Prior to the incubation with IL4 + IL13, Raw 264.7 cells were treated with either SGT‐53 or scL‐vec nanocomplex for 24 hr. (a) The shift between M1/M2 phenotypes was evaluated 24 hr later by flow cytometry (M1 = CD86+, M2 = CD206+ using CD45+CD11b+F4/80+ macrophage gate). (b) The ratio of M1/M2 macrophages. *p < 0.001, **p < 0.05, t‐test with ANOVA (n = 4). (c) Expression of genes associated with macrophage polarization was assessed by RT‐qPCR in Raw 264.7 cells. *p < 0.001, **p < 0.05, t‐test with ANOVA (n = 5). (d) Mice bearing GL261 tumor were treated with SGT‐53 and changes in expression of M1‐ and M2‐related genes were assessed using NanoString analysis. (e) Expression of genes associated with macrophage polarization was assessed by RT‐qPCR in tumor. *p < 0.05 compared to untreated group, Student's t‐test (n = 3). (f) Representative immunohistochemical staining and (g) quantifications of NOS2 and ARG1 are shown. Scale bars, 100 μm. *p < 0.001, compared to untreated group, Student's t‐test (n = 20).
Discussion
In recent years, immune checkpoint blockade has made exciting progress as an approach to treat cancers. Specifically, monoclonal antibodies targeting PD‐1, PD‐L1 and CTLA‐4 have been successfully demonstrated to elicit durable antitumor immune responses in a range of tumor types, and the FDA has approved several such antibodies. However, not all patients have exhibited durable responses, and patients have been observed to acquire the resistance to ICIs.26 The recent failure of late‐phase clinical trials evaluating anti‐PD‐1 in patients with recurrent glioblastoma reflects the continued challenges for immunotherapy of brain cancer.2, 27 In our preclinical models of glioblastoma, anti‐PD‐1 treatment also failed to show any significant therapeutic efficacy as a single modality immunotherapy.
The failure of ICIs in glioblastoma might be in part due to these tumors being immunologically “cold,” meaning that in glioblastoma there is, generally speaking, a paucity of tumor‐infiltrating immune effector cells. The lack of glioblastoma‐infiltrating T cells as well as T‐cell dysfunction likely contributes to the unresponsiveness of these tumors to single modality immunotherapy. To overcome this unresponsiveness, a large number of studies have been conducted that seek to turn immunologically “cold” tumors into “hot” tumors.28, 29 Our approach combines the tumor‐targeting nanomedicine (SGT‐53) to promote antitumor immune responses and to mitigate against the immunologically “cold” status of glioblastoma. Previously we have demonstrated that concurrent SGT‐53 treatment can convert otherwise unresponsive breast or head and neck cancers to anti‐PD‐1‐responsive tumors.30, 31 Here, we demonstrate increased tumor responses to concomitant anti‐PD‐1 and SGT‐53 treatment in mouse syngeneic models of glioblastoma tumor. Mice receiving this combination therapy demonstrated a significantly decreased tumor growth rate and a survival benefit was conferred compared to untreated mice or mice receiving either SGT‐53 or anti‐PD‐1 as monotherapies. Extrapolating our observations in syngeneic murine tumors to human cancers, the conversion of glioblastoma to being responsive to checkpoint blockade has the potential to convert glioblastoma patients that do not respond to anti‐PD‐1 therapy into responders. This conversion of nonresponders would allow checkpoint inhibitors to benefit a larger percentage of the total glioblastoma patient population.
Immune responses are modulated in a multifaceted way by enhanced p53 expression in that SGT‐53 treatment triggered changes in both innate and adaptive immune responses. Treatment with SGT‐53 markedly modified the previously immunosuppressive microenvironment of these tumors. We have observed that SGT‐53 treatment caused immunogenic changes in GL261 tumors including elevated surface expression of CRT (reflective of ICD), as well as FAS, PD‐L1, ICAM1 and MHC I. Notably, the increased CRT expression appears to promote immunogenic phagocytosis by the innate immune cells that would elevate tumor antigen presentation to turn on the cancer immunity cycle.32 These multiple changes likely combine to enhance the immune responses to the glioblastoma tumors. Indeed, both immunohistochemical and flow cytometry analyses demonstrated a significantly elevated tumor‐infiltration of lymphocytes and myeloid cells in the mice receiving SGT‐53 plus anti‐PD‐1. Collectively, our results indicate that tumors treated with combination of SGT‐53 and anti‐PD‐1 are more immunologically “hot,” and this change would be expected to result in increased antitumor immune responses. Importantly, the SGT‐53 plus anti‐PD‐1 combination treatment restored T‐cell effector function and increased CTLs within tumor, which was positively correlated with a survival benefit in mice bearing intracranial glioblastoma tumors. Consistent with our findings, there is increasing evidence indicating that p53 can regulate the cell‐mediated adaptive immune response to tumors and ultimately promote CTL‐induced cancer cell killing.33, 34, 35, 36 Introduction of p53 into tumor cells has also been shown to enhance induction of apoptosis after exposure to CTL‐mediated cytotoxic insults,35 and p53 accumulation in tumor cells is an indispensable component in the GzmB‐induced apoptotic signaling pathway.37
Generally speaking, cancer cells appear to create a tumor microenvironment that is relatively enriched in signals that polarize the TAMs toward the tumor‐promoting M2 (alternatively activated) phenotype that can suppress antitumor immune responses.38 Skewing the M1/M2 ratio toward the tumor‐inhibiting M1 phenotype would be beneficial in the treatment of cancer. We have observed that SGT‐53 treatment results in such a shift in macrophages from the M2 toward M1 phenotype suggesting a role for p53 as a “brake” to the alternative activation of macrophages. In agreement with our observation, others have shown that p53 activation after nutlin‐3a treatment (which inhibits the interaction between p53 and mdm2 that blunts p53 activity) resulted in downregulation of M2 gene expression in macrophages, while loss of p53 showed increased expression of M2 genes and enhanced proliferation of M2 macrophages.39 Transcriptome profiling and RT‐qPCR analyses revealed that SGT‐53 treatment can significantly downmodulate Myc, a proto‐oncogene whose deregulated expression is associated with tumor development in many human cancers.40 Myc has been previously proposed to regulate M2 macrophage activation, and Myc inhibition could lead to reduced expression of M2 markers.39, 41 Another relevant gene is Snail that plays a role in epithelial to mesenchymal transition and metastasis.42 It has been reported that the overexpression of Snail increased tumor infiltration by M2 macrophages, while depletion of Snail led to more functional M1 macrophages.43 Of note, we have found a significant downmodulation of Snail in tumors treated with SGT‐53. Collectively, these observations suggest that it may be possible to use SGT‐53 treatment as a new means of reversing the tumor‐promoting and immunosuppressive condition mediated by macrophages in the tumor microenvironment, although further investigations on underlying mechanisms are needed.
Although the elements involved in tumor response are complex, studies seeking biomarkers that might be used to predict response to anti‐PD‐1 have found that tumor expression of PD‐L1 is the single feature most highly correlated with response thus far.24, 44 It has been suggested that cancer patients who do not respond to treatment with anti‐PD‐1 are those having tumors with relatively low expression of PD‐L1.44, 45, 46 Our experiments revealed that SGT‐53 treatment upregulated PD‐L1 expression in both cultured GL261 cells and in syngeneic GL261 tumors in vivo. This PD‐L1 upregulation has also been observed in multiple human glioblastoma cell lines (data not shown). Thus, it is our hypothesis that SGT‐53 treatment will also elevate expression of PD‐L1 on human tumors and expand the fraction of patients who respond to anti‐PD‐1 antibodies. The ability of SGT‐53 to elevate tumor PD‐L1 in multiple syngeneic mouse models,30, 31 together with the fact that the treatment with SGT‐53 plus the checkpoint inhibitor results in enhanced infiltration of tumors by TILs provides a clear rationale for a trial involving the combination of SGT‐53 plus ICIs.
In summary, we report that introducing functional wtp53 gene via tumor‐targeted p53 gene therapy (SGT‐53) was able to augment anti‐PD‐1 immune checkpoint blockade and convert mouse syngeneic glioblastoma tumors that were unresponsive (immunologically “cold” tumors) into tumors that were more responsive to anti‐PD‐1 immunotherapy. These findings suggest that SGT‐53 has potential to enhance the efficacy of ICIs and thereby provide for improved outcomes in the treatment of glioblastoma. Our observations provide compelling motivation to move to test in the context of a glioblastoma clinical trial the combination of SGT‐53 together with an immune checkpoint blockade agent.
Acknowledgements
This work was supported by the NCI grant (5R01CA132012‐02 to E.H.C.); and a research grant from SynerGene Therapeutics, Inc. This study was conducted at the Lombardi Cancer Center Shared Resource facilities that are partially supported by NCI (P30‐CA051008).
Conflict of interest: E.H.C. is the one of inventors of the described technology, for which several patents owned by Georgetown University have been issued. The patents have been licensed to SynerGene Therapeutics, Inc. for commercial development. E.H.C. owns an equity interest in SynerGene Therapeutics, Inc. and serves as a non‐paid scientific consultant to the company. S.K. and J.B.H. are salaried employees of SynerGene Therapeutics, Inc. and owns stock/stock option in same. No potential conflicts of interest were disclosed by the other authors.
References
- 1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359:492–507. [DOI] [PubMed] [Google Scholar]
- 2. Lim M, Xia Y, Bettegowda C, et al. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 2018;15:422–42. [DOI] [PubMed] [Google Scholar]
- 3. Buerki RA, Chheda ZS, Okada H. Immunotherapy of primary brain tumors: facts and hopes. Clin Cancer Res 2018;24:5198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim ES, Kim JE, Patel MA, et al. Immune checkpoint modulators: an emerging Antiglioma armamentarium. J Immunol Res 2016;2016:4683607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Donnell JS, Smyth MJ, Teng MW. Acquired resistance to anti‐PD1 therapy: checkmate to checkpoint blockade? Genome Med 2016;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ott PA, Hodi FS, Kaufman HL, et al. Combination immunotherapy: a road map. J Immunother Cancer 2017;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015;14:561–84. [DOI] [PubMed] [Google Scholar]
- 11. Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol 2017;51:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell 2007;12:303–12. [DOI] [PubMed] [Google Scholar]
- 13. Allred DC, Clark GM, Elledge R, et al. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node‐negative breast cancer. J Natl Cancer Inst 1993;85:200–6. [DOI] [PubMed] [Google Scholar]
- 14. Valente JFA, Queiroz JA, Sousa F. p53 as the focus of gene therapy: past, present and future. Curr Drug Targets 2018;19:1801–17. [DOI] [PubMed] [Google Scholar]
- 15. Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol 2013;25:85–92. [DOI] [PubMed] [Google Scholar]
- 16. Cui Y, Guo G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int J Mol Sci 2016;17:E1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim SS, Rait A, Kim E, et al. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014;8:5494–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Senzer N, Nemunaitis J, Nemunaitis D, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther 2013;21:1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pirollo KF, Nemunaitis J, Leung PK, et al. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: a phase 1b study. Mol Ther 2016;24:1697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu L, Huang CC, Huang W, et al. Systemic tumor‐targeted gene delivery by anti‐transferrin receptor scFv‐immunoliposomes. Mol Cancer Ther 2002;1:337–46. [PubMed] [Google Scholar]
- 21. Kim SS, Harford JB, Moghe M, et al. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res 2018;46:1424–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu F, Lang R, Zhao J, et al. CD8(+) cytotoxic T cell and FOXP3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat 2011;130:645–55. [DOI] [PubMed] [Google Scholar]
- 23. Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47‐signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA 2012;109:6662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taube JM, Klein A, Brahmer JR, et al. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti‐PD‐1 therapy. Clin Cancer Res 2014;20:5064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sica A, Larghi P, Mancino A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol 2008;18:349–55. [DOI] [PubMed] [Google Scholar]
- 26. Ribas A, Hu‐Lieskovan S. What does PD‐L1 positive or negative mean? J Exp Med 2016;213:2835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reardon DA, Omuro A, Brandes AA, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of Nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol 2017;19:iii21. [Google Scholar]
- 28. Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016;44:343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaur P, Asea A. Radiation‐induced effects and the immune system in cancer. Front Oncol 2012;2:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moore EC, Sun L, Clavijo PE, et al. Nanocomplex‐based TP53 gene therapy promotes anti‐tumor immunity through TP53‐ and STING‐dependent mechanisms. Oncoimmunology 2018;7:e1404216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim SS, Harford JB, Moghe M, et al. Combination with SGT‐53 overcomes tumor resistance to a checkpoint inhibitor. Oncoimmunology 2018;7:e1484982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 33. Wang B, Niu D, Lai L, et al. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun 2013;4:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu K, Wang J, Zhu J, et al. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 1999;18:7740–7. [DOI] [PubMed] [Google Scholar]
- 35. Thiery J, Abouzahr S, Dorothee G, et al. p53 potentiation of tumor cell susceptibility to CTL involves Fas and mitochondrial pathways. J Immunol 2005;174:871–8. [DOI] [PubMed] [Google Scholar]
- 36. Braun MW, Iwakuma T. Regulation of cytotoxic T‐cell responses by p53 in cancer. Transl Cancer Res 2016;5:692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meslin F, Thiery J, Richon C, et al. Granzyme B‐induced cell death involves induction of p53 tumor suppressor gene and its activation in tumor target cells. J Biol Chem 2007;282:32991–9. [DOI] [PubMed] [Google Scholar]
- 38. Kulkarni A, Chandrasekar V, Natarajan SK, et al. A designer self‐assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat Biomed Eng 2018;2:589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li L, Ng DS, Mah WC, et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ 2015;22:1081–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pello OM, De Pizzol M, Mirolo M, et al. Role of c‐MYC in alternative activation of human macrophages and tumor‐associated macrophage biology. Blood 2012;119:411–21. [DOI] [PubMed] [Google Scholar]
- 41. He XY, Xiang C, Zhang CX, et al. p53 in the myeloid lineage modulates an inflammatory microenvironment limiting initiation and invasion of intestinal tumors. Cell Rep 2015;13:888–97. [DOI] [PubMed] [Google Scholar]
- 42. Wang Y, Shi J, Chai K, et al. The role of snail in EMT and tumorigenesis. Curr Cancer Drug Targets 2013;13:963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brenot A, Knolhoff BL, DeNardo DG, et al. SNAIL1 action in tumor cells influences macrophage polarization and metastasis in breast cancer through altered GM‐CSF secretion. Oncogene 2018;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 2015;372:2018–28. [DOI] [PubMed] [Google Scholar]
- 45. Daud AI, Wolchok JD, Robert C, et al. Programmed death‐ligand 1 expression and response to the anti‐programmed death 1 antibody Pembrolizumab in melanoma. J Clin Oncol 2016;34:4102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jing W, Li M, Zhang Y, et al. PD‐1/PD‐L1 blockades in non‐small‐cell lung cancer therapy. Onco Targets Ther 2016;9:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.