

Abstract
Objective
Smoking is associated with an increased risk of rheumatoid arthritis (RA) in subsets of patients defined according to the presence or absence of anti–citrullinated protein antibodies (ACPAs) and rheumatoid factors (RFs). Moreover, an interaction between smoking and the HLA–DRB1 shared epitope (SE) has been demonstrated to be a risk factor for seropositive RA. The aim of this study was to investigate the interplay between smoking and the HLA–DRB1 SE with regard to risk of RA in different patient subsets based on ACPA and RF status.
Methods
Incident cases of RA (3,645 cases, 5,883 matched controls) were divided into 4 subgroups based on the presence or absence of RF and anti–cyclic citrullinated peptide 2 (anti‐CCP2) antibodies. The influence of smoking on the risk of disease was determined in each RA subgroup, using logistic regression models with calculation of odds ratios and 95% confidence intervals (95% CIs). The potential interaction between smoking and HLA–DRB1 SE genes was evaluated by calculating the attributable proportion due to interaction (AP).
Results
In the RF+/anti‐CCP2+ subset of RA patients, both smoking and the presence of the HLA–DRB1 SE conferred independent disease risks, and there was a strong interaction between the 2 risk factors (AP 0.4, 95% CI 0.3, 0.5). In the RF−/anti‐CCP2+ patient subset, the HLA–DRB1 SE conferred an increased risk of RA, whereas the independent influence of smoking was limited. However, there was a significant interaction between the HLA–DRB1 SE and smoking (AP 0.2, 95% CI 0.02, 0.5). In the RF+/anti‐CCP2− patient subset, there was an increased risk of disease among smokers, which was only marginally affected by the presence of the HLA–DRB1 SE, and no interaction between the 2 factors was observed (AP 0.002, 95% CI −0.3, 0.3). In the RF−/anti‐CCP2− patient subset, neither smoking nor the presence of the HLA–DRB1 SE conferred an increased risk of RA.
Conclusion
These findings demonstrate different effects of smoking and HLA–DRB1 in the 4 serologically defined RA subsets.
Introduction
Rheumatoid arthritis (RA) is an immune‐mediated inflammatory disease resulting from the complex interaction between genetic constitution and environmental triggers. The most important genetic risk factor for RA defined to date is the shared epitope (SE) of HLA–DRB1 1, 2, 3, and smoking has been identified as the most important environmental factor in the development of RA 4, 5, 6.
The effects of these 2 risk factors, the HLA–DRB1 SE and smoking, and the interaction between them have been shown to be confined to the subset of RA patients whose disease is defined by the presence of anti–citrullinated protein antibodies (ACPAs) and/or rheumatoid factors (RFs), and a hypothesis regarding the etiology of this subset has been formulated based on the interaction between the HLA–DRB1 SE and smoking, as well as between the HLA–DRB1 SE and other airway exposures 7, 8. However, the potential roles of RF and ACPAs in the pathogenesis of different subsets of RA have not yet been fully elucidated. We used an updated version of the Swedish population‐based case–control study Epidemiological Investigation of Rheumatoid Arthritis (EIRA) to investigate the interplay between smoking and the HLA–DRB1 SE with regard to risk of RA in different serologically defined patient subsets grouped according to ACPA and RF status.
Patients and Methods
Study design and study subjects
The present study investigated data from the ongoing EIRA project, which is a population‐based case–control study comprising subjects ages 18–70 years in the middle and southern parts of Sweden. All hospital‐based and most privately run rheumatology units in the study area participated in recruiting incident RA cases to the study. All patients identified as an incident case fulfilled the American College of Rheumatology 1987 classification criteria for RA 9. During the study period (November 1996 to September 2014), completed questionnaires were obtained from 3,724 RA cases and 5,935 matched healthy controls. Subjects who could not provide detailed information on smoking habits were excluded, as were patients whose ACPA or RF status was not available. A flow chart depicting the distribution of subjects is presented in Supplementary Figure 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40852/abstract). For each case recruited between November 1996 and October 2005, 1 control subject was randomly selected from the national population register, matched by age in 5‐year age strata, by sex, and by residential area (EIRA I). For each case recruited between October 2005 and September 2014, 2 control subjects were selected using the same matching criteria (EIRA II). The response proportion was 92% for the cases and 75% for the controls. All aspects of the study were approved by the ethics committee of the Karolinska Institutet.
Anti–cyclic citrullinated peptide 2 (anti‐CCP2) and RF analyses
Cases were categorized into either anti‐CCP2 positive or anti‐CCP2 negative based on the results of an Immunoscan‐RA Mark2 enzyme‐linked immunosorbent assay (anti‐CCP2 test). An antibody level exceeding 25 AU/ml was regarded as a positive result. RF positivity or RF negativity was determined locally by the unit entering the case into the study.
Data collection and definition of smoking status
Information regarding lifestyle factors and different environmental exposures was collected using a standardized questionnaire. Detailed information on smoking was obtained by asking each subject about current and previous smoking habits, including duration of smoking, average number of cigarettes smoked per day, and type of cigarettes. For each case, the time of the initial appearance of RA symptoms was used as an estimate of the date of disease onset, and the year in which this occurred was defined as the index year. The corresponding controls were given the same index year. Information regarding smoking was considered prior to or during the index year in the cases and during the same period of time in the corresponding controls. Subjects who had smoked during the index year were defined as current smokers, those who had stopped smoking prior to the index year were defined as past smokers, and those who had never smoked before or during the index year were defined as never smokers.
Genotyping
Blood samples were available from participants who answered the questionnaire between November 1996 and May 2012. HLA–DRB1 genotypes were obtained using a previously described method 10. Data on genotypes were available for 3,355 cases (63%) and 2,840 controls (48%). The HLA–DRB1*01, HLA–DRB1*04, and HLA–DRB1*10 alleles were classified as the SE alleles.
Statistical analysis
Using logistic regression analyses with calculation of odds ratios (ORs) and 95% confidence intervals (95% CIs), the risk of occurrence of each RA serologic subset in patients with different smoking habits was compared with that in never smokers. The occurrence of RA among those who had started and stopped smoking in different life periods was compared with that among never smokers. A trend test for a dose‐response relationship regarding cumulative dose of smoking and risk of each subset of RA was performed using a continuous variable for cumulative dose of smoking (expressed in pack‐years) in a logistic regression model.
In addition, we investigated the interaction between smoking and SE genes with regard to each RA subset. The potential interaction was analyzed using departure from additivity of effects as the criterion for interaction and was evaluated by calculating the attributable proportion due to interaction (with 95% CIs) 11, 12.
All analyses were adjusted for age, sex, residential area, ancestry, and study. Assessment of ancestry was based on whether or not the subject was born in Sweden, and whether or not either of the subject's parents had immigrated to Sweden. A subject who was born in Sweden and whose parents had not immigrated was classified as Swedish. Adjustments were also made for educational level (university degree or no university degree), exposure to passive smoking (yes or no), alcohol consumption (number of standardized drinks per week at study inclusion), and body mass index at inclusion in the study (≤25 kg/m2 or >25 kg/m2). However, these factors had only a minor influence on the results and were therefore not retained in the final analyses. All analyses were conducted using SAS software version 9.4 (SAS Institute).
Results
The majority of the patients with incident RA were both RF positive and ACPA positive (57%), whereas 25% were negative for both classes of antibodies. Nine percent of patients were ACPA positive only, and 9% were RF positive only. The characteristics of the cases and controls are presented in Supplementary Table 1 (on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40852/abstract). There were no significant differences between the RA subsets with regard to age, sex, or ancestry.
Among the RA patients, ACPA positivity independently correlated with both the HLA–DRB1 SE status and the RF status (Table 1). Smoking habits did not significantly influence these correlations, but there was a significant dose‐dependent relationship between smoking and RF positivity irrespective of anti‐CCP2 and HLA–DRB1 SE status (P < 0.0001).
Table 1.
Rate of anti‐CCP2 positivity among RA cases categorized by RF status, number of SE alleles, and smoking statusa
| No. of RA cases | Anti‐CCP2 positive, no. (%) | |
|---|---|---|
| RF‐negative | ||
| 0 alleles | ||
| Never smoker | 179 | 18 (10.1) |
| Ever smoker | 254 | 23 (9.1) |
| 1 allele | ||
| Never smoker | 209 | 59 (28.2) |
| Ever smoker | 317 | 104 (32.8) |
| 2 alleles | ||
| Never smoker | 73 | 38 (52.1) |
| Ever smoker | 111 | 60 (54.1) |
| RF‐positive | ||
| 0 alleles | ||
| Never smoker | 128 | 86 (67.2) |
| Ever smoker | 296 | 205 (69.3) |
| 1 allele | ||
| Never smoker | 313 | 265 (84.7) |
| Ever smoker | 808 | 701 (86.8) |
| 2 alleles | ||
| Never smoker | 192 | 177 (92.2) |
| Ever smoker | 496 | 473 (95.4) |
Anti‐CCP2 = anti–cyclic citrullinated peptide 2; RA = rheumatoid arthritis; RF = rheumatoid factor; SE = shared epitope.
Compared with never smokers, the overall OR for developing RF−/anti‐CCP2− RA among ever smokers was 1.1 (95% CI 0.96–1.3). The corresponding ORs for the other subsets of patients who were ever smokers were as follows: for RF−/anti‐CCP2+ RA, OR 1.2 (95% CI 0.98−1.6); for RF+/anti‐CCP2− RA, OR 1.6 (95% CI 1.2–1.9); for RF+/anti‐CCP2+ RA, OR 2.0 (95% CI 1.8–2.2) (Table 2). The association between smoking and risk of RA increased numerically with increasing exposure to smoking (i.e., increasing pack‐years of smoking) in all 3 antibody‐dependent subsets, but was largest in the subsets positive for RF (P for trend < 0.0001 in the RF‐positive subsets).
Table 2.
Odds of developing rheumatoid arthritis, stratified by serologic subset, according to different categories of smokers compared with never smokers, in total and by cumulative dose of smokinga
| Anti‐CCP2−, RF− | Anti‐CCP2+, RF− | Anti‐CCP2−, RF+ | Anti‐CCP2+, RF+ | |||||
|---|---|---|---|---|---|---|---|---|
| No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | |
| Total | ||||||||
| Never smoker | 367/2,655 | 1.0 (reference) | 133/2,655 | 1.0 (reference) | 111/2,655 | 1.0 (reference) | 594/2,655 | 1.0 (reference) |
| Past smoker | 330/1,909 | 1.1 (0.9–1.4) | 109/1,909 | 1.2 (0.98–1.6) | 116/1,909 | 1.3 (1.02–1.8) | 774/1,909 | 1.8 (1.6–2.1) |
| Current smoker | 218/1,319 | 1.1 (0.9–1.3) | 89/1,319 | 1.3 (1.004–1.7) | 109/1,319 | 1.8 (1.3–2.3) | 695/1,319 | 2.4 (2.1–2.7) |
| <10 pack‐years | ||||||||
| Never smoker | 367/2,655 | 1.0 (reference) | 133/2,655 | 1.0 (reference) | 111/2,655 | 1.0 (reference) | 594/2,655 | 1.0 (reference) |
| Past smoker | 179/1,062 | 1.1 (0.9–1.4) | 58/1,062 | 1.1 (0.8–1.5) | 62/1,062 | 1.2 (0.9–1.7) | 320/1,062 | 1.2 (1.001–1.5) |
| Current smoker | 68/476 | 1.1 (0.8–1.5) | 28/476 | 1.0 (0.7–1.3) | 28/476 | 1.2 (0.8–1.8) | 148/476 | 1.3 (1.1–1.5) |
| 10–20 pack‐years | ||||||||
| Never smoker | 367/2,655 | 1.0 (reference) | 133/2,655 | 1.0 (reference) | 111/2,655 | 1.0 (reference) | 594/2,655 | 1.0 (reference) |
| Past smoker | 79/495 | 1.0 (0.7–1.2) | 30/495 | 1.3 (0.9–2.0) | 25/495 | 1.2 (0.8–1.9) | 205/495 | 1.9 (1.6–2.3) |
| Current smoker | 52/325 | 1.1 (0.8–1.5) | 26/325 | 1.6 (1.04–2.5) | 37/325 | 2.4 (1.6–3.6) | 201/325 | 2.7 (2.2–3.3) |
| >20 pack‐years | ||||||||
| Never smoker | 367/2,655 | 1.0 (reference) | 133/2,655 | 1.0 (reference) | 111/2,655 | 1.0 (reference) | 594/2,655 | 1.0 (reference) |
| Past smoker | 72/352 | 1.2 (0.9–1.6) | 21/352 | 1.5 (0.9–2.4) | 29/352 | 1.9 (1.3–2.7) | 249/352 | 3.1 (2.6–3.7) |
| Current smoker | 98/518 | 1.2 (0.9–1.5) | 35/518 | 1.6 (1.05–2.4) | 44/518 | 2.0 (1.2–3.1) | 346/518 | 3.6 (2.9–4.4) |
All analyses were adjusted for age, sex, residential area, and ancestry. Anti‐CCP2 = anti–cyclic citrullinated peptide 2; RF = rheumatoid factor; OR = odds ratio; 95% CI = 95% confidence interval.
Values are the number of exposed cases and controls.
The influence of smoking on the risk of developing RF+/anti‐CCP2+ RA increased significantly with the number of SE alleles (P for trend < 0.0001) (Table 3).
Table 3.
Odds of developing rheumatoid arthritis, stratified by serologic subset, according to HLA SE status and different categories of smokers compared with never smokersa
| Anti‐CCP2−, RF− | Anti‐CCP2+, RF− | Anti‐CCP2−, RF+ | Anti‐CCP2+, RF+ | |||||
|---|---|---|---|---|---|---|---|---|
| No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | |
| HLA SE negative | ||||||||
| Never smoker | 161/592 | 1.0 (reference) | 18/592 | 1.0 (reference) | 42/592 | 1.0 (reference) | 86/592 | 1.0 (reference) |
| Past smoker | 142/501 | 1.0 (0.7–1.3) | 12/501 | 1.0 (0.4–1.8) | 43/501 | 1.1 (0.7–1.7) | 117/501 | 1.6 (1.2–2.2) |
| Current smoker | 89/263 | 1.2 (0.9–1.6) | 11/263 | 1.3 (0.6–2.8) | 48/263 | 2.4 (1.5–3.7) | 88/263 | 2.3 (1.6–3.2) |
| HLA SE heterozygote | ||||||||
| Never smoker | 151/521 | 1.0 (reference) | 59/521 | 1.0 (reference) | 49/521 | 1.0 (reference) | 265/521 | 1.0 (reference) |
| Past smoker | 125/464 | 0.9 (0.7–1.1) | 58/464 | 1.2 (0.8–1.8) | 58/464 | 1.6 (0.9–2.0) | 369/464 | 1.6 (1.3–2.0) |
| Current smoker | 95/272 | 1.1 (0.8–1.5) | 46/272 | 1.5 (0.97–2.3) | 49/272 | 1.7 (1.1–2.7) | 336/272 | 2.4 (1.9–2.9) |
| HLA SE homozygote | ||||||||
| Never smoker | 35/137 | 1.0 (reference) | 38/137 | 1.0 (reference) | 15/137 | 1.0 (reference) | 177/137 | 1.0 (reference) |
| Past smoker | 35/92 | 1.3 (0.9–2.6) | 29/92 | 1.3 (0.7–2.2) | 11/92 | 1.1 (0.5–2.5) | 238/92 | 2.2 (1.6–3.1) |
| Current smoker | 16/60 | 1.0 (0.6–2.0) | 31/60 | 1.9 (1.04–3.4) | 12/60 | 1.8 (0.8–3.9) | 235/60 | 3.2 (2.2–4.6) |
All analyses were adjusted for age, sex, residential area, and ancestry. SE = shared epitope; anti‐CCP2 = anti–cyclic citrullinated peptide 2; RF = rheumatoid factor; OR = odds ratio; 95% CI = 95% confidence interval.
Values are the number of exposed cases and controls.
The risk of RA conferred by HLA–DRB1 SE seropositivity was mainly observed in anti‐CCP2+ RA patients, irrespective of RF status (Table 4). The interaction between smoking and HLA–DRB1 SE genes, measured as the attributable proportion due to interaction, was highest in the subset positive for both RF and anti‐CCP2, but a notable interaction was observed also in the RF−/anti‐CCP2+ RA subset for the group consisting of individuals who had smoked more than 10 pack‐years (Table 4). This interaction was also stronger among HLA–DRB1 SE homozygotes than among HLA–DRB1 SE heterozygotes (Table 5). No significant interaction was observed between smoking and HLA–DRB1 SE genes with regard to the risk of anti‐CCP2− RA, regardless of RF status (Table 4).
Table 4.
Interaction between the HLA SE and smoking in relation to odds of developing rheumatoid arthritisa
| Anti‐CCP2−, RF− | Anti‐CCP2+, RF− | Anti‐CCP2−, RF+ | Anti‐CCP2+, RF+ | |||||
|---|---|---|---|---|---|---|---|---|
| No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | No. cases/no. controlsb | OR (95% CI) | |
| Total | ||||||||
| HLA SE negative | ||||||||
| Never smoker | 161/592 | 1.0 (reference) | 18/592 | 1.0 (reference) | 42/592 | 1.0 (reference) | 86/592 | 1.0 (reference) |
| Ever smoker | 231/764 | 1.1 (0.9, 1.4) | 23/764 | 1.1 (0.6, 2.0) | 91/764 | 1.8 (1.2, 2.6) | 205/764 | 1.9 (1.5, 2.5) |
| HLA SE positive | ||||||||
| Never smoker | 184/636 | 1.0 (0.8, 1.3) | 97/636 | 5.0 (3.0, 8.4) | 63/636 | 1.4 (0.9, 2.1) | 441/636 | 4.8 (3.7, 6.1) |
| Ever smoker | 261/848 | 1.1 (0.9, 1.4) | 160/848 | 6.7 (4.0, 11.1) | 126/848 | 2.2 (1.5, 3.1) | 1166/848 | 10.0 (7.8, 12.8) |
| APc | 0.03 (−0.3, 0.3) | 0.2 (0.02, 0.5) | 0.002 (−0.3, 0.3) | 0.4 (0.3, 0.5) | ||||
| <10 pack‐years of smoking | ||||||||
| HLA SE negative | ||||||||
| 0 pack‐years | 161/592 | 1.0 (reference) | 18/592 | 1.0 (reference) | 42/592 | 1.0 (reference) | 86/592 | 1.0 (reference) |
| <10 pack‐years | 101/340 | 1.1 (0.8, 1.4) | 10/340 | 1.0 (0.5, 2.0) | 35/340 | 1.4 (0.8, 2.2) | 62/340 | 1.2 (0.8, 1.7) |
| HLA SE positive | ||||||||
| 0 pack‐years | 184/636 | 1.0 (0.8, 1.3) | 97/636 | 5.0 (3.0, 8.4) | 63/636 | 1.4 (0.9, 2.1) | 441/636 | 4.8 (3.7, 6.2) |
| <10 pack‐years | 119/402 | 1.1 (0.8, 1.4) | 68/402 | 5.1 (3.0, 8.8) | 52/402 | 1.7 (1.1, 2.6) | 374/402 | 6.3 (4.8, 8.3) |
| APc | −0.06 (−0.5, 0.3) | 0.04 (−0.3, 0.4) | −0.02 (−0.5, 0.5) | 0.2 (0.05, 0.4) | ||||
| 10–20 pack‐years of smoking | ||||||||
| HLA SE negative | ||||||||
| 0 pack‐years | 161/592 | 1.0 (reference) | 18/592 | 1.0 (reference) | 42/592 | 1.0 (reference) | 86/592 | 1.0 (reference) |
| 10–20 pack‐years | 59/198 | 1.1 (0.8, 1.5) | 6/198 | 1.2 (0.5, 3.0) | 25/198 | 2.0 (1.2, 3.5) | 59/198 | 2.3 (1.6, 3.4) |
| HLA SE positive | ||||||||
| 0 pack‐years | 184/636 | 1.0 (0.8, 1.3) | 97/636 | 5.0 (3.0, 8.4) | 63/636 | 1.4 (0.9, 2.1) | 441/636 | 4.8 (3.7, 6.3) |
| 10–20 pack‐years | 61/220 | 1.0 (0.7, 1.4) | 47/220 | 8.5 (4.8, 15.3) | 33/220 | 2.4 (1.4, 3.9) | 317/220 | 11.4 (8.5, 15.3) |
| APc | −0.09 (−0.6, 0.4) | 0.4 (0.1, 0.7) | −0.06 (−0.6, 0.5) | 0.5 (0.3, 0.6) | ||||
| >20 pack‐years of smoking | ||||||||
| HLA SE negative | ||||||||
| 0 pack‐years | 161/592 | 1.0 (reference) | 18/592 | 1.0 (reference) | 42/592 | 1.0 (reference) | 86/592 | 1.0 (reference) |
| >20 pack‐years | 71/226 | 1.2 (0.8, 1.6) | 7/226 | 1.5 (0.6, 3.6) | 31/226 | 2.5 (1.5, 4.1) | 84/226 | 3.3 (2.3, 4.6) |
| HLA SE positive | ||||||||
| 0 pack‐years | 184/636 | 1.1 (0.8, 1.3) | 97/636 | 5.0 (3.0, 8.4) | 63/636 | 1.4 (0.9, 2.1) | 441/636 | 4.8 (3.7, 6.2) |
| >20 pack‐years | 81/226 | 1.3 (0.98, 1.8) | 45/226 | 9.2 (5.1, 16.6) | 41/226 | 3.1 (2.0, 5.0) | 475/226 | 18.2 (14, 24) |
| APc | 0.09 (−0.3, 0.5) | 0.4 (0.1, 0.7) | 0.09 (−0.4, 0.5) | 0.6 (0.5, 0.7) | ||||
All analyses were adjusted for age, sex, residential area, and ancestry. Anti‐CCP2 = anti–cyclic citrullinated peptide 2; RF = rheumatoid factor; OR = odds ratio; 95% CI = 95% confidence interval.
Values are the number of exposed cases and controls.
Attributable proportion (AP) due to interaction between the HLA shared epitope (SE) and smoking.
Table 5.
Odds of developing rheumatoid arthritis, stratified by serologic subset, in subjects categorized by the number of HLA SE alleles and smoking statusa
| Anti‐CCP2+, RF− | Anti‐CCP2+, RF+ | |||||
|---|---|---|---|---|---|---|
| No. cases/no. controlsb | OR (95% CI) | APc | No. cases/no. controlsb | OR (95% CI) | APc | |
| HLA SE | ||||||
| 0 alleles | ||||||
| Never smoker | 18/592 | 1.0 (reference) | 86/592 | 1.0 (reference) | ||
| Ever smoker | 23/764 | 1.1 (0.6–2.0) | 205/764 | 1.8 (1.4–2.4) | ||
| 1 allele | ||||||
| Never smoker | 59/501 | 4.0 (2.3–6.8) | 264/501 | 3.6 (2.7–4.7) | ||
| Ever smoker | 100/697 | 5.3 (3.2–8.8) | 0.2 (0.02–0.5) | 693/697 | 6.9 (5.3–8.8) | 0.4 (0.2–0.5) |
| 2 alleles | ||||||
| Never smoker | 38/135 | 7.8 (5.4–17.8) | 177/135 | 8.8 (6.4–12.1) | ||
| Ever smoker | 60/151 | 14.0 (8.0–24.3) | 0.3 (0.04–0.7) | 473/151 | 21.5 (16.0–28.8) | 0.6 (0.4–0.7) |
Anti‐CCP2 = anti–cyclic citrullinated peptide 2; RF = rheumatoid factor; OR = odds ratio; 95% CI = 95% confidence interval.
Values are the number of exposed cases and controls.
Attributable proportion (AP) due to interaction between the HLA shared epitope (SE) and ever smoking.
A summary of the risk of RA conferred by smoking and the presence of the HLA–DR SE in the 4 different serologically defined subsets of RA patients is provided in Figure 1.
Figure 1.
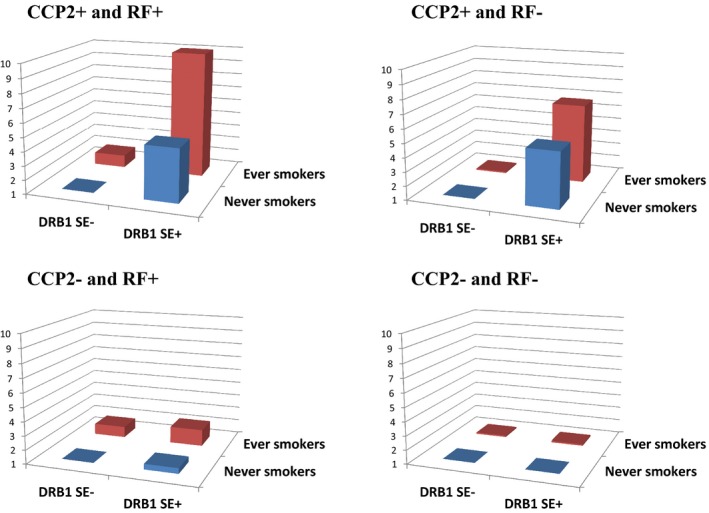
Odds ratios for the different serologic subsets of rheumatoid arthritis stratified by positivity or negativity for rheumatoid factor (RF) and anti–cyclic citrullinated peptide 2 (anti‐CCP2) antibodies, according to different combinations of HLA–DRB1 shared epitope (SE) and smoking status. Data are shown in Table 4.
Discussion
The main finding of the present study is that the impact of smoking and HLA–DRB1 genes and their interaction with regard to risk of RA varied between the 4 serologically defined subsets of RA. It has previously been difficult to distinguish the association between the HLA–DRB1 SE and ACPAs from the unique association between smoking and RF, due to the fact that sample sizes have been limited. By using well‐defined serologic subsets of RA, we have demonstrated that smoking is indeed a prominent risk factor for RF+/anti‐CCP2− RA, whereas the effect of smoking is more limited, but still existing, in RF−/anti‐CCP2+ RA. Our results are consistent with those of several previous studies in which it was suggested that smoking may generate RF and ACPAs as well as other autoantibodies in RA 13. The situation with regard to the association with the HLA–DRB1 SE was even more obvious. A clear association with the class II genes was observed for both anti‐CCP2–positive RA subsets irrespective of RF status, whereas no association was observed for the RF+/anti‐CCP2− RA subset.
Another recent study showed that RF levels are associated with ACPA positivity irrespective of smoking history, and noted that there seemed to be a difference in the importance of the number of SE alleles in determining ACPA positivity between RF‐negative and RF‐positive RA patients 10. The results of the present study confirmed a correlation between RF and anti‐CCP2 positivity in RA patients that was independent of smoking habits. However, the correlation was present regardless of HLA–DRB1 SE status (P < 0.0001).
A question that can now be addressed more distinctly than before is how smoking may be involved in the induction of RF and ACPAs (herein measured as anti‐CCP2 antibodies) in individuals with different genetic setups. Our study in the RF+/anti‐CCP2− RA subset clearly showed that smoking may induce RF independent of both the HLA–DRB1 SE and presence/induction of ACPAs. This is consistent with findings in previous studies in healthy individuals, which showed that smoking can induce RF production 14. This lack of relationship with the HLA–DRB1 SE status is also compatible with the notion of T cell–independent triggering mechanisms, as demonstrated recently by the findings of a low number of T cell–mediated somatic mutations in single RF‐producing B cells from RA patients 15. The situation was substantially different in the RF−/anti‐CCP2+ RA group, in whom there was a major and gene dose–dependent effect of HLA–DRB1 and a more limited, but still visible, effect of smoking, particularly in heavy smokers. This finding is compatible with prior reports of high numbers of T cell–dependent somatic mutations in genes coding for anticitrulline‐reactive antibodies 15, 16, 17.
The situation in the major subset of RA patients who were positive for both RF and anti‐CCP2 was also different, with major effects of both smoking and the HLA–DRB1 SE and with a pronounced interaction between the 2 risk factors. Thus, the challenge is to understand the mechanisms that explain why the gene–environment interaction between the HLA–DRB1 SE and smoking is most pronounced in conjunction with the presence of both RF and anti‐CCP2 antibodies. As previously described, the data suggest that class II major histocompatibility complex (MHC)–dependent immunity may be triggered at sites in which smoke primarily interacts with the immune system, i.e., in the lungs and related mucosal tissues 18, 19. A more precise molecular definition with regard to which structures in the HLA–DRB1 molecule are involved in this interaction has also been provided recently 20.
An obvious hypothesis for a triggering scenario would be that RF generated by T cell–independent effects of smoking 14 would enhance class II MHC–dependent T cell activation against citrullinated proteins at mucosal sites where smoke encounters the immune system. Such enhancing effects on antigen presentation from RF and other mechanisms that generate immune complexes are well known 21. This scenario may be further strengthened from the generation of RF due to reactivity of T cells against antigens present in local immune complexes 22. Taken together, these data suggest that the dramatic interaction between the HLA–DRB1 SE and smoking in conferring a risk of RF‐positive RA 8 and ACPA‐positive RA 6, 23 requires the simultaneous presence of both of these antibodies.
Interestingly, the presence of both RF and ACPAs also appears to provide the highest risk for subsequent development of RA in antibody‐positive, but still nonarthritic, individuals 24, 25. The synergizing effects between ACPAs, RF, and immune complexes have also been described in models of effector phases of joint inflammation in RA 26, 27, 28. All of these prior studies addressed in vitro–formed immune complexes, but the first report of an evaluation of ACPA‐containing immune complexes obtained in vivo was recently published 29.
Our study was designed as a case–control study with incident RA cases, and information regarding smoking habits and exposure to passive smoking was collected retrospectively. Recall bias was minimized by using incident cases of RA. We took great effort to obtain information on lifestyle factors and environmental exposures from the RA patients in a way that was identical to that used for the controls. Furthermore, the questionnaire contained a wide range of questions regarding many potential environmental risk factors, and no section in the questionnaire was given prime focus.
A potential selection bias may arise when recruiting cases and controls. The proportion of respondents with regard to participation in the EIRA study was 92% for cases and 75% for controls. Since the structure of the Swedish public health care system provides equal access to medical services for all Swedish citizens, it is most likely that almost all cases of RA are referred to public rheumatology units, and it is not likely that the few unidentified cases would cause a substantial bias in our calculations. Selection bias among controls is likely to be modest, since the prevalence of smoking among controls, seen as an indicator of lifestyle, was consistent with that observed in the general population at equivalent ages 30.
In summary, our findings describe how smoking and the HLA–DRB1 SE may play different roles in the pathogenesis of different serologically defined subsets of RA, and that RF and ACPAs appear to act together in both the triggering and the effector phases in the major RF+/ACPA+ subset of RA.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Hedström had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Rönnelid, Klareskog, Alfredsson.
Acquisition of data
Klareskog, Alfredsson.
Analysis and interpretation of data
Hedström.
Supporting information
The EIRA study was supported by the Swedish Medical Research Council, the Swedish Society for Medical Research, the Swedish Council for Health, Working Life and Welfare, the 80‐Year Foundation of King Gustaf V, the Swedish Rheumatism Foundation, Stockholm County Council, AFA, and IMU‐supported BeTheCure projects.
1Anna Karin Hedström, MD, PhD, Lars Alfredsson, PhD: Karolinska Institutet, Stockholm, Sweden; 2Johan Rönnelid, MD, PhD: Uppsala University, Uppsala, Sweden; 3Lars Klareskog, MD, PhD: Karolinska Institutet at Karolinska University Hospital, Stockholm, Sweden.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205–13. [DOI] [PubMed] [Google Scholar]
- 2. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012;44:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okada Y, Kim K, Han B, Pillai NE, Ong RT, Saw WY, et al. Risk for ACPA‐positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum Mol Genet 2014;23:6916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case‐control study, using incident cases. Ann Rheum Dis 2003;62:835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee YH, Bae SC, Song GG. Gene‐environmental interaction between smoking and shared epitope on the development of anti‐cyclic citrullinated peptide antibodies in rheumatoid arthritis: a meta‐analysis. Int J Rheum Dis 2014;17:528–35. [DOI] [PubMed] [Google Scholar]
- 6. Sparks JA, Karlson EW. The roles of cigarette smoking and the lung in the transitions between phases of preclinical rheumatoid arthritis. Curr Rheumatol Rep 2016;18:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klareskog L, Al Catrina. Autoimmunity: lungs and citrullination. Nat Rev Rheumatol 2015;11:261–2. [DOI] [PubMed] [Google Scholar]
- 8. Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene–environment interaction between smoking and shared epitope genes in HLA–DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum 2004;50:3085–92. [DOI] [PubMed] [Google Scholar]
- 9. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 10. Murphy D, Mattey D, Hutchinson D. Anti‐citrullinated protein antibody positive rheumatoid arthritis is primarily determined by rheumatoid factor titre and the shared epitope rather than smoking per se. PLoS One 2017;12:e0180655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rothman KJ. Epidemiology: an introduction. New York (NY): Oxford University Press; 2002. [Google Scholar]
- 12. Andersson T, Alfredsson L, Källberg H, Zdravkovic S, Ahlbom A. Calculating measures of biological interaction. Eur J Epidemiol 2005;20:575–9. [DOI] [PubMed] [Google Scholar]
- 13. Van Wesemael TJ, Ajeganova S, Humphreys J, Terao C, Muhammad A, Symmons DP, et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti‐citrullinated protein antibodies per se: a multicenter cohort study. Arthritis Res Ther 2016;18:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jónsson T, Thorsteinsson J, Valdimarsson H. Does smoking stimulate rheumatoid factor production in non‐rheumatic individuals? APMIS 1998;106:970–4. [DOI] [PubMed] [Google Scholar]
- 15. Lu DR, McDavid AN, Kongpachith S, Lingampalli N, Glanville J, Ju CH, et al. T cell–dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol 2018;70:1732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steen J, Forsström B, Sahlström P, Odowd V, Israelsson L, Krishnamurthy A, et al. Recognition of amino acid motifs, rather than specific proteins, by human plasma cell–derived monoclonal antibodies to posttranslationally modified proteins in rheumatoid arthritis. Arthritis Rheumatol 2018;71:196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lloyd KA, Steen J, Amara K, Titcombe PJ, Israelsson L, Lundström SL, et al. Variable domain N‐linked glycosylation and negative surface charge are key features of monoclonal ACPA: implications for B‐cell selection. Eur J Immunol 2018;48:1030–45. [DOI] [PubMed] [Google Scholar]
- 18. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol 2018;14:542–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kharlamova N, Jiang X, Sherina N, Potempa B, Israelsson L, Quirke AM, et al. Antibodies to porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol 2016;68:604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim K, Jiang X, Cui J, Lu B, Costenbader KH, Sparks JA, et al. Interactions between amino acid–defined major histocompatibility complex class II variants and smoking in seropositive rheumatoid arthritis. Arthritis Rheumatol 2015;67:2611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen‐antibody complexes by rheumatoid factor B cells. J Exp Med 1991;173:487–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tarkowski A, Czerkinsky C, Nilsson LA. Simultaneous induction of rheumatoid factor‐ and antigen‐specific antibody‐secreting cells during the secondary immune response in man. Clin Exp Immunol 1985;61:379–87. [PMC free article] [PubMed] [Google Scholar]
- 23. Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006;54:38–46. [DOI] [PubMed] [Google Scholar]
- 24. Ten Brinck RM, van Steenbergen HW, van Delft MA, Verheul MK, Toes RE, Trouw LA, et al. The risk of individual autoantibodies, autoantibody combinations and levels for arthritis development in clinically suspect arthralgia. Rheumatology (Oxford) 2017;56:2145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lingampalli N, Sokolove J, Lahey LJ, Edison JD, Gilliland WR, Holers VM, et al. Combination of anti‐citrullinated protein antibodies and rheumatoid factor is associated with increased systemic inflammatory mediators and more rapid progression from preclinical to clinical rheumatoid arthritis. Clin Immunol 2018;195:119–26. [DOI] [PubMed] [Google Scholar]
- 26. Sokolove J, Johnson DS, Lahey LJ, Wagner CA, Cheng D, Thiele GM, et al. Rheumatoid factor as a potentiator of anti–citrullinated protein antibody–mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol 2014;66:813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor‐ and complement‐dependent effector functions of the disease‐specific anti‐citrullinated protein autoantibodies. J Immunol 2015;194:3664–74. [DOI] [PubMed] [Google Scholar]
- 28. Laurent L, Anquetil F, Clavel C, Ndongo‐Thiam N, Offer G, Miossec P, et al. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis‐specific immune complexes containing anticitrullinated protein antibodies. Ann Rheum Dis 2015;74:1425–31. [DOI] [PubMed] [Google Scholar]
- 29. Sohrabian A, Mathsson‐Alm L, Hansson M, Knight A, Lysholm J, Cornillet M, et al. Number of specific ACPA in immune complexes from synovial fluid, but not conventional anti‐CCP levels, associate with inflammation and joint destruction in rheumatoid arthritis. Ann Rheum Dis 2018;77:1345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. SCB . Statistics Sweden; 2018. URL: http://www.scb.se.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials