

Abstract
Toxoplasma gondii causes retinitis and encephalitis. Avoiding targeting by autophagosomes is key for its survival because T. gondii cannot withstand lysosomal degradation. During invasion of host cells, T. gondii triggers epidermal growth factor receptor (EGFR) signalling enabling the parasite to avoid initial autophagic targeting. However, autophagy is a constitutive process indicating that the parasite may also use a strategy operative beyond invasion to maintain blockade of autophagic targeting. Finding that such a strategy exists would be important because it could lead to inhibition of host cell signalling as a novel approach to kill the parasite in previously infected cells and treat toxoplasmosis. We report that T. gondii induced prolonged EGFR autophosphorylation. This effect was mediated by PKCα/PKCβ ➔ Src because T. gondii caused prolonged activation of these molecules and their knockdown or incubation with inhibitors of PKCα/PKCβ or Src after host cell invasion impaired sustained EGFR autophosphorylation. Addition of EGFR tyrosine kinase inhibitor (TKI) to previously infected cells led to parasite entrapment by LC3 and LAMP‐1 and pathogen killing dependent on the autophagy proteins ULK1 and Beclin 1 as well as lysosomal enzymes. Administration of gefitinib (EGFR TKI) to mice with ocular and cerebral toxoplasmosis resulted in disease control that was dependent on Beclin 1. Thus, T. gondii promotes its survival through sustained EGFR signalling driven by PKCα/β ➔ Src, and inhibition of EGFR controls pre‐established toxoplasmosis.
Keywords: infection, microbial–cell interaction, protozoa
1. INTRODUCTION
Pathogens can manipulate host cell signalling cascades as strategies to promote their survival. Studies on how microbes accomplish this task not only advance our understanding of host–pathogen interactions, but, importantly, they open up the possibility of novel therapeutic strategies based on pharmacologic approaches to block these cascades. Toxoplasma gondii is an obligate intracellular protozoan that is the most common cause of infectious retinitis in the world and can cause encephalitis in immunosuppressed patients. Current antibiotic regimens against ocular toxoplasmosis are suboptimal because they have no effect on visual outcome or recurrence rates (Bosch‐Driessen, Berrendschot, Ongkosuwito, & Rothova, 2002; Rothova et al., 1993). In addition, regimens against ocular and cerebral toxoplasmosis can have significant side effects (Neville et al., 2015). Identification of host‐directed therapies against T. gondii could lead to a combined approach of antimicrobial agents plus manipulation of host cell signalling to improve treatment against toxoplasmosis.
T. gondii cannot withstand the lysosomal environment within host cells. Thus, avoidance of the lysosomal compartment is central to survival of the parasite. One mechanism that leads pathogens to lysosomal degradation follows the classical pathway of fusion with early endosomes, late endosomes, and lysosomes (Pauwels, Trost, Beyaert, & Hoffmann, 2017). T. gondii avoids this pathway of lysosomal degradation by blocking host type I transmembrane proteins from entering the membrane of the parasitophorous vacuole (PV) that contains intracellular tachyzoites (Besteiro, Dubremetz, & Lebrun, 2011; Mordue, Desai, Dustin, & Sibley, 1999). Thus, endosomes and lysosomes are not recruited, and the PV does not follow the path of classical lysosomal degradation (Besteiro et al., 2011; Mordue et al., 1999).
Macroautophagy (called herein autophagy) is another pathway that targets pathogens to lysosomal degradation (Levine, Mizushima, & Virgin, 2011). Autophagy is a homeostatic mechanism whereby an autophagosome encircles a portion of the cytoplasm or an organelle (Klionsky & Emr, 2000; Mizushima, Ohsumi, & Yoshimori, 2002, Yoshimori, 2004). The autophagosome delivers its cargo to the lysosomal compartment for degradation (Klionsky & Emr, 2000). Ligation of CD40 causes targeting of T. gondii by autophagosomes leading to parasite killing mediated by lysosomal degradation. CD40 and CD154 (CD40 ligand) are required for resistance against cerebral and ocular toxoplasmosis (Portillo et al., 2010; Reichmann et al., 2000; Subauste, Wessendarp, Sorensen, & Leiva, 1999). CD40 expressed in endothelial cells diminishes parasite invasion of the retina and brain by inducing autophagic killing of T. gondii (Portillo et al., 2019). In addition, CD40–CD154 also likely protect via autophagic degradation of the parasite in microglia/macrophages (Portillo et al., 2010). Although IFN‐γ, a key mediator of protection against acute and chronic toxoplasmosis, requires some autophagy proteins to disrupt the membrane of the PV in mouse cells and kill the parasite, this process does not act via lysosomal degradation and, thus, has not been found to be bona fide autophagy (Choi et al., 2014).
CD40 ligation acts on ULK1, a central initiator of autophagy in mammalian cells (Chan, Longatti, McKnight, & Tooze, 2009, Itakura & Mizushima, 2010, Russell et al., 2013), causing its S555 phosphorylation (marker of ULK1 activation; Liu, Lopez Corcino, Portillo, Miao, & Subauste, 2016), induces dissociation of Beclin 1 from Bcl‐2 (Liu et al., 2016), a process that enables Beclin 1 to stimulate autophagosome formation (Pattingre et al., 2005), and activates PKR (Ogolla et al., 2013), a protein that stimulates autophagy (Talloczy, Virgin, & Levine, 2006). As a result, CD40 increases autophagosome formation and maturation to autolysosomes (autophagy flux; Ogolla et al., 2013, Van Grol, Muniz‐Feliciano, Portillo, Bonilha, & Subauste, 2013, Liu et al., 2016). CD40 ligation in haematopoietic and nonhaematopoietic cells from humans and mice leads to accumulation of the autophagy protein LC3 around the parasite followed by recruitment of the lysosomal marker LAMP‐1 and killing of T. gondii that is dependent on ULK1 (the kinase that triggers autophagy) as well as PKR, the autophagy proteins Beclin 1, ATG5, ATG7, and lysosomal enzymes (Andrade, Wessendarp, Gubbels, Striepen, & Subauste, 2006; Liu et al., 2016; Ogolla et al., 2013; Portillo et al., 2010; Van Grol et al., 2013).
Autophagy occurs constitutively indicating that T. gondii must deploy strategies to avoid being targeted by autophagosomes. In studies conducted during the early phase of T. gondii infection, we showed that invasion by T. gondii causes epidermal growth factor receptor (EGFR) signalling in various cell types from humans and rodents (epithelial cells, endothelial cells, and microglia), and two signalling pathways downstream of EGFR signalling enable the parasite to activate Akt and STAT3, negative regulators of autophagy that protect the parasite from initial targeting by autophagy (Muniz‐Feliciano et al., 2013; Portillo et al., 2017). In the first pathway, parasite micronemal proteins (MICs) with EGF domains (MIC3 and MIC6) induce EGFR autophosphorylation that in turn activates Akt (Muniz‐Feliciano et al., 2013). In the second pathway, focal adhesion kinase (FAK) becomes activated at the level of the moving junction (site of penetration into the host cell), causing Src‐mediated transactivation of EGFR (Y845 phosphorylation) and activation of STAT3, that in turn prevents PKR from becoming activated leading to inhibition of autophagic targeting of the parasite (Portillo et al., 2017). The Akt and STAT3 signalling pathways become activated independently (Portillo et al., 2017). Blockade of these signalling cascades prior to infection results in accumulation of double membrane structures, LC3, and LAMP‐1 around the PV and killing of T. gondii dependent on ULK1, Beclin 1, ATG7, and lysosomal enzymes (Muniz‐Feliciano et al., 2013; Portillo et al., 2017).
The mechanisms described above occur during invasion of host cells. Finding that T. gondii uses a strategy to persistently block autophagic targeting beyond host cell invasion would be important because it would indicate that pharmacologic approaches that prevent the parasite from activating this survival strategy could represent a novel host‐directed therapy that would cause lysosomal degradation of T. gondii in previously infected cells without the need for immune activation of these cells. Herein, we report that T. gondii induces prolonged autophosphorylation of EGFR in host cells that is driven by prolonged activation of the PKCα/PKCβ ➔ Src pathway. Addition of the EGFR tyrosine kinase inhibitor (TKI) gefitinib to cells already infected with T. gondii results in parasite killing dependent on the autophagy machinery. Moreover, treatment with gefitinib leads to control of ocular and cerebral toxoplasmosis in mice with pre‐established disease, a protective effect that is not dependent on enhanced cellular or humoral immunity but that requires normal expression of the autophagy protein Beclin 1. These studies identified EGFR as a potential therapeutic target against toxoplasmosis.
2. RESULTS
2.1. T. gondii causes prolonged EGFR autophosphorylation
In studies that were conducted between 2 to 30 min postchallenge with T. gondii tachyzoites, we reported that the parasite causes EGFR autophosphorylation dependent on MIC3 and MIC6, parasite adhesins deployed during invasion of host cells (Muniz‐Feliciano et al., 2013). To determine whether EGFR autophosphorylation can persist beyond the process of host cell invasion, mammalian cells were infected with the uracil auxotroph cps strain of T. gondii. This parasite strain is unable to replicate in the absence of exogenous uracil in culture medium (Fox & Bzik, 2010). Thus, the use of nonreplicating tachyzoites (no exogenous uracil) would prevent reactivation of EGFR due to potential new cycles of invasion. Human retinal pigment epithelial (RPE) cells were challenged with cps T. gondii followed by extensive washing to remove extracellular tachyzoites. T. gondii caused prolonged phosphorylation of Y1068 EGFR, a major residue that is autophosphorylated during EGFR activation (Figure 1a). In contrast to T. gondii, incubation with EGF caused short‐lived Y1068 phosphorylation of EGFR (Figure 1a). Next, we examined whether intracellular replication affected prolonged phosphorylation of Y1068 EGFR. Addition of uracil after challenge with T. gondii caused prolonged Y1068 EGFR phosphorylation that appeared similar to that observed in infected RPE cells incubated without uracil (Figure 1a). Prolonged EGFR Y1068 phosphorylation was also observed in mouse endothelial cells infected with cps T. gondii as well as RPE cells infected with PTG T. gondii (type II strain) and BV‐2 cells infected with RH T. gondii (type I strain; Figure 1b). T. gondii also caused prolonged phosphorylation of Y1173, another major residue autophosphorylated during EGFR activation (Figure 1c). Altogether, T. gondii causes sustained EGFR autophosphorylation in various mammalian cells.
Figure 1.
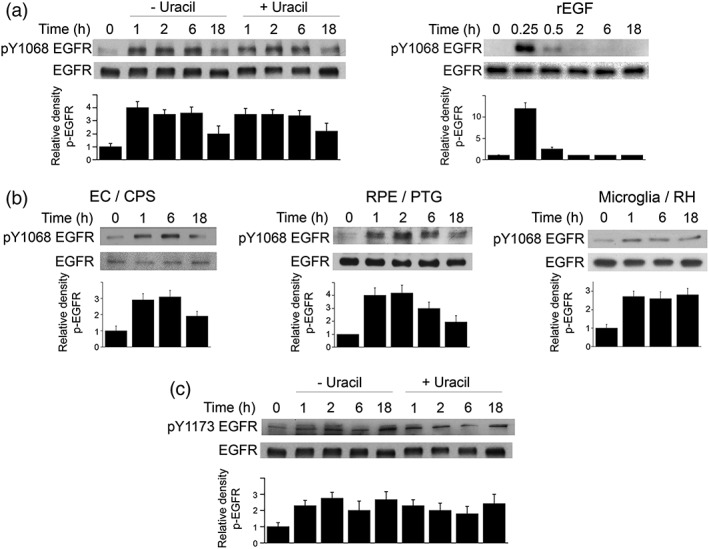
Toxoplasma gondii induces prolonged EGFR autophosphorylation in mammalian cells. (a) Human retinal pigment epithelial (RPE) cells were challenged with tachyzoites of the cps strain of T. gondii and cultured in the absence or presence of uracil. RPE cells were also incubated with recombinant human EGF. Cell lysates were obtained at the indicated time points and used to probe for total EGFR and phospho‐Y1068 EGFR. Relative density of phospho‐EGFR signal was obtained by normalisation to total EGFR signal followed by normalisation relative to the uninfected control samples (0‐min time point). Relative density of phospho‐EGFR for uninfected samples was given a value of 1. (b) Mouse endothelial cells (mHEVc), RPE cells, and microglia (BV‐2) were challenged with tachyzoites of cps, PTG (type II strain), or RH (type I strain) T. gondii as indicated. Cell lysates were analysed as above. (c) Lysates from RPE cells challenged with cps T. gondii were probed for total EGFR and phospho‐Y1173 EGFR. Data shown are representative of three to four independent experiments. Densitometry data represent mean + SEM of three to four independent experiments
2.2. Src promotes prolonged EGFR autophosphorylation in T. gondii‐infected cells
Ligand‐dependent EGFR activation can be induced not only by EGF but also by other ligands that include heparin‐binding EGF‐like growth factor, amphiregulin, and TGF‐α (Gschwind, Fischer, & Ullrich, 2004; Higashiyama et al., 2008; Liebmann, 2010). These ligands exist as membrane‐bound proteins and are released as soluble proteins when cleaved by a disintegrin and metalloproteinases (ADAMs; Gschwind et al., 2004, Higashiyama et al., 2008, Liebmann, 2010). Parasite MIC3 and MIC6 but not host cell ADAMs drive EGFR activation in the early stages after T. gondii infection (Muniz‐Feliciano et al., 2013). However, it is possible that ADAM‐mediated release of endogenous EGFR ligands may drive prolonged EGFR activation. One hour prior to infection with T. gondii RPE cells were incubated with vehicle or the broad‐spectrum ADAM inhibitor GM6001, a potent inhibitor of release of autologous EGFR ligands. GM6001 did not impair prolonged phosphorylation of Y1068 EGFR induced by T. gondii (Figure 2a). In contrast, GM6001 inhibited EGFR activation in RPE cells incubated with lysophosphatidic acid, an inducer of EGFR ligands release (Muniz‐Feliciano et al., 2013; Figure 2a).
Figure 2.
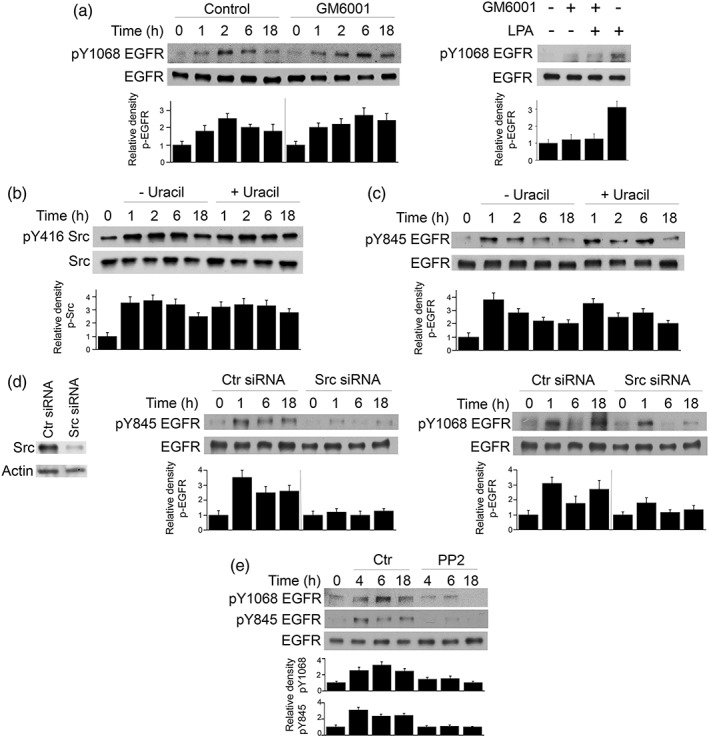
Src promotes prolonged EGFR autophosphorylation during Toxoplasma gondii infection. (a) RPE cells were treated with or without the ADAM inhibitor GM6001 beginning 1 hr before challenge with cps T. gondii. Cell lysates were obtained at the indicated time points and used to examine total EGFR and phospho‐Y1068 EGFR by immunoblot. A vertical line was inserted between densitometry data from control and GM6001‐treated cells to indicate that band densities from infected cells treated with or without GM6001 were compared with bands from their respective uninfected cells, which were given an arbitrary number of 1. RPE cells were also treated with or without lysophosphatidic acid in the presence or absence of GM6001. Cell lysates were obtained at 15 min and subjected to immunoblotting. (b, c) RPE cells were challenged with cps T. gondii, cultured with or without uracil and lysed at the indicated time points. Cell lysates were used to examine expression of total Src and phospho‐Y416 Src by immunoblot (b) or total EGFR and phospho‐Y845 EGFR (c). Relative densities of phospho‐Src and phospho‐EGFR for uninfected samples were given a value of 1. (d) RPE cells transfected with control or Src siRNA were incubated with cps T. gondii. Expression of total EGFR, phospho‐Y845 EGFR, and phospho‐Y1068 was assessed by immunoblot. A vertical line was inserted between densitometry data of lysates from control and Src siRNA‐treated cells to indicate that relative densities of phospho‐EGFR from infected cells transfected with control or Src siRNA were compared with bands from their respective uninfected cells. Relative density of phospho‐EGFR for uninfected samples was given a value of 1. (e) RPE cells were incubated with PP2 or vehicle 2 hr after challenge with cps T. gondii. Cell lysates were obtained and used to probe for total EGFR, phospho‐Y845 EGFR, and phospho‐Y1068. Data shown are representative of three to four independent experiments. Densitometry data represent mean + SEM of three to four experiments
EGFR can also be transactivated in a ligand‐independent manner, a process that can be mediated by Src (Drube, Stirnweiss, Valkova, & Liebmann, 2006; Tice, Biscardi, Nickles, & Parsons, 1999). To determine if Src mediates prolonged EGFR activation, we first examined whether T. gondii induced prolonged Src activation. RPE cells incubated with cps T. gondii exhibited prolonged phosphorylation of Y416 of Src (a hallmark of Src activation; Figure 2b). Similar to EGFR autophosphorylation, Src activation occurred in the absence of parasite replication (cells infected with cps T. gondii and incubated without uracil) and did not appear to be affected by addition of uracil (Figure 2b). To begin to determine whether Src modulates prolonged EGFR signalling, we first examined whether T. gondii causes prolonged EGFR phosphorylation at the unique Y845 residue, the hallmark of EGFR transactivation caused by binding of Src to EGFR (Biscardi et al., 1999; Kloth et al., 2003; Sato, Nagao, Iwasaki, Nishihira, & Fukami, 2003; Tice et al., 1999). Indeed, T. gondii caused prolonged phosphorylation of Y845 of EGFR (Figure 2c). Moreover, knockdown of Src ablated sustained Y845 phosphorylation of EGFR (Figure 2d). Interestingly, Src knockdown also markedly reduced Y1068 EGFR autophosphorylation, an effect that appeared to be more pronounced at 6 and 18 hr postchallenge compared with 1 hr postchallenge (Figure 2d). These results could be compatible with the report that MIC3 and MIC6 mediate Y1068 EGFR autophosphorylation observed during at least 30 min postinfection with T. gondii (Muniz‐Feliciano et al., 2013). Finally, addition of the Src inhibitor PP2 2 hr postinfection with T. gondii inhibited Y1068 autophosphorylation of EGFR (Figure 2e). Taken together, T. gondii causes prolonged Src activation, and sustained EGFR autophosphorylation is largely dependent on Src.
2.3. T. gondii‐induced PKCα and PKCβ activation promotes prolonged Src and EGFR activation
We previously reported that FAK mediates Src activation during the process of host cell invasion by T. gondii (Portillo et al., 2017). Indeed, addition of a FAK inhibitor prior to infection ablates phosphorylation of Y416 of Src (Portillo et al., 2017). In contrast, addition of the FAK inhibitor after invasion did not affect prolonged Src Y416 phosphorylation (Figure 3a). Thus, studies were conducted to identify an alternative explanation for sustained Src activation. Protein kinase C (PKC) are widely expressed cytosolic serine/threonine kinases that can activate Src and function though this signalling molecule (Brandt et al., 2003; Brandt, Gimona, Hillmann, Haller, & Mischak, 2002; Li et al., 2015). Moreover, studies performed within 30 min after challenge with T. gondii revealed that PKCα and PKCβ translocate to the membrane of host cells, an event associated with PKC activation (Masek et al., 2006). To begin to determine whether PKCα and PKCβ promote prolonged Src and EGFR phosphorylation, we examined whether T. gondii causes sustained phosphorylation of T638 and T641 of PKCα and in PKCβ, respectively, classical markers of PKC activation. RPE cells infected with cps T. gondii exhibited prolonged phosphorylation of PKCα and PKCβ (Figure 3b). Knockdown of PKCα or PKCβ partially reduced phosphorylation of Y416 Src in T. gondii‐infected cells (Figure 3c). We examined the effect of combined knockdown of both PKCα and PKCβ because both isoforms can function cooperatively (Preiss, Namgaladze, & Brune, 2007; Rudkouskaya, Chernoguz, Haskew‐Layton, & Mongin, 2008). Indeed, knockdown of PKCα plus PKCβ markedly inhibited phosphorylation of Y416 Src (Figure 3c). Knockdown of PKCα plus PKCβ impaired Y1068 autophosphorylation of EGFR (Figure 3d). Moreover, addition of Gö 6976 (selective inhibitor PKCα and PKCβ) 2 hr after challenge with T. gondii inhibited not only phosphorylation of Y416 Src but also phosphorylation of Y1068 EGFR (Figure 3e). The role of PKCα and PKCβ also applies to a type II strain of T. gondii because similar results were obtained when Gö 6976 was added 2 hr after challenge with PTG T. gondii (Figure 3f). Altogether, T. gondii causes prolonged PKCα/PKCβ‐dependent Src signalling that in turn promotes prolonged EGFR autophosphorylation.
Figure 3.
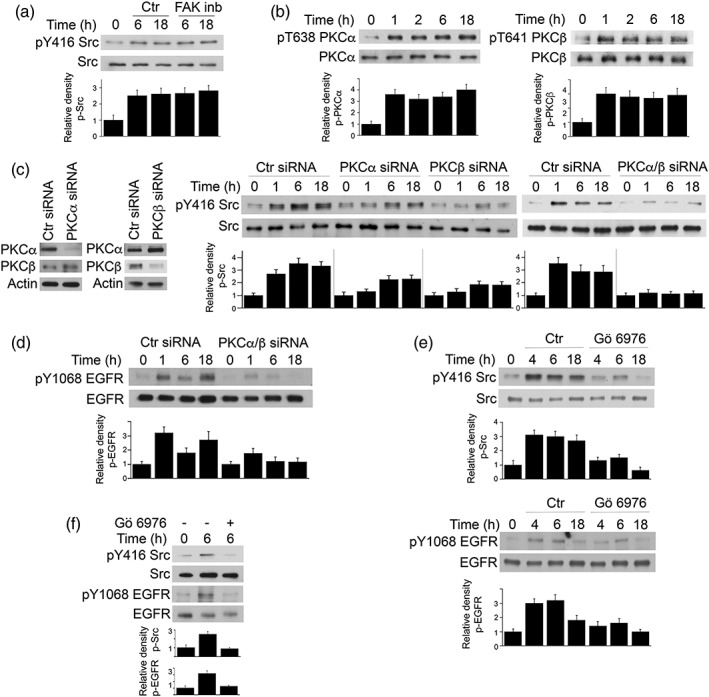
PKCα and PKCβ mediate prolonged Src and EGFR activation in Toxoplasma gondii‐infected cell. (a) RPE cells were incubated with FAK inhibitor PF‐573228 or vehicle 2 hr after challenge with cps T. gondii. Cell lysates were obtained and used to probe for total Src and phospho‐Y416 Src. Relative density of phospho‐Src for uninfected samples was given a value of 1. (b) RPE cells were challenged with cps T. gondii. Total PKCα, phospho‐T638 PKCα, total PKCβ, and phospho‐T641 PKCβ were assessed by immunoblot. Relative density of phospho‐PKC for uninfected samples was assessed as above. (c, d) RPE cells transfected with control, PKCα, and/or PKCβ siRNA were incubated with cps T. gondii. Expression of total Src and phospho‐Y416 Src (c) and total EGFR and phospho‐Y1068 EGFR (d) was assessed by immunoblot. (e, f) RPE cells were incubated with Gö 6976 or vehicle 2 hr after challenge with cps (e) or PTG T. gondii (f). Cell lysates were obtained and used to probe for total Src and phospho‐Y416 Src, total EGFR, and phospho‐Y1068 EGFR. Data shown are representative of three to four independent experiments. Densitometry data represent mean + SEM of three to four independent experiments
2.4. Pharmacologic inhibition of EGFR causes autophagic killing of T. gondii in cells previously infected with the parasite
EGFR is a well‐characterised therapeutic target in oncology, and EGFR inhibitors have been used in clinical practice for over 10 years (Lurje & Lenz, 2009). Thus, in order to examine the functional relevance of our findings, we tested whether inhibition of EGFR signalling in cells previously infected with T. gondii affected the survival of the parasite. Addition of EGFR TKI AG1478 or gefitinib 2 hr after challenge with T. gondii impaired phosphorylation of EGFR Y1068 (autophosphorylation) and EGFR Y845 (transactivation of EGFR; Figure 4a). AG1478 or gefitinib added 2 hr postinfection caused a dose‐dependent reduction in parasite load (Figure 4b). AG1478 at 0.1 μM and gefitinib at 1 μM still exhibited potent anti‐T. gondii activity, concentrations that are frequently reported to yield diminished cytostatic activity against various cancer cell lines (Di Gennaro et al., 2003; Fung, Yu, Ye, & Tannock, 2013). Similar results were observed in cells infected with a type II strain of T. gondii (Figure S1A). Anti‐T. gondii activity was detected not only in RPE but also in endothelial cells and microglia, even when the EGFR TKI was added 6 hr postinfection (Figure 4c). The EGFR TKI reduced the percentages of infected cells, parasite containing vacuoles per 100 cells and tachyzoites per 100 cells, effects that were similar to those observed when the inhibitor was added prior to infection with T. gondii (Figure 4c). However, the EGFR TKI did not significantly affect the number of tachyzoites per vacuole in the cells that remained infected (Figure 4c). These results indicate that the effect of pharmacologic inhibition of EGFR was to induce parasite killing rather than reduce intracellular replication. The effect on EGFR TKI was specific because incubation with EGFR TKI enhanced killing of T. gondii only in EGFR+ but not in EGFR− cells (Figure S1B). Similarly, a Src inhibitor (PP2) and a PKCα/PKCβ inhibitor (Gö 6976) enhanced killing of T. gondii only in cells with normal expression of Src and PKCα/PKCβ, respectively (Figure S1C, D). The kinase inhibitors did not affect parasite replication (tachyzoites/vacuole) arguing against a direct effect on T. gondii. Taken together, addition of relatively low concentrations of EGFR TKI after T. gondii infection caused parasite killing in various cell types.
Figure 4.
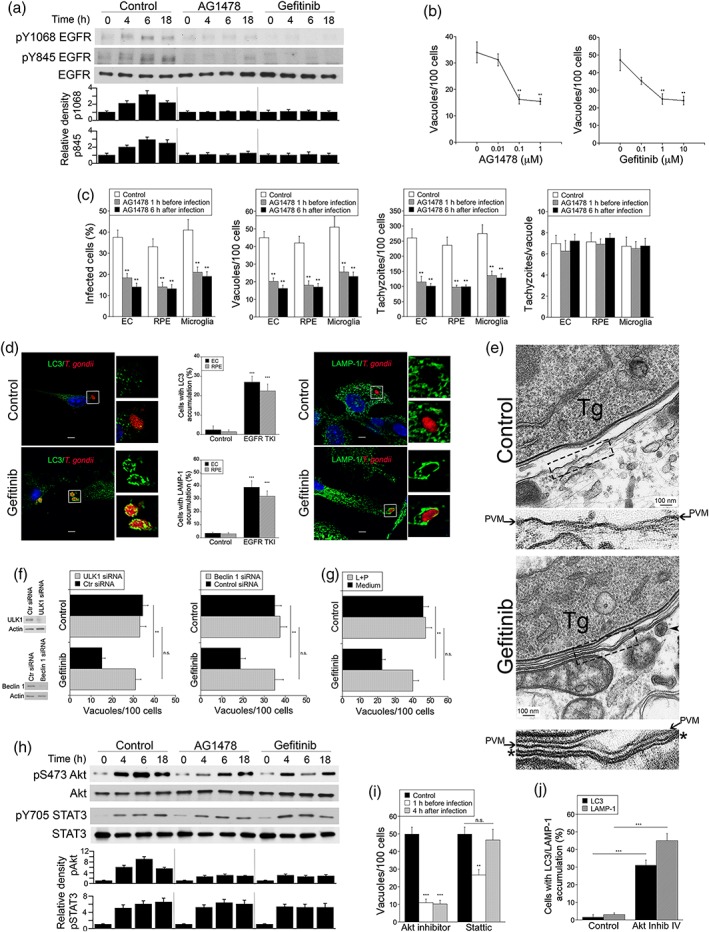
EGFR TKI added after infection with Toxoplasma gondii inhibit parasite‐induced Akt activation and trigger parasite killing. (a) RPE cells were challenged with cps T. gondii. AG1478, gefitinib, or vehicle were added 2 hr after infection. Cell lysates were probed for total EGFR, phospho‐Y1068, and phospho‐Y845 EGFR. Relative densities for uninfected samples were given a value of 1. (b) RPE cells were challenged with RH T. gondii. Different concentrations of AG1478 or gefitinib were added 2 hr after challenge with T. gondii. Monolayers were examined at 24 hr to determine the numbers of T. gondii‐containing vacuoles per 100 cells. (c) Human brain microvascular endothelial cells (EC), RPE, and the microglia cell line BV‐2 were challenged with RH T. gondii. AG1478 (0.1 μM) or vehicle were added either 1 hr prior or 6 hr after challenge with T. gondii as indicated. Monolayers were examined at 24 hr to determine the percentages of infected cells, the numbers of T. gondii‐containing vacuoles, and tachyzoites per 100 cells as well as the numbers of parasites per vacuole. (d) Human brain microvascular endothelial cells and RPE cells were challenged with T. gondii‐RFP (RH) and incubated with or without gefitinib beginning at 2 hr postchallenge. Expression of LC3 and LAMP‐1 was examined by immunofluorescence 6 and 8 hr after addition of gefitinib, respectively. Original magnification 600×. Bar, 5 μm. Images shown represent endothelial cells. Bar graphs depict the percentages of vacuoles in endothelial cells and RPE cells that were surrounded by LC3 or LAMP‐1. (e) Human brain microvascular endothelial cells were treated with or without gefitinib beginning 2 hr after challenge with T. gondii (Tg) and processed for electron microscopy after 6 hr. Areas within the boxes are magnified at the bottom. Arrows indicate the PVM; asterisk (*) indicates the double membrane structure around the vacuole. Arrowhead indicates a likely lysosome. Bar, 100 nm. (f) RPE cells were transfected with control siRNA, ULK1 siRNA, or Beclin siRNA followed by treatment with or without gefitinib starting 2 hr postchallenge with RH T. gondii. Monolayers were examined by light microscopy 24 hr postinfection. (g) RPE cells were infected with RH T. gondii. Gefitinib with or without leupeptin plus pepstatin (L + P) were added postinfection, and monolayers were examined microscopically 24 hr postchallenge. (h) RPE cells were challenged with RH T. gondii. AG1478, gefitinib, or vehicle were added 2 hr after infection. Cell lysates were probed for total Akt, phospho‐S473 Akt, total STAT3, and phospho Y705 STAT3. Relative densities for uninfected samples were given a value of 1. (i) RPE cells were challenged with RH T. gondii. Akt inhibitor IV, Stattic, or vehicle were added either 1 hr prior or 2 hr after challenge with T. gondii as indicated. Monolayers were examined as above. (j) RPE cells were challenged with T. gondii‐RFP (RH) and incubated with or without Akt inhibitor IV beginning at 2 hr postchallenge. Accumulation of LC3 and LAMP‐1 around the parasite was assessed as above. Results are shown as the mean + SEM of a representative experiment out of two to three independent experiments. **P < .01; ***P < .001
Addition of gefitinib to endothelial cells or RPE postinfection resulted in accumulation of the autophagosome marker LC3 around intracellular tachyzoites followed by the lysosome marker LAMP‐1 (Figure 4d). By electron microscopy, autophagosomes are described as a double membrane structure characterised by two parallel electron‐dense membrane layers separated by an electron‐translucent space that entraps cytoplasm or organelles (Eskelinen, 2008). Indeed, a double membrane structure with those characteristics was noted around the PVs in cells treated with gefitinib (Figure 4e). Moreover, knockdown of ULK1 or Beclin 1, critical inducers of autophagy, as well as incubation with inhibitors of lysosomal enzymes (leupeptin plus pepstatin) ablated toxoplasmacidal activity induced by addition of EGFR TKI postinfection (Figure 4f,g). Altogether, pharmacologic inhibition of EGFR that is delayed until after infection with T. gondii causes killing of the parasite dependent on the autophagy machinery.
EGFR autophosphorylation and EGFR transactivation triggered during invasion of host cells function via Akt and STAT3 respectively to avoid initial autophagic targeting of the parasite (Muniz‐Feliciano et al., 2013; Portillo et al., 2017). EGFR transactivation induced during invasion is responsible for the early stages of STAT3 signalling detected within the first hour postinfection (Portillo et al., 2017), whereas the parasite kinase ROP16 mediates STAT3 activation at later stages postinfection (Butcher et al., 2011; Saeij et al., 2007). EGFR TKI added postinfection inhibited S473 phosphorylation of Akt (Figure 4h). In contrast, addition of EGFR TKI after challenge with T. gondii failed to inhibit STAT3 Y705 phosphorylation (Figure 4h), consistent with the known role of ROP16 as driver of STAT3 signalling at time points beyond 1 hr postinfection (Butcher et al., 2011). Next, we treated host cells with inhibitors of Akt or STAT3 to examine the role of these signalling molecules in survival of the parasite at a stage postinvasion of host cells. Addition of Akt inhibitor IV before or after challenge with T. gondii resulted in a similar anti‐T. gondii activity (Figure 4i). The STAT3 inhibitor Stattic induced parasite killing if added prior to infection but not when added postparasite challenge (Figure 4i). Similar to the other inhibitors used, the effects of Akt inhibitor IV and Stattic appeared specific to their respective targets and not directed against the parasite itself (Figure S1E, F). Addition of Akt inhibitor IV postinfection resulted in accumulation of the autophagosome marker LC3 and the lysosome marker LAMP‐1 around intracellular tachyzoites (Figure 4j). Together, these results indicate that Akt but not STAT3 is a downstream signalling molecule that plays a major role in promoting parasite survival in later stages of intracellular parasitism.
2.5. Gefitinib protects against ocular and cerebral toxoplasmosis in a manner dependent on expression of the autophagy protein Beclin 1
Gefitinib reaches neural tissue and has been utilised extensively in mice including in mouse models of neurodegeneration (Wang et al., 2012). B6 mice infected with ME49 T. gondii are susceptible to ocular and cerebral toxoplasmosis and exhibit elevated rates of mortality after long‐term infection (Gazzinelli, Brezin, Li, Nussenblatt, & Chan, 1994; Gazzinelli, Eltoum, Wynn, & Sher, 1993). B6 mice were infected with 30 tissue cysts of ME49 T. gondii followed by treatment with vehicle or gefitinib (10 mg kg−1 day−1 i.p. once a day) beginning at 6 days postinfection, a time when T. gondii has already reached the CNS (Courret et al., 2006). The dose of gefitinib used is approximately 1/4–1/15 of dosages frequently used in mouse models of cancer (Ciardiello et al., 2000, Sirotnak, Zakowski, Miller, Scher, & Kris, 2000, Ciardiello & Tortora, 2001, Naruse et al., 2002, Wen et al., 2015, Liu et al., 2017). At 2 weeks after initiation of treatment, mice that received gefitinib had significantly lower T. gondii load and diminished histopathology in the eye and brain (Figure 5). Protection was not accompanied by increased ocular and cerebral mRNA levels of mediators of resistance against cerebral and ocular toxoplasmosis (Figure 6a). Similarly, systemic expression of mediators of protective cell‐mediated immunity and serum levels of anti‐T. gondii IgG were not increased in gefitinib‐treated mice (Figure 6b–f).
Figure 5.
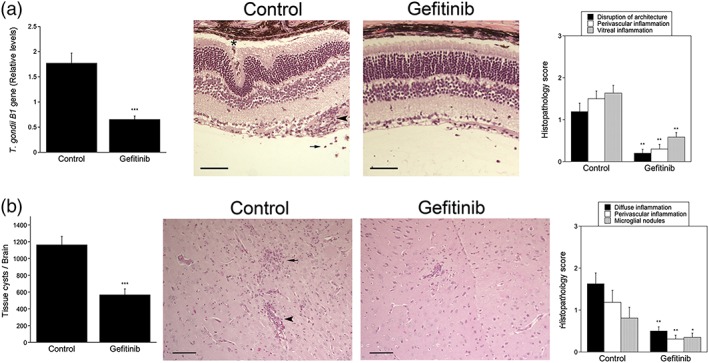
Gefitinib diminishes Toxoplasma gondii load in the eye and brain and enhances resistance to ocular and cerebral toxoplasmosis. B6 mice were infected with T. gondii tissue cysts i.p. Beginning at day 6 postinfection, mice were treated with gefitinib (10 mg kg−1 i.p. 5 days week−1) or vehicle for 14 days. The eyes and brains were collected 20 days postinfection. (a) T. gondii B1 gene was examined using qPCR. Levels were compared with those of one control mouse that was given an arbitrary value of 1. Bar graph represents mean + SEM of nine to 10 mice pooled from two experiments. The eye from infected control mouse shows more prominent disruption of retinal architecture including the formation of folds and invasion by RPE (asterisk), as well as perivascular (arrowhead) and vitreal inflammation (arrow). H&E; 200×. Bar, 50 μm. Histopathology scores are shown in a bar graph as mean + SEM of nine to 10 mice. (b) Number of T. gondii tissue cysts per brain. The brain from control mouse shows more prominent parenchymal (arrow) and perivascular inflammation (arrowhead). Periodic acid Schiff haematoxylin original magnification 200×. Bar, 50 μm. Bar graphs represent mean + SEM of nine to 10 mice. *P < .05; **P < .01; ***P < .001
Figure 6.
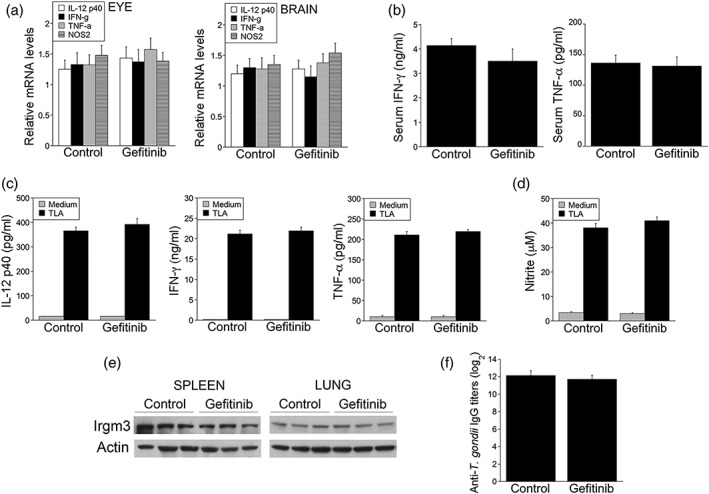
Effect of gefitinib on expression of IL‐12, IFN‐γ, TNF‐α, and NOS2 in the eye and brain as well as on the induction of systemic cellular and humoral immunity. (a) B6 mice were infected with ME49 Toxoplasma gondii, treated for 14 days with gefitinib or vehicle beginning on day 6 postinfection. Levels of IL‐12 p40, IFN‐γ, TNF‐α, and NOS2 mRNA in the eyes and brains were examined using qPCR. Each group contained nine to 10 mice pooled from two experiments. Results are shown as the mean + SEM. (b) Serum levels of IFN‐γ and TNF‐α at day 14 postinfection. (c, d) Splenocytes collected at day 14 postinfection were incubated with or without T. gondii lysate antigens (TLA), and supernatants were collected to measure IL‐12 p40, IFN‐γ, and TNF‐α (c) or measure nitric oxide by Griess reaction (d). Bars are mean + SEM of nine to 10 samples per group. (e) Spleen and lung lysates were subjected to immunoblot using Ab to IRGM3 and actin. (f) Serum titres of anti‐T. gondii IgG were measured by ELISA. Bars are mean + SEM of nine to 10 samples per group
We examined whether gefitinib was also protective when administered to mice with pre‐established ocular and cerebral toxoplasmosis and whether the protective effect of gefitinib was dependent on the expression of the autophagy protein Beclin 1. B6 mice infected with ME49 tissue cysts were treated with or without gefitinib beginning at 2 or 4 weeks postinfection, time points where retinal and cerebral histopathology has already developed (Lopez Corcino, Portillo, & Subauste, 2019). Gefitinib reduced parasite load in the eye and brain as well as the histopathology scores (Figure 7). Finally, autophagy deficient Becn1 +/− mice or autophagy sufficient Becn1 +/+ mice (B6) infected with ME49 tissue cysts were treated with or without gefitinib. In marked contrast to Becn1 +/+ mice, Becn1 +/− mice did not exhibit reduction in parasite load or histopathology in response to gefitinib administration (Figure 8). Taken together, gefitinib protects against cerebral and ocular toxoplasmosis independently of modulation of immunity against T. gondii but in a manner that requires normal levels of expression of the autophagy protein Beclin 1.
Figure 7.
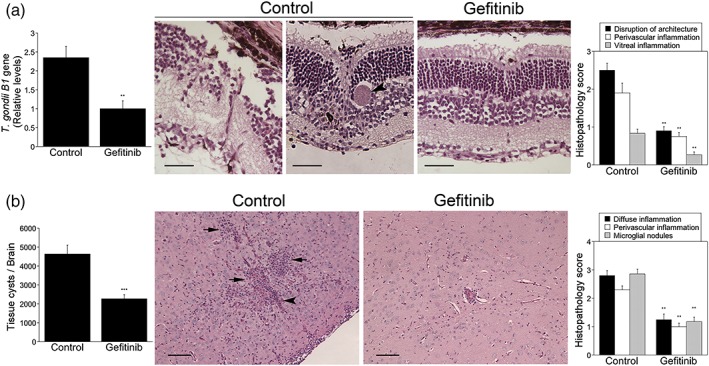
Gefitinib enhances resistance to ocular and cerebral toxoplasmosis in mice with pre‐established disease. B6 mice were infected with Toxoplasma gondii tissue cysts i.p. (a, b) Beginning at 4 weeks postinfection, mice were treated with gefitinib (10 mg kg−1 i.p. 5 days week−1) or vehicle and were euthanised after 14 days. The eyes and brains were collected 6 weeks postinfection. (a) T. gondii B1 gene was examined in the eye by using qPCR. Bar graph represents mean + SEM of 12 mice pooled from three experiments. The eyes from infected control mouse show marked disruption of retinal architecture, as well as occasional tissue cysts (arrowhead in the image to the right). The eye from the gefitinib‐treated mouse shows reduced disruption in retinal architecture. H&E; 200×. Bar, 50 μm. Histopathology scores are shown in a bar graph as mean + SEM of 12 samples per group pooled from three experiments. (b) Numbers of T. gondii tissue cysts per brain. The brain from control mouse shows more prominent parenchymal (arrows) and perivascular inflammation (arrowhead). Periodic acid Schiff haematoxylin original magnification 200×. Bar, 50 μm. Histopathology scores are shown in bar graph as mean + SEM of 12 samples per group pooled from three experiments. Bar graphs represent mean + SEM of nine mice pooled from two experiments. ** P < .01; *** P < .001
Figure 8.
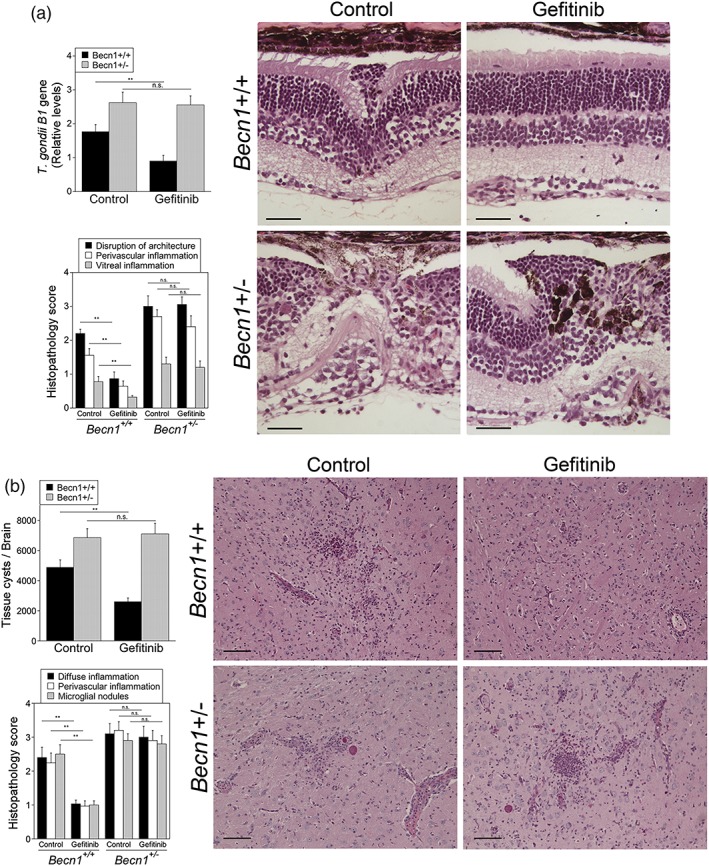
Gefitinib fails to control ocular and cerebral toxoplasmosis in Beclin 1‐deficient mice. Becn1 +/+ and Becn1 +/− mice were infected with T. gondii tissue cysts i.p., and at 2 weeks postinfection, they were treated with gefitinib or vehicle for 2 weeks. The eyes and brains were collected 4 weeks postinfection. (a) Toxoplasma gondii B1 gene was examined in the eye by using qPCR. Bar graph represents mean + SEM of nine mice pooled from two experiments. Image from gefitinib‐treated Becn1 +/+ mouse shows better preservation in retinal architecture. Gefitinib does not improve retinitis in Becn1 +/− mice. Retina from gefitinib‐treated mouse shows invasion by retinal pigment epithelial cells (arrowhead). H&E; 200×. Bar, 50 μm. Histopathology scores are shown in a bar graph as mean + SEM of nine samples per group pooled from two experiments. (b) Numbers of T. gondii tissue cysts per brain. Gefitinib does not improve encephalitis in Becn1 +/− mice. Periodic acid Schiff haematoxylin original magnification 200×. Bar, 50 μm. Bar graphs represent mean + SEM of nine mice pooled from two experiments. **P < .01; n.s., not significant
3. DISCUSSION
Approaches to manipulate host cell signalling have been proposed as novel avenues for treatment of infectious diseases (Napier et al., 2011; Reeves et al., 2005). We previously reported that T. gondii induces EGFR signalling during invasion of host cells, and inhibition of EGFR prior to invasion leads to autophagic killing of the parasite (Muniz‐Feliciano et al., 2013). However, the potential utility of pharmacologic inhibition of EGFR for control of toxoplasmosis will depend on finding whether T. gondii causes sustained EGFR signalling that enables the parasite to continually evade killing by host cells. We report that T. gondii causes prolonged autophosphorylation of EGFR in host cells. This response is driven by sustained Src activation that in turn is dependent on parasite‐induced activation of PKCα and PKCβ. Pharmacologic inhibition of EGFR signalling in cells previously infected with T. gondii leads to killing of the parasite dependent on the autophagy machinery. Killing occurred without the need for immune activation of these cells. In vivo administration of a relatively low dose of gefitinib to mice with pre‐established ocular and cerebral toxoplasmosis resulted in control of the disease that was dependent on the expression of the autophagy protein Beclin 1. Thus, these studies uncovered molecular events by which T. gondii induces sustained EGFR signalling to promote its survival and provide proof of principle that EGFR could become an adjunctive therapeutic target in these diseases, especially considering that a relatively low dose of an EGFR TKI was sufficient to induce control ocular and cerebral toxoplasmosis.
EGFR is expressed on various cells including epithelial, endothelial, and microglia (Real et al., 1986). Engagement of EGFR by ligands causes its autophosphorylation (Basu, Biswas, & Das, 1984). In the case of T. gondii, parasite MICs with EGF‐like domains (MIC3 and MIC6) cause initial EGFR autophosphorylation leading to recruitment of signalling molecules PI3K and Akt and subsequent initial inhibition of autophagic targeting of the parasite (Muniz‐Feliciano et al., 2013). However, autophagy is a constitutive process indicating that T. gondii likely utilises a strategy to persistently avoid the autophagy pathway. Our studies revealed that T. gondii caused prolonged EGFR autophosphorylation and uncovered Src as a mediator of this event. Inhibition of prolonged EGFR signalling by EGFR TKI led to recruitment of LC3 and LAMP‐1 around T. gondii. It also led to encirclement of the PV by a double membrane structure with features compatible with an autophagosome. Although autophagosomes usually measure less than 1.5 μm, they can at times be much larger and can occasionally sequester structures as large as a nucleus (Kovacs, Rez, Palfia, & Kovacs, 2000). Whether EGFR inhibition triggers the formation of a single autophagosome that encircles the PV or triggers the fusion of several autophagosomes that coalesce around the vacuole remains to be determined. Future studies that include immuno‐electron microscopy or correlative light‐electron microscopy can address these possibilities. Regardless of how an autophagosome may be formed around the PV, the recruitment of LC3 and LAMP‐1 together with the demonstration that EGFR TKI caused pathogen killing dependent on ULK1, Beclin 1, and lysosomal enzymes support that killing is mediated by autophagy.
The parasite also avoids initial autophagic targeting by triggering FAK‐dependent Src activation at the site of invasion (Portillo et al., 2017). This in turn leads to Y845 phosphorylation of EGFR and activation of STAT3, a molecule that mediates inhibition of autophagic targeting (Portillo et al., 2017). The EGFR TKI also inhibited sustained EGFR Y845 phosphorylation. However, studies of inhibition of STAT3 suggest that the role of this molecule in avoidance of autophagic killing is restricted to the early stages postinfection. STAT3 activation in T. gondii‐infected cells is biphasic (Butcher et al., 2011; Saeij et al., 2007). The initial phase is mediated by EGFR Y845 phosphorylation (Portillo et al., 2017), whereas the second phase (after 1–1.5 hr postinfection) is dependent on T. gondii ROP16 (Butcher et al., 2011; Saeij et al., 2007). Blockade of EGFR Y845‐STAT3 cascade promotes autophagic targeting of T. gondii by allowing transient (within the first hour postinfection) activation PKR‐eIF2α signalling (Portillo et al., 2017). The time frame when PKR‐eIF2α signalling is operative may explain why the ROP16–STAT3 pathway does not appear to be an important inhibitor of autophagic targeting of T. gondii (Portillo et al., 2017).
Taken together, there are three mechanisms by which T. gondii prevents targeting by autophagosomes; two that are triggered during invasion of the host cell: (a) MIC3/MIC6‐dependent EGFR autophosphorylation resulting in Akt activation (Muniz‐Feliciano et al., 2013), (b) FAK‐mediated activation of Src at the level of the moving junction leading to Src‐mediated transactivation of EGFR (Y845 phosphorylation) and STAT3 signalling (Portillo et al., 2017), and (c) a third mechanism that operates at later stages of the intracellular stage of T. gondii whereby prolonged PKCα/PKCβ‐Src signalling maintains EGFR autophosphorylation that in turn appears to function via Akt (Figure 9). Although EGFR TKI markedly impaired T. gondii‐induced sustained EGFR autophosphorylation, these agents had a partial inhibitory effect on sustained activation of Akt. These findings raise the possibility that there may be additional mechanisms besides EGFR signalling by which T. gondii causes prolonged activation of Akt, a major negative regulator of autophagy.
Figure 9.
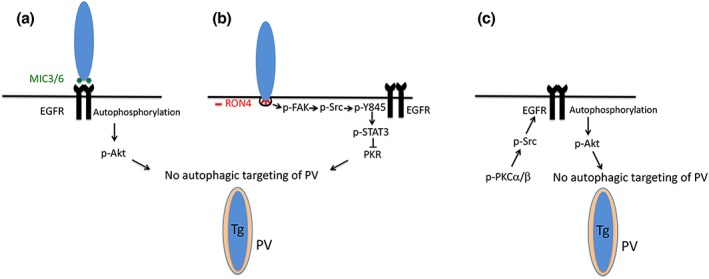
Toxoplasma gondii activates signalling cascades both during invasion of host cells and during its intracellular stage that enable the parasite to avoid autophagic targeting. (a) Parasite adhesins with EGF‐like domains (MIC3 and MIC6) induce phosphorylation in tyrosine residues in the C‐terminal end of EGFR (autophosphorylation) and activation of Akt, an inhibitor of autophagy. (b) During penetration of mammalian cells, the formation of the moving junction (characterised by the presence of rhoptry neck proteins including RON4) is accompanied by activation of FAK that leads to Src signalling. Src transactivates EGFR (Y845 phosphorylation) triggering early STAT3 signalling. This prevents activation of PKR, thus inhibiting autophagic targeting of the parasite. (c) During its intracellular stage, T. gondii causes activation of PKCα/PKCβ–Src signalling that sustains EGFR autophosphorylation maintaining blockade of autophagic targeting, an effect that appears to be mediated via Akt
Src can act downstream of PKC (Brandt et al., 2002; Brandt et al., 2003; Li et al., 2015). A study reported evidence compatible with activation of PKCα and PKCβ in mammalian cells shortly after challenge with T. gondii (Masek et al., 2006). Another study that used FRET‐based assessment of PKC activity in a reporter cell line provided evidence compatible with PKC activation in later stages of T. gondii infection, although the isoforms activated were not identified (Millholland et al., 2013). We report that T. gondii causes activation of PKCα and PKCβ as assessed by phosphorylation of T638 and T641, respectively, an effect that persists for at least 18 hr postchallenge. Moreover, PKCα and PKCβ promoted sustained Src–EGFR signalling. A burst of PKC activation detected after four cycles of parasite replication has been linked to parasite egress from host cells (Millholland et al., 2013). Our results revealed that, at earlier stages postinfection, activation of PKCα and PKCβ is important because it promotes parasite survival by triggering a signalling cascade that enables T. gondii to avoid killing by the autophagy machinery.
EGFR autophosphorylation at late time points postinfection appeared weaker than that observed at early time points. However, lower level EGFR autophosphorylation was still relevant because addition of EGFR TKI 6 hr postinfection caused parasite killing. Of relevance, MIC3/6 ko strains of T. gondii can still block being targeted by basal autophagy despite inducing reduced levels of EGFR autophosphorylation (~25% compared with WT tachyzoites; Muniz‐Feliciano et al., 2013). These results support the role of EGFR as a potent regulator of T. gondii survival within host cells.
We recently reported that expression of a dominant negative EGFR in endothelial cells reduced CNS invasion by T. gondii, an effect that was likely mediated by autophagic targeting of the parasite within neural endothelial cells (Lopez Corcino et al., 2019). Our studies now revealed that gefitinib, a clinically approved EGFR TKI, protected against ocular and cerebral toxoplasmosis in mice with pre‐established disease, further supporting that EGFR is likely an important regulator of pathogenesis in these forms of toxoplasmosis. Gefitinib likely exerts its protective effect via autophagy because (a) normal levels of Beclin 1 are required in order for gefitinib to be effective against ocular and cerebral toxoplasmosis; (b) autophagy proteins (i.e., Beclin 1 and ATG7) are necessary for protection against these forms of toxoplasmosis (Portillo et al., 2010); and (c) gefitinib killed T. gondii in previously infected cells through the autophagy pathway.
In summary, although T. gondii MICs are an early switch for EGFR autophosphorylation, this work uncovered that PKCα/β ➔ Src signalling in T. gondii‐infected cells leads to prolonged EGFR autophosphorylation and, thus, maintains the nonfusogenic nature of the PV ensuring parasite survival. These findings are of in vivo relevance because inhibition of EGFR signalling promotes control of toxoplasmosis. This work also links biological discovery to potential therapeutic design. Current antibiotic regimens against toxoplasmosis are not proven to be effective in the case of ocular toxoplasmosis and can cause significant side effects. A combination of antibiotics plus EGFR TKI, potentially at low doses, may result in an optimised regimen against toxoplasmosis. The fact that other pathogens cause EGFR activation in host cells (Eierhoff, Hrincius, Rescher, Ludwig, & Ehrhardt, 2010; Frank et al., 2013; Keates et al., 2001; Koff, Shao, Kim, Ueki, & Nadel, 2006; Zhang et al., 2004) and pharmacologic inhibition of EGFR reduces the growth of Mycobacterium tuberculosis in the lungs of infected mice (Stanley et al., 2014) indicates that EGFR may prove to be a therapeutic target against various pathogens.
4. EXPERIMENTAL PROCEDURES
All data generated and analysed during this study are included in this article.
4.1. Ethics statement
This study was performed in accordance with regulations of the Guide for the Care and Use of Laboratory Animals and the National Institute of health. The protocol received approval from the Institutional Animal Care and Use Committee of Case Western Reserve University (Protocol Number: 2015‐0130).
4.2. Mice
C57BL/6 mice (Jackson Laboratories) and mice with heterozygous depletion of Beclin 1 (Becn1 +/−; B6 background; gift from Beth Levine, University of Texas Southwestern) were bred at the Animal Resource Center of Case Western Reserve University. Female mice from 6 to 10 weeks of age were infected i.p. with 30 cysts of the ME49 strain of T. gondii. Mice were treated with vehicle or gefitinib (10 mg kg−1 i.p.) 5 days a week as indicated.
4.3. Histology and immunohistochemistry
Five‐micron sections from the brain and eye were stained by periodic acid Schiff haematoxylin and haematoxylin and eosin, respectively. Histopathology (brain: microglial nodules, perivascular, and diffuse inflammation; retina: disruption of architecture, perivascular, and vitreal inflammation) was scored as previously described (Gazzinelli et al., 1993; Gazzinelli et al., 1994).
4.4. Real‐time quantitative PCR
Genomic DNA was isolated with DNeasy kit (QIAGEN) and utilised as template to measure T. gondii B1 gene expression using SYBR green PCR Master Mix. L32 was used as housekeeping gene. RNA was isolated with RNeasy kit (QIAGEN). RNA was reverse transcribed with Quantitect Reverse Transcription kit (QIAGEN) and was utilised as template for RT‐PCR using the SYBR Green PCR Master Mix and primers for IFN‐γ, IL‐12 p40, TNF‐α, NOS2, and 18S rRNA (Portillo et al., 2010). Samples were normalised to 18S rRNA. Samples were run in triplicates using a StepOne Real‐Time PCR system (Applied Biosystems). Gene expression was calculated by the ΔΔCt method.
4.5. ELISA and nitric oxide assay
Splenocytes (2 × 106 ml−1) were incubated with T. gondii lysate antigens (10 μg ml−1), and the supernatants were collected at 24 hr to measure IL‐12 p40 and at 72 hr to measure IFN‐γ and TNF‐α. The concentration of nitrite was measured in supernatants collected at 72 hr using the Griess Reaction Assay (Promega Corporation). Cytokine concentrations were also measured in sera. Anti‐T. gondii IgG was detected in sera by ELISA and the antibody titre was calculated at the highest dilution of serum that yielded a reading higher than the mean plus 2 SD in the sera from uninfected mice.
4.6. Mammalian cells and in vitro infection with T. gondii
Human retinal pigment epithelial cells (ARPE‐19; American Type Culture Collection; Manassas, VA), primary human brain microvascular endothelial cells (ScienceCell Research Laboratories), mouse high endothelial venule cells (gift from Joan Cook‐Mills; Northwestern University), previously described mouse high endothelial venule cells transduced with Src shRNA or control shRNA encoding lentiviral vectors (Portillo et al., 2017), the mouse microglia cell line BV‐2, and parental CHO cells and CHO with stable expression of EGFR (Muniz‐Feliciano et al., 2013) were cultured in complete media following standard procedures. Mammalian cells were infected with tachyzoites of the RH, PTG, or cps (uracil auxotroph; Fox & Bzik, 2010) strains of T. gondii. The percentages of infection, the numbers of vacuoles and tachyzoites per 100 cells, and the numbers of tachyzoites per vacuole were determined using light microscopy by counting at least 200 cells per monolayer. In specified experiments, mammalian cells were treated with gefitinib (LC Laboratories), PP2 (2 μM; Sigma Chemical), AG1478, GM6001 (10 μM), Gö 6976 (1 μM), Akt inhibitor IV (1.25 μM), lysosomal inhibitors leupeptin and pepstatin (both 10 μM; all from EMD Millipore), Stattic (5 μM; Tocris), FAK inhibitor PF‐573228 (1 μM; Pfizer, Inc.), or lysophosphatidic acid (30 μM; Sigma Chemical) as indicated. T. gondii tachyzoites were maintained in human foreskin fibroblasts.
4.7. RNAi‐mediated silencing
siRNA for human PKCα (MISSION® siRNA1, SIHK1818), PKCβ (MISSION® siRNA3, SIHK1823), and their corresponding nontargeting siRNA (MISSION® siRNA Universal negative control #1, SIC001) were obtained from Sigma‐Aldrich. siRNA for human ULK1 (Assay ID 118261) and its control (Negative Control #1 siRNA; AM4611) were from Thermo Fisher. Cells were transfected using Lipofectamine 2000 (Thermo Fisher). RNAi‐mediated silencing of Src, Beclin 1, Akt, and STAT3 was reported previously (Muniz‐Feliciano et al., 2013; Portillo et al., 2017). Cells were transfected with siRNA (10 nM), and immunoblot or infection with T. gondii was performed after 2 days.
4.8. Immunofluorescence
Mammalian cells were infected with RH‐RFP T. gondii tachyzoites and fixed with 4% paraformaldehyde at 6 or 8 hr. LC3 and LAMP‐1 accumulation around the parasite were detected with anti‐LC3 (MBL international) and anti‐LAMP‐1 (Developmental Studies Hybridoma Bank, University of Iowa) antibodies and a secondary antibody conjugated to Alexa Fluor 488 (Jackson Immuno‐Research Laboratories). Accumulation of LC3 or LAMP‐1 around T. gondii was defined as the presence of a ring‐like structure that surrounds the parasite (Andrade et al., 2006; Van Grol et al., 2013). At least 50 cells per well (duplicate or triplicate wells per group per experiment) were counted manually. Slides were analysed using an Olympus FV1200 IX‐83 confocal microscope equipped with an UPlanSApa 60× objective, N.A. 1.35 oil. Images were acquired using FLUOVIEW v4.2b. Z‐stack images were deconvolved using MetaMorph v7.8.1.0. Images were processed in Photoshop CC 19.1.1. using similar linear adjustments for all samples.
4.9. Transmission electron microscopy
Endothelial cells were seeded onto a sterilised Aclar Embedding Film (Electron Microscopy Sciences), incubated with or without T. gondii tachyzoites followed by addition of gefitinib or vehicle after 2 hr. After 6 hr, the cells were fixed with glutaraldehyde, postfixed in ferrocyanide‐reduced 1% osmium tetroxide, and embedded in Poly/Bed (Polysciences) as described (Muniz‐Feliciano et al., 2013). Thin sections were stained with acidified uranyl acetate followed by triple lead stain of Sato. These sections were examined in a FEI Tecnai Spirit (T12) transmission electron microscope with a Gatan US4000 4k × 4k CCD.
4.10. Immunoblot
Protein samples from lysed cells were loaded at equal concentrations in an SDS‐PAGE gel and transferred to a PDVF membrane. The membranes were probed with antibodies for EGFR, Irgm3, actin (all from Santa Cruz Biotechnologies), p‐Y1068 EGFR (Thermo Fisher), p‐Y1173 EGFR (Santa Cruz Biotechnologies), p‐Y845 EGFR, Src and p‐Y416 Src, Akt, p‐S473 Akt (all from Cell Signaling), PKCα, PKCβ and p‐T642 PKCβ1 (all from Genetex), p‐T638 PKCα (BioLegend), ULK1 (Sigma‐Aldrich), and Beclin 1 (BD Biosciences). Images were generated using Photoshop. When needed, change in brightness and/or contrast was applied equally to the whole image.
4.11. Statistics
Paired Student's t test and analysis of variance were used to determine the statistical significance of the data. Results were considered statistically significant when P < .05.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Y. L. C. and C. S. S. conceived and designed the experiments. Y. L. C., S. G. F., L. E. M., S. T., J. S. Y., S. K., S. H., and J. A. C. P. performed the experiments. Y. L. C., S. G. F., J. A. C. P., and C. S. S. analysed the data. Y. L. C. and C. S. S. wrote the manuscript. All authors reviewed the manuscript.
FUNDING
This work was supported by the National Institutes of Health/National Eye Institute Grants EY018341 (C. S. S.), REY018341B (Y. L. C.), and P30 EY11373, as well as Prevent Blindness Ohio (Y. L. C.).
Supporting information
Figure S1. Effects of kinase inhibitors on parasite survival.
ACKNOWLEDGEMENTS
We thank Catherine Doller and Anthony Gardella (Case Western Reserve University) for tissue processing and image collection.
Lopez Corcino Y, Gonzalez Ferrer S, Mantilla LE, et al. Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: Perspectives for in vivo control of the parasite. Cellular Microbiology. 2019;21:e13084 10.1111/cmi.13084
REFERENCES
- Andrade, R. M. , Wessendarp, M. , Gubbels, M. J. , Striepen, B. , & Subauste, C. S. (2006). CD40 induces macrophage anti‐Toxoplasma gondii activity by triggering autophagy‐dependent fusion of pathogen‐containing vacuoles and lysosomes. The Journal of Clinical Investigation, 116, 2366–2377. 10.1172/JCI28796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, M. , Biswas, R. , & Das, M. (1984). 42,000‐molecular weight EGF receptor has protein kinase activity. Nature, 311, 477–480. 10.1038/311477a0 [DOI] [PubMed] [Google Scholar]
- Besteiro, S. , Dubremetz, J. F. , & Lebrun, M. (2011). The moving junction of apicomplexan parasites: A key structure for invasion. Cellular Microbiology, 13, 797–805. 10.1111/j.1462-5822.2011.01597.x [DOI] [PubMed] [Google Scholar]
- Biscardi, J. S. , Maa, M. C. , Tice, D. A. , Cox, M. E. , Leu, T. H. , & Parsons, J. T. (1999). c‐Src‐mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. The Journal of Biological Chemistry, 274, 8335–8343. 10.1074/jbc.274.12.8335 [DOI] [PubMed] [Google Scholar]
- Bosch‐Driessen, L. E. H. , Berrendschot, T. T. J. M. , Ongkosuwito, J. V. , & Rothova, A. (2002). Ocular toxoplasmosis. Clinical features and prognosis of 154 patents. Ophthalmology, 109, 869–878. 10.1016/S0161-6420(02)00990-9 [DOI] [PubMed] [Google Scholar]
- Brandt, D. , Gimona, M. , Hillmann, M. , Haller, H. , & Mischak, H. (2002). Protein kinase C induces actin reorganization via a Src‐ and Rho‐dependent pathway. The Journal of Biological Chemistry, 277, 20903–20910. 10.1074/jbc.M200946200 [DOI] [PubMed] [Google Scholar]
- Brandt, D. T. , Goerke, A. , Heuer, M. , Gimona, M. , Leitges, M. , Kremmer, E. , … Mischak, H. (2003). Protein kinase Cδ induces Src kinase activity via activation of the protein tyrosine phosphatase PTPα. The Journal of Biological Chemistry, 278, 34073–34078. 10.1074/jbc.M211650200 [DOI] [PubMed] [Google Scholar]
- Butcher, B. A. , Fox, B. A. , Rommereim, L. M. , Kim, S. G. , Maurer, K. J. , Yarovinsky, F. , … Denkers, E. Y. (2011). Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase‐1‐dependent growth control. PLoS Pathogens, 7, e1002236 10.1371/journal.ppat.1002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, E. Y. , Longatti, A. , McKnight, N. C. , & Tooze, S. A. (2009). Kinase‐inactivated ULK proteins inhibit autophagy via their conserved C‐terminal domains using an Atg13‐independent mechanism. Molecular and Cellular Biology, 29, 157–171. 10.1128/MCB.01082-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. , Park, S. , Biering, S. B. , Selleck, E. , Liu, C. Y. , Zhang, X. , … Virgin, H. W. (2014). The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin‐like conjugation systems of autophagy. Immunity, 40, 924–935. 10.1016/j.immuni.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardiello, F. , Caputo, R. , Bianco, R. , Damiano, V. , Pomatico, G. , De Placido, S. , … Tortora, G. (2000). Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD‐1839 (Iressa), an epidermal growth factor receptor‐selective tyrosine kinase inhibitor. Clinical Cancer Research, 6, 2053–2063. [PubMed] [Google Scholar]
- Ciardiello, F. , & Tortora, G. (2001). A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clinical Cancer Research, 7, 2958–2970. [PubMed] [Google Scholar]
- Courret, N. , Darche, S. , Sonigo, P. , Milon, G. , Buzoni‐Gatel, D. , & Tardieux, I. (2006). CD11c‐ and CD11b‐expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood, 107, 309–316. 10.1182/blood-2005-02-0666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gennaro, E. , Barbarino, M. , Bruzzese, F. , De Lorenzo, S. , Caraglia, M. , Abbruzzese, A. , … Budillon, A. (2003). Critical role of both p27KIP1 and p21CIP1/WAF1 in the antiproliferative effect of ZD1839 (‘Iressa’), an epidermal growth factor receptor tyrosine kinase inhibitor, in head and neck squamous carcinoma cells. Journal of Cellular Physiology, 195, 139–150. 10.1002/jcp.10239 [DOI] [PubMed] [Google Scholar]
- Drube, S. , Stirnweiss, J. , Valkova, C. , & Liebmann, C. (2006). Ligand‐independent and EGF receptor‐supported transactivation: Lessons from β2‐adrenergic receptor signalling. Cellular Signalling, 18, 1633–1646. 10.1016/j.cellsig.2006.01.003 [DOI] [PubMed] [Google Scholar]
- Eierhoff, T. , Hrincius, E. R. , Rescher, U. , Ludwig, S. , & Ehrhardt, C. (2010). The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathogens, 6, e1001099 10.1371/journal.ppat.1001099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen, E. L. (2008). Fine structure of the autophagosome. Methods in Molecular Biology, 445, 11–28. 10.1007/978-1-59745-157-4_2 [DOI] [PubMed] [Google Scholar]
- Fox, B. A. , & Bzik, D. J. (2010). Avirulent uracil auxotrophs based on disruption of orotidine‐5′‐monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii . Infection and Immunity, 78, 3744–3752. 10.1128/IAI.00287-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, C. G. , Reguerlo, V. , Rother, M. , Moranta, D. , Maeurer, A. P. , Garmendia, J. , … Bengoechea, J. A. (2013). Klebsiella pneumoniae targets an EGF receptor‐dependent pathway to subvert inflammation. Cellular Microbiology, 15, 1212–1233. 10.1111/cmi.12110 [DOI] [PubMed] [Google Scholar]
- Fung, A. S. , Yu, M. , Ye, Q. J. , & Tannock, I. F. (2013). Scheduling of paclitaxel and gefitinib to inhibit repopulation for optimal treatment of human cancer cells and xenografts that overexpress the epidermal growth factor receptor. Cancer Chemotherapy and Pharmacology, 72, 585–595. 10.1007/s00280-013-2229-3 [DOI] [PubMed] [Google Scholar]
- Gazzinelli, R. T. , Brezin, A. , Li, Q. , Nussenblatt, R. B. , & Chan, C. C. (1994). Toxoplasma gondii: Acquired ocular toxoplasmosis in the murine model, protective role of TNF‐α and IFN‐γ. Experimental Parasitology, 78, 217–229. 10.1006/expr.1994.1022 [DOI] [PubMed] [Google Scholar]
- Gazzinelli, R. T. , Eltoum, I. , Wynn, T. A. , & Sher, A. (1993). Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF‐α and correlates with the down‐regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. Journal of Immunology, 151, 3672–3681. [PubMed] [Google Scholar]
- Gschwind, A. , Fischer, O. M. , & Ullrich, A. (2004). The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nature Reviews. Cancer, 4, 361–370. 10.1038/nrc1360 [DOI] [PubMed] [Google Scholar]
- Higashiyama, S. , Iwabuki, H. , Morimoto, C. , Hieda, M. , Inoue, H. , & Matsushita, N. (2008). Membrane‐anchored growth factors, the epidermal growth factor family: Beyond receptor ligands. Cancer Science, 99, 214–220. 10.1111/j.1349-7006.2007.00676.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura, E. , & Mizushima, N. (2010). Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy, 6, 764–776. 10.4161/auto.6.6.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keates, S. , Sougioultzis, S. , Keates, A. C. , Zhao, D. , Peek, R. M. J. , Shaw, L. M. , & Kelly, C. P. (2001). cag + Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS epithelial cells. The Journal of Biological Chemistry, 276, 48127–48134. 10.1074/jbc.M107630200 [DOI] [PubMed] [Google Scholar]
- Klionsky, D. J. , & Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science, 290, 1717–1721. 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloth, M. T. , Laughlin, K. K. , Biscardi, J. S. , Boerner, J. L. , Parsons, S. J. , & Silva, C. M. (2003). STAT5b, a mediator of synergism between c‐Src and the epidermal growth factor receptor. The Journal of Biological Chemistry, 278, 1671–1679. 10.1074/jbc.M207289200 [DOI] [PubMed] [Google Scholar]
- Koff, J. L. , Shao, M. X. G. , Kim, S. , Ueki, I. F. , & Nadel, J. A. (2006). Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. Journal of Immunology, 177, 8693–8700. 10.4049/jimmunol.177.12.8693 [DOI] [PubMed] [Google Scholar]
- Kovacs, A. L. , Rez, G. , Palfia, Z. , & Kovacs, J. (2000). Autophagy in the epithelial cells of murine seminal vesicle in vitro. Formation of large sheets of nascent isolation membranes, sequestration of the nucleus and inhibition by wortmannin and 3‐ethyladenine. Cell and Tissue Research, 302, 253–261. [DOI] [PubMed] [Google Scholar]
- Levine, B. , Mizushima, N. , & Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature, 469, 323–335. 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Yang, B. , Chen, M. , Klein, J. D. , Sands, J. M. , & Chen, G. (2015). Activation of protein kinase C‐α and Src kinase increases urea transporter A1 α‐2, 6 sialylation. Journal of the American Society of Nephrology, 26, 926–934. 10.1681/ASN.2014010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebmann, C. (2010). EGF receptor activation by GPCRs: An universal pathway reveals different versions. Molecular and Cellular Endocrinology, 331, 222–231. [DOI] [PubMed] [Google Scholar]
- Liu, E. , Lopez Corcino, Y. , Portillo, J.‐A. C. , Miao, Y. , & Subauste, C. S. (2016). Identification of signaling pathways by which CD40 stimulates autophagy and anti‐microbial activity against Toxoplasma gondii in macrophages. Infection and Immunity, 84, 2616–2626. 10.1128/IAI.00101-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , He, K. , Ma, Q. , Yu, Q. , Liu, C. , Ndege, I. , … Yu, Z. (2017). Autophagy inhibitor facilitates gefitinib sensitivity in vitro and in vivo by activating mitochondrial apoptosis in triple negative breast cancer. PLoS One, 12, e0177694 10.1371/journal.pone.0177694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Corcino, Y. , Portillo, J.‐A. C. , & Subauste, C. S. (2019). Epidermal growth factor receptor promotes cerebral and retinal invasion by Toxoplasma gondii . Scientific Reports, 9, 669 10.1038/s41598-018-36724-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurje, G. , & Lenz, H.‐J. (2009). EGFR signaling and drug discovery. Oncology, 77, 400–410. 10.1159/000279388 [DOI] [PubMed] [Google Scholar]
- Masek, K. S. , Fiore, J. , Leitges, M. , Yan, S. F. , Freedman, B. D. , & Hunter, C. A. (2006). Host cell Ca2+ and protein kinase C regulate innate recognition of Toxoplasma gondii . Journal of Cell Science, 119, 4565–4573. 10.1242/jcs.03206 [DOI] [PubMed] [Google Scholar]
- Millholland, M. G. , Mishra, S. , Dupont, C. D. , Love, M. S. , Patel, B. , Shilling, D. , … Greenbaum, D. C. (2013). A host GPCR signaling network required for the cytolysis of infected cells facilitates release of apicomplexan parasites. Cell Host & Microbe, 13, 15–28. 10.1016/j.chom.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , Ohsumi, Y. , & Yoshimori, T. (2002). Autophagosome formation in mammalian cells. Cell Structure and Function, 27, 421–429. 10.1247/csf.27.421 [DOI] [PubMed] [Google Scholar]
- Mordue, D. G. , Desai, N. , Dustin, M. , & Sibley, L. D. (1999). Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. The Journal of Experimental Medicine, 190, 1783–1792. 10.1084/jem.190.12.1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz‐Feliciano, L. , Van Grol, J. , Portillo, J.‐A. C. , Liew, L. , Liu, B. , Carlin, C. R. , … Subauste, C. S. (2013). Toxoplasma gondii‐induced activation of EGFR prevents autophagy protein‐mediated killing of the parasite. PLoS Pathogens, 9, e1003809 10.1371/journal.ppat.1003809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier, R. J. , Rafi, W. , Cheruvu, M. , Powell, K. R. , Zaunbrecher, M. A. , Bornmann, W. , … Kalman, D. (2011). Imatinib‐sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host & Microbe, 10, 475–485. 10.1016/j.chom.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse, I. , Ohmori, T. , Ao, Y. , Fukumoto, H. , Kuroki, T. , Mori, M. , … Nishio, K. (2002). Antitumor activity of the selective epidermal growth factor receptor‐tyrosine kinase inhibitor (EGFR‐TKI) Iressa (ZD1839) in an EGFR‐expressing multidrug‐resistant cell line in vitro and in vivo. International Journal of Cancer, 98, 310–315. 10.1002/ijc.10173 [DOI] [PubMed] [Google Scholar]
- Neville, A. J. , Zach, S. J. , Wang, X. , Larson, J. J. , Judge, A. K. , Davis, L. A. , … Davis, P. H. (2015). Clinically available medicines demonstrating anti‐toxoplasma activity. Antimicrobial Agents and Chemotherapy, 59, 7161–7169. 10.1128/AAC.02009-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogolla, P. , Portillo, J.‐A. C. , White, C. L. , Patel, K. , Lamb, B. , Sen, G. C. , & Subauste, C. S. (2013). The protein kinase double‐stranded RNA‐dependent (PKR) enhances protection against disease cause by a non‐viral pathogen. PLoS Pathogens, 9, e100557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre, S. , Tassa, A. , Qu, X. , Garuti, R. , Liang, X. H. , Mizushima, N. , … Levine, B. (2005). Bcl‐2 antiapoptotic proteins inhibit Beclin 1‐dependent autophagy. Cell, 122, 927–939. 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- Pauwels, A. M. , Trost, M. , Beyaert, R. , & Hoffmann, E. (2017). Patterns, receptors, and signals: Regulation of phagosome maturation. Trends in Immunology, 38, 407–422. 10.1016/j.it.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo, J.‐A. C. , Okenka, G. , Reed, E. , Subauste, A. , Van Grol, J. , Gentil, K. , … Subauste, C. S. (2010). The CD40‐autophagy pathway is needed for host protection despite IFN‐γ‐dependent immunity and CD40 induces autophagy via control of p21 levels. Plos One, 5(12), e14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo, J.‐A. C. , Van Grol, J. , Saffo, S. , Lopez Corcino, Y. , Rodriguez, M. , Fox, B. , … Toosi, Z. (2019). CD40 in endothelial cells restricts neural tissue invasion by Toxoplasma gondii . Infection and Immunity, May 20, Epub ahead of print. 10.1128/IAI.00868-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo, J. C. , Muniz‐Feliciano, L. , Lopez Corcino, Y. , Lee, S. J. , Van Grol, J. , Parsons, S. J. , … Subauste, C. S. (2017). Toxoplasma gondii induces FAK‐Src‐STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PLoS Pathogens, 13, e1006671 10.1371/journal.ppat.1006671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss, S. , Namgaladze, D. , & Brune, B. (2007). Critical role for classical PKC in activating Akt by phospholipase A2‐modified LDL in monocytic cells. Cardiovascular Research, 73, 833–840. 10.1016/j.cardiores.2006.12.019 [DOI] [PubMed] [Google Scholar]
- Real, F. X. , Rettig, W. J. , Chesa, P. G. , Melamed, M. R. , Old, L. J. , & Mendelsohn, J. (1986). Expression of epidermal growth factor receptor in human cultured cells and tissues: Relationship to cell lineage and stage of differentiation. Cancer Research, 46, 4726–4731. [PubMed] [Google Scholar]
- Reeves, P. M. , Bommarius, B. , Lebeis, S. , McNulty, S. , Christensen, J. , Swimm, A. , … Kalman, D. (2005). Disabling poxvirus pathogenesis by inhibition of Abl‐family tyrosine kinases. Nature Medicine, 11, 731–739. 10.1038/nm1265 [DOI] [PubMed] [Google Scholar]
- Reichmann, G. , Walker, W. , Villegas, E. N. , Craig, L. , Cai, G. , Alexander, J. , & Hunter, C. A. (2000). The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infection and Immunity, 68, 1312–1318. 10.1128/IAI.68.3.1312-1318.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothova, A. , Meenken, C. , Buitenhuis, H. J. , Brinkman, C. J. , Baarsma, G. S. , Boen‐Tan, T. N. , … Kijlstra, A. (1993). Therapy for ocular toxoplasmosis. American Journal of Ophthalmology, 115, 517–523. 10.1016/S0002-9394(14)74456-3 [DOI] [PubMed] [Google Scholar]
- Rudkouskaya, A. , Chernoguz, A. , Haskew‐Layton, R. E. , & Mongin, A. A. (2008). Two conventional protein kinase C isoforms, α and βI, are involved in the ATP‐induced activation of volume‐regulated anion channel and glutamate release in cultured astrocytes. Journal of Neurochemistry, 105, 2260–2270. 10.1111/j.1471-4159.2008.05312.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, R. C. , Tian, Y. , Yuan, H. , Park, H. W. , Chang, Y. Y. , Kim, J. , … Guan, K. L. (2013). ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nature Cell Biology, 15, 741–750. 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij, J. P. , Coller, S. , Boyle, J. P. , Jerome, M. E. , White, M. W. , & Boothroyd, J. C. (2007). Toxoplasma co‐opts host gene expression by injection of a polymorphic kinase homologue. Nature, 445, 324–327. 10.1038/nature05395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K. , Nagao, T. , Iwasaki, T. , Nishihira, Y. , & Fukami, Y. (2003). Src‐dependent phosphorylation of the EGF receptor Tyr‐845 mediates Stat‐p21waf1 pathway in A431 cells. Genes to Cells, 8, 995–1003. 10.1046/j.1356-9597.2003.00691.x [DOI] [PubMed] [Google Scholar]
- Sirotnak, F. M. , Zakowski, M. F. , Miller, V. A. , Scher, H. I. , & Kris, M. G. (2000). Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clinical Cancer Research, 6, 4885–4892. [PubMed] [Google Scholar]
- Stanley, S. A. , Barczak, A. K. , Silvis, M. R. , Luo, S. S. , Sogi, K. , Vokes, M. , … Hung, D. T. (2014). Identification of host‐targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathogens, 10, e1003946 10.1371/journal.ppat.1003946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste, C. S. , Wessendarp, M. , Sorensen, R. U. , & Leiva, L. (1999). CD40–CD40 ligand interaction is central to cell‐mediated immunity against Toxoplasma gondii: Patients with hyper IgM syndrome have a defective type‐1 immune response which can be restored by soluble CD40L trimer. Journal of Immunology, 162, 6690–6700. [PubMed] [Google Scholar]
- Talloczy, Z. , Virgin, H. W. , & Levine, B. (2006). PKR‐dependent autophagic degradation of herpes simplex virus type 1. Autophagy, 2, 24–29. 10.4161/auto.2176 [DOI] [PubMed] [Google Scholar]
- Tice, D. A. , Biscardi, J. S. , Nickles, A. L. , & Parsons, S. J. (1999). Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proceedings of the National Academy of Sciences of the United States of America, 96, 1415–1420. 10.1073/pnas.96.4.1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Grol, J. , Muniz‐Feliciano, L. , Portillo, J.‐A. C. , Bonilha, V. L. , & Subauste, C. S. (2013). CD40 induces anti‐Toxoplasma gondii activity in non‐hematopoietic cells dependent on autophagy proteins. Infection and Immunity, 81, 2002–2011. 10.1128/IAI.01145-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Chiang, H.‐C. , Wu, W. W. , Liang, B. , Xie, Z. , Yao, X. , … Zhong, Y. (2012). Epidermal growth factor receptor is a preferred target for treating amyloid‐β‐induced memory loss. Proceedings of the National Academy of Sciences of the United States of America, 109, 16743–16748. 10.1073/pnas.1208011109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, W. , Wu, J. , Liu, L. , Tian, Y. , Buettner, R. , Hsieh, M. Y. , … Yim, J. H. (2015). Synergistic anti‐tumor effect of combined inhibition of EGFR and JAK/STAT3 pathways in human ovarian cancer. Molecular Cancer, 14, 100 10.1186/s12943-015-0366-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori, T. (2004). Autophagy: A regulated bulk degradation process inside cells. Biochemical and Biophysical Research Communications, 313, 453–458. 10.1016/j.bbrc.2003.07.023 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Li, H. , Wang, J. , Dong, Z. , Mian, S. , & Yu, F.‐S. X. (2004). Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa‐infected human corneal epithelial cells. Investigative Ophthalmology & Visual Science, 45, 2569–2576. 10.1167/iovs.03-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of kinase inhibitors on parasite survival.