

Abstract
Introduction
Ambulatory individuals with spinal muscular atrophy (SMA) experience muscle weakness, gait impairments, and fatigue that affect their walking ability. Improvements have been observed in motor function in children treated with nusinersen, but its impact on fatigue has not been studied.
Methods
Post hoc analyses were used to examine changes in 6‐minute walk test (6MWT) distance and fatigue in children and adolescents with SMA type II and III who received their first dose of nusinersen in the phase Ib/IIa, open‐label CS2 study and were ambulatory during CS2 or the extension study, CS12.
Results
Fourteen children performed the 6MWT. Median (25th, 75th percentile) distance walked increased over time by 98.0 (62.0, 135.0) meters at day 1050, whereas median fatigue changed by −3.8% (−19.7%, 1.4%).
Discussion
These results support previous studies demonstrating clinically meaningful effects of nusinersen on motor function in children and adolescents with later‐onset SMA.
Keywords: 6‐minute walk test, fatigue, neuromuscular junction, nusinersen, spinal muscular atrophy
Abbreviations
- 6MWT
6‐minute walk test
- NMJ
neuromuscular junction
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
1. INTRODUCTION
Spinal muscular atrophy (SMA) is a chronic autosomal recessive neuromuscular disorder characterized by progressive muscle atrophy and weakness caused by biallelic disruption of the survival motor neuron 1 (SMN1) gene.1, 2 SMA manifests in different phenotypes defined by age of symptom onset and maximum motor milestone achievement.2, 3 SMA type II and III are milder forms than type I, with symptom onset at age 7–18 months and >18 months, respectively.4 Individuals with SMA type III attain the ability to walk unaided at some point; however, muscle weakness causes gait impairment and reduced endurance.5 Over time, patients may lose the ability to walk independently.2
Tools assessing walking ability and fatigue are clinically relevant in individuals with milder forms of SMA.5 The 6‐minute walk test (6MWT) is a valid and clinically meaningful measure of ambulatory function and has demonstrated sensitivity to fatigue‐related changes in individuals with SMA.6, 7, 8, 9 Fatigue measured during the 6MWT may represent underlying disease‐specific mechanisms,10 and improvements may represent treatment benefit. Neuromuscular junction (NMJ) development and maturation abnormalities, including immaturity, denervation, and neurofilament accumulation, as well as endocytosis and axonal transport abnormalities, have been identified in animal models and in individuals with SMA.11, 12, 13, 14 Data suggesting a relationship between NMJ dysfunction and fatigability have been reported.11, 15
Nusinersen is the first drug approved for the treatment of SMA,16, 17 and has demonstrated significant and clinically meaningful efficacy by increasing event‐free survival, motor function, and achievement of motor milestones in infantile‐onset SMA,18 and improving/maintaining motor function in later‐onset SMA.19, 20 Nusinersen has resulted in clinically meaningful improvements in walking ability (defined as ≥30‐meter increase from baseline in 6MWT distance) in children/adolescents with later‐onset SMA6, 20, 21; however, the effects of nusinersen treatment on fatigability are unknown. In this post hoc analysis we further examined 6MWT performance and the relationship between distance walked and fatigue in the subgroup of nusinersen‐treated children and adolescents with SMA who were ambulatory during the CS2 dose‐escalation or CS12 extension studies and completed the 6MWT. Given that participants of ambulatory age were followed for ~3 years, these studies provide the opportunity to evaluate longer‐term nusinersen treatment effects on 6MWT fatigability in individuals with SMA.
2. METHODS
This analysis included children and adolescents with SMA type II and III, 2–15 years of age, who received their first nusinersen dose in the CS2 study, were ambulatory during the CS2 or CS12 studies, and performed the 6MWT. Institutional review board approval and participant/parent/legal guardian informed consent were obtained. Both study designs and results of an integrated analysis have been described in previous work.20 The list of CS2 and CS12 Study Group investigators and site personnel is provided in Data S1 online. Briefly, the CS2 study (NCT01703988) was an 85‐day (+168‐day follow‐up), open‐label, phase Ib/IIa, multicenter, multiple‐dose, dose‐escalation study examining multiple ascending nusinersen doses in children with SMA type II and III. Four dose levels were evaluated sequentially in 4 separate cohorts: cohort 1 received nusinersen 3 mg on days 1, 29, and 85; cohort 2 received nusinersen 6 mg on days 1, 29, and 85; cohort 3 received nusinersen 9 mg on days 1 and 85; and cohort 4 received nusinersen 12 mg on days 1, 29, and 85. The cohort 2 participants who were ambulatory did not receive their first dose of nusinersen in the CS2 study, and therefore were not included in the analysis. After varying treatment breaks, participants could continue treatment in the open‐label extension study CS12 (NCT02052791), a 533‐day (+182‐day follow‐up) single‐dose‐level study in which participants received 4 doses of nusinersen 12 mg on days 1, 169, 351, and 533. To allow for an integrated analysis and to account for differing times between CS2 study end and CS12 study initiation between participants, a windowing approach was utilized in which study days were derived from the first day of CS2 dosing for all CS2 and CS12 study visits.
Changes in 6MWT distance and fatigue over 253 and 1050 days were evaluated. The 6MWT measures the distance a person can walk quickly in 6 minutes; details of test administration have been described elsewhere.7 Briefly, participants were instructed to walk as quickly as possible along a 25‐meter linear course, turn around a cone, return in the opposite direction, and repeat the loop as many times as possible for 6 minutes. Tests were administered by trained physical therapists; distance walked at each minute was recorded. Fatigue was calculated by subtracting the distance walked in the sixth minute from the distance walked in the first minute. Measurements are expressed as percentages, where a positive value represents fatigue. Correlations between 6MWT distance and fatigue at baseline and day 1050 were evaluated using Pearson correlation analysis. Subgroup analyses compared results in participants aged ≤11 years with those aged >11 years; these age groups are similar to those reported previously.5, 22
3. RESULTS
Fourteen children were included in the analyses. All participants had SMA type III, except for 1 participant with SMA type II who achieved the ability to walk independently and performed the 6MWT during the CS12 study (Table 1). Baseline 6MWT distances and fatigue are listed in Table 2. At baseline, participants aged ≤11 years walked farther and had less 6MWT fatigue than those aged >11 years (Table 2). Median (25th, 75th percentile) distance walked increased over time by 17.0 (0.0, 51.0) meters at day 253 and by 98.0 (62.0, 135.0) meters at day 1050 (Figure 1). Median (25th, 75th percentile) change in fatigue was −0.1% (−5.0%, 11.3%) at day 253 and − 3.8% (−19.7%, 1.4%) at day 1050 (Figure 1). There was an inverse relationship between 6MWT distance and fatigue at baseline (n = 14; Pearson r = −0.804; P = .0005) and at day 1050 (n = 9; Pearson r = −0.773; P = .0147).
Table 1.
Baseline characteristics in the CS2 study
| Characteristic | Total cohorta (N = 14) | By age group at screening | |
|---|---|---|---|
| ≤11 years (n = 9) | >11 years (n = 5) | ||
| Male, n (%) | 6 (43) | 4 (44) | 2 (40) |
| Age at symptom onset, months | |||
| Mean (SD) | 23.9 (13.6) | 18.2 (7.3) | 34.2 (16.9) |
| Median (range) | 18.0 (11–60) | 18.0 (11–36) | 36.0 (15–60) |
| Age at SMA diagnosis, months | |||
| Mean (SD) | 47.4 (34.6) | 36.7 (23.9) | 66.6 (44.9) |
| Median (range) | 33.5 (15–144) | 27.0 (15–96) | 56.0 (29–144) |
| SMN2 copy number, n (%) | |||
| 3 copies | 9 (64) | 7 (78) | 2 (40) |
| 4 copies | 5 (36) | 2 (22) | 3 (60) |
| SMA type, n (%) | |||
| Type II | 1 (7) | 1 (11) | 0 |
| Type III | 13 (93) | 8 (89) | 5 (100) |
| Ambulatory, n (%) | 13 (93) | 8 (89) | 5 (100) |
| Nonambulatory, n (%) | 1 (7) | 1 (11) | 0 |
| Dose level in CS2, n (%) | |||
| 3 mg | 2 (14) | 0 | 2 (40) |
| 6 mg | 0 | 0 | 0 |
| 9 mg | 6 (43) | 5 (56) | 1 (20) |
| 12 mg | 6 (43) | 4 (44) | 2 (40) |
| Time from end of CS2 to first dose in CS12, days | |||
| Mean (SD) | 119.3 (79.9) | 113.0 (69.8) | 129.4 (102.1) |
| Median (range) | 118.0 (19–233) | 104.0 (26–196) | 168.0 (19–233) |
Abbreviations: SMA, spinal muscular atrophy; SMN, survival motor neuron.
All children who received their first nusinersen dose in the CS2 study, were ambulatory during CS2 or CS12, and performed the 6MWT were included in these analyses.
Table 2.
Baseline motor function in the CS2 study
| Characteristic | Total cohorta (N = 14) | By age group at screening | |
|---|---|---|---|
| ≤11 years (n = 9) | >11 years (n = 5) | ||
| Motor function at screening, n (%) | |||
| Sitting without support | 14 (100) | 9 (100) | 5 (100) |
| Walking with support | 14 (100) | 9 (100) | 5 (100) |
| Standing without support | 12 (86) | 8 (89) | 4 (80)b |
| Walking independently | 13 (93) | 8 (89) | 5 (100) |
| 6MWT distance, meters | |||
| Mean (SD) | 235.2 (188.2) | 259.8 (155.5) | 191.0 (250.9) |
| Median (range) | 250.5 (0–563) | 295.0 (0–563)c | 45.0 (0–550)d |
| 6MWT fatigue, % | |||
| Mean (SD) | 38.2 (47.1) | 23.9 (41.7) | 64.0 (49.3) |
| Median (range) | 14.8 (−16 to 100) | 0.0 (−16 to 100) | 100 (8–100) |
Abbreviations: 6MWT, 6‐minute walk test.
All children who received their first nusinersen dose in the CS2 study, were ambulatory during CS2 or CS12, and performed the 6MWT were included in these analyses.
This motor milestone history item was reported as “no” in 1 case, but the participant was able to stand independently according to the Hammersmith Functional Motor Scale Expanded item “Stand unsupported.”
One participant with SMA type II gained the ability to walk independently during the course of the study.
Two participants were able to walk a short distance or <25 meters, and did not have 6MWT performed at baseline; the 6MWT total distance was imputed as 0 meters at baseline.
Figure 1.
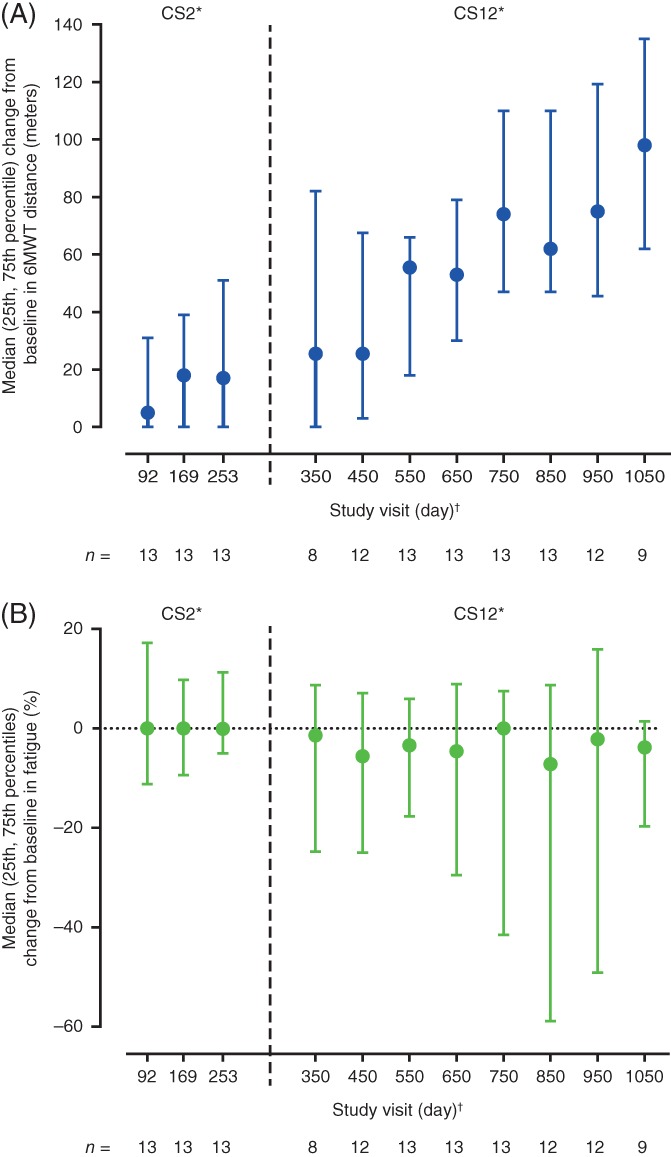
Median (25th, 75th percentile) change from baseline in 6MWT (A) distance and (B) fatigue over time. *Study visits up to and including day 253 occurred during the CS2 study, subsequent study visits occurred during the CS12 study. The day of the first study visit attended in the CS12 study depended upon the time between the end of the CS2 study and first dose in the CS12 study and the windowing of study visits described as follows. Median (range) time from the end of the CS2 study to first dose in the CS12 study was 118.0 (19–233) days. †Study days were derived from the first day of CS2 dosing for all CS2 and CS12 study visits and were assigned visit days as follows: study visits that took place >50 and ≤131 days from the first dose in the CS2 study were labeled day 92, visits >131 and ≤211 days from the first dose in CS2 were labeled day 169, those >211 and ≤302 days were labeled day 253, those >302 and ≤400 days were labeled day 350, and study visits >X − 50 to ≤X + 50 were labeled day X (starting at day 450 and increasing by 100 until day 1050), times between visit days were not equal, but were similar. 6MWT, 6‐minute walk test
4. DISCUSSION
Ambulatory children and adolescents treated with nusinersen in the CS2 multiple‐dose and CS12 extension studies demonstrated improvements in ambulatory function (6MWT), with clinically meaningful increases in walking distance and modest decreases or stabilization in fatigue. Although there is no precedent for improvements like these in individuals with SMA, changes of ≥30 meters in 6MWT distance are considered clinically meaningful and to impact everyday activities in other pediatric neuromuscular disorders.6, 21
Older children and adolescents with SMA are more at risk for declining ambulatory function.5 A longitudinal natural history study of ambulatory patients with SMA type III showed that 6MWT trajectories were age‐dependent.5 The mean rate of change in 6MWT distance improved by 9.8 meters/year in patients <6 years of age, but declined by 20.8 meters/year in adolescents and young adults 11–19 years of age, regardless of age at time of diagnosis.5 In this study, 5 participants were >11 years old at baseline and 3 additional participants became ≥11 years old during the study. For individuals with SMA, preserving, restoring, and improving ambulation, as well as reducing fatigue and improving endurance, are considered extremely important factors that can affect quality of life.23 Importantly, improvement or stabilization in 6MWT distance is unexpected in these older participants based on SMA natural history.5
NMJ dysfunction has been identified in SMA mouse models and patients.11, 14, 15 In individuals with SMA, fatigue on the 6MWT was associated with a decrement on low‐rate repetitive nerve stimulation, possibly due to developmental NMJ abnormalities or NMJ function alterations.11, 15 Although the mechanism remains unknown, 6MWT fatigue implicates dysfunction at the NMJ in SMA, and has not been demonstrated by individuals with other neuromuscular disorders who exhibit weakness.10, 15 The observed decrease or stabilization of 6MWT fatigue may represent a nusinersen treatment effect. Further understanding of the underlying mechanisms of fatigue may help explain modest treatment responses. Laboratory evidence suggests that muscle mitochondria may be vulnerable to SMN deficiency.24, 25 Fatigue may also be the clinical consequence of these laboratory observations in addition to NMJ dysfunction. Results of this analysis suggest that fatigue may serve as an informative outcome measure in ambulatory patients and should be considered as an endpoint in future SMA clinical trials. Moreover, nonambulant individuals with SMA similarly exhibit fatigability on upper extremity endurance tasks, further demonstrating that fatigue may be a general characteristic of SMA.26
This study has some limitations. CS2 and CS12 were open‐label, uncontrolled studies, and the sample size of the post hoc analyses was relatively small. Differences in CS2 doses could have led to early efficacy result variations. These studies were designed before the approval of nusinersen, and participants received nusinersen maintenance doses at 6‐month intervals instead of the approved 4‐month intervals.16, 27 The time period between completion of the CS2 study and the beginning of treatment in the CS12 study was longer for children receiving lower nusinersen doses in the CS2 study; therefore, children in the CS2 9‐ and 12‐mg dosing cohorts may have reached steady‐state levels of nusinersen more quickly than those who received 3‐ and 6‐mg doses. In addition, the clinically meaningful effect size for reduction in or stabilization of fatigue remains to be established. Longitudinal natural history studies that evaluate normal variability and trajectories in fatigue over time are needed.
This post hoc analysis has reported on levels of fatigue after nusinersen treatment and further extends and supports previous results demonstrating the clinically meaningful effects of nusinersen on motor function in children and adolescents with later‐onset SMA (ie, those with or most likely to develop SMA type II or III).20
CONFLICTS OF INTEREST
J.M. has been an advisory board member for Astellas, Biogen, Cytokinetics, Roche, and Scholar Rock; a consultant to Biogen and Ionis Pharmaceuticals, Inc; and has received research support from the Muscular Dystrophy Association and the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health. S.D.Y. has been an advisory board member for Biogen, Roche, and Scholar Rock; received personal compensation for activities with Biogen, Cure SMA, and Scholar Rock as a consultant; and received research support from the SMA Foundation. E.S.M. has been an advisory board member for Biogen, Roche, and Scholar Rock; and been a consultant for AveXis, Cytokinetics, Ionis Pharmaceuticals, Inc, and Roche. A.P. has received research support from the SMA Foundation; serves as a consultant for Biogen; and has been an advisory board member for AveXis, Biogen, Roche, and Scholar Rock. A.M.G. received travel/lodging and compensation from Biogen to serve on an advisory board; licensing fees from Children's Hospital of Philadelphia for development of the CHOP INTEND motor scale; institutional support from AveXis, Biogen, and Roche for trial training; and personal compensation from ATOM International and Mallinckrodt for trial training. R.S.F. received grants and advisor fees from Biogen and Ionis Pharmaceuticals, Inc during ENDEAR and CHERISH and from Biogen during NURTURE and SHINE, and grants from Cytokinetics; outside the submitted work, he has received advisor fees and grants from AveXis and Roche, and has served as an advisor to Novartis; he has served in an advisory capacity for the nonprofit organizations Cure SMA, SMA Europe, the SMA Foundation, and SMA Reach (UK); he has been on the data safety monitoring board for the AveXis AVXS‐101 phase I gene transfer study and the Roche Moonfish phase IIb study; received personal compensation for serving as coeditor of Swaiman's Pediatric Neurology text (2018); received licensing fees from Children's Hospital of Philadelphia for development of the CHOP INTEND motor scale; and was recipient of grants from AveXis, Cytokinetics, Ionis Pharmaceuticals, Inc/Biogen, and Roche. B.T.D. has been an advisory board member for AveXis, Biogen, Cytokinetics, Dynacure, PTC, Roche, and Sarepta; received research support from Cure SMA, the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the Slaney Family Fund for SMA, the SMA Foundation, and the Working on Walking Fund; and received grants from Biogen and Ionis Pharmaceuticals, Inc during the ENDEAR, CHERISH, CS2, CS12, and CS11 studies, and also from Cytokinetics, Fibrogen, PTC, Roche, Santhera, Sarepta, and Summit, and reports no personal financial interests in these companies. F.M. is principal investigator for ongoing AveXis, Ionis Pharmaceuticals, Inc/Biogen, and Roche clinical trials; has received funding from Biogen, SMA Europe, and the SMA Trust; and has been an advisory board member for AveXis, Biogen, Novartis, Pfizer, PTC, Roche, Sarepta, Summit, and Wave, and has no financial interests in these companies. E.M. has been a an advisory board member for SMA studies for AveXis, Biogen, Ionis Pharmaceuticals, Inc, Novartis, and Roche; is principal investigator for ongoing AveXis, Ionis Pharmaceuticals, Inc/Biogen, and Roche clinical trials; and received funding from Famiglie SMA Italy, Italian Telethon, and SMA Europe. D.C.D. has been an advisor/consultant for AveXis, Biogen, Cytokinetics, Ionis Pharmaceuticals, Inc, Metafora, Roche, Sanofi, Sarepta, and the SMA Foundation, with no financial interests in these companies; received grants from the Department of Defense, Glut1 Deficiency Foundation, Hope for Children Research Foundation, the National Institutes of Health, and the SMA Foundation; and received clinical trial funding from Biogen, Mallinckrodt, PTC, Sarepta, Scholar Rock, and Ultragenyx. K.M.B. is an employee of Otonomy, Inc; and is an unpaid advisor to the nonprofit organizations Myotonic Dystrophy Foundation and SMA Foundation. E.S. and C.F.B. are employees of and hold stock/stock options in Ionis Pharmaceuticals, Inc. R.F. and W.F. are employees of and hold stock/stock options in Biogen.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
The authors thank the patients who participated in the CS2 and CS12 studies and their parents/guardians and family members. The authors also thank the CS2 and CS12 study investigators and all the contributors to CS2 and CS12, including the medical monitors, data safety and monitoring board members, study coordinators, physical therapists, pharmacists and laboratory technicians, and patient advocacy groups. The CS2 and CS12 studies were funded by Biogen and Ionis Pharmaceuticals, Inc. This analysis was funded by Biogen. Biogen provided funding for medical writing support in the development of this article. Rebecca Jarvis, PhD, and Allison Green, PhD, from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript according to journal requirements. Biogen and Ionis Pharmaceuticals, Inc reviewed and provided feedback on the manuscript to the authors. The authors had full editorial control of the manuscript and provided their final approval of all content.
Montes J, Dunaway Young S, Mazzone ES, et al. Nusinersen improves walking distance and reduces fatigue in later‐onset spinal muscular atrophy. Muscle Nerve. 2019;60:409–414. 10.1002/mus.26633
Present address Kathie M. Bishop, Otonomy, Inc, 4796 Executive Drive, San Diego, California 92121.
Portions of the material in this study were presented as a poster at the 70th Annual Meeting of the American Academy of Neurology, April 2018, Los Angeles, California, and as a podium presentation at the Cure SMA Annual Conference, June 2018, Dallas, Texas.
Funding information Biogen
REFERENCES
- 1. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell. 1995;80:155‐165. [DOI] [PubMed] [Google Scholar]
- 2. Darras BT, Monani UR, De Vivo DC. Genetic disorders affecting the motor neuron: spinal muscular atrophy In: Swaiman KF, Ashwal S, Ferriero DM, et al., eds. Swaiman's Pediatric Neurology: Principles and Practice. 6th ed. Edinburgh, UK: Elsevier; 2017:1057‐1064. [Google Scholar]
- 3. Finkel R, Bertini E, Muntoni F, Mercuri E, ENMC SMA Workshop Study Group . 209th ENMC International Workshop: Outcome measures and clinical trial readiness in spinal muscular atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25:593‐602. [DOI] [PubMed] [Google Scholar]
- 4. D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montes J, McDermott MP, Mirek E, et al. Ambulatory function in spinal muscular atrophy: age‐related patterns of progression. PLoS One. 2018;13:e0199657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dunaway Young S, Montes J, Kramer SS, et al. Six‐minute walk test is reliable and valid in spinal muscular atrophy. Muscle Nerve. 2016;54:836‐842. [DOI] [PubMed] [Google Scholar]
- 7. Montes J, McDermott MP, Martens WB, et al. Six‐minute walk test demonstrates motor fatigue in spinal muscular atrophy. Neurology. 2010;74:833‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montes J, Dunaway S, Montgomery MJ, et al. Fatigue leads to gait changes in spinal muscular atrophy. Muscle Nerve. 2011;43:485‐488. [DOI] [PubMed] [Google Scholar]
- 9. Montes J, Dunaway S, Garber CE, Chiriboga CA, De Vivo DC, Rao AK. Leg muscle function and fatigue during walking in spinal muscular atrophy type 3. Muscle Nerve. 2014;50:34‐39. [DOI] [PubMed] [Google Scholar]
- 10. Montes J, Blumenschine M, Dunaway S, et al. Weakness and fatigue in diverse neuromuscular diseases. J Child Neurol. 2013;28:1277‐1283. [DOI] [PubMed] [Google Scholar]
- 11. Wadman RI, Vrancken AF, van den Berg LH, van der Pol WL. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology. 2012;79:2050‐2055. [DOI] [PubMed] [Google Scholar]
- 12. Boido M, Vercelli A. Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front Neuroanat. 2016;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dimitriadi M, Derdowski A, Kalloo G, et al. Decreased function of survival motor neuron protein impairs endocytic pathways. Proc Natl Acad Sci USA. 2016;113:E4377‐E4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kariya S, Park GH, Maeno‐Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pera MC, Luigetti M, Pane M, et al. 6MWT can identify type 3 SMA patients with neuromuscular junction dysfunction. Neuromuscul Disord. 2017;27:879‐882. [DOI] [PubMed] [Google Scholar]
- 16. Spinraza [Prescribing information]. Cambridge, MA: Biogen; 2018.
- 17. Hoy SM. Nusinersen: first global approval. Drugs. 2017;77:473‐479. [DOI] [PubMed] [Google Scholar]
- 18. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med. 2017;377:1723‐1732. [DOI] [PubMed] [Google Scholar]
- 19. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later‐onset spinal muscular atrophy. N Engl J Med. 2018;378:625‐635. [DOI] [PubMed] [Google Scholar]
- 20. Darras BT, Chiriboga CA, Iannaccone ST, et al. Nusinersen in later‐onset spinal muscular atrophy: long‐term results from the phase 1/2 studies. Neurology. 2019;92:e2492‐e2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonald CM, Henricson EK, Abresch RT, et al. The 6‐minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord. 2016;26:126‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rouault F, Christie‐Brown V, Broekgaarden R, et al. Disease impact on general well‐being and therapeutic expectations of European Type II and Type III spinal muscular atrophy patients. Neuromuscul Disord. 2017;27:428‐438. [DOI] [PubMed] [Google Scholar]
- 24. Berger A, Mayr JA, Meierhofer D, et al. Severe depletion of mitochondrial DNA in spinal muscular atrophy. Acta Neuropathol. 2003;105:245‐251. [DOI] [PubMed] [Google Scholar]
- 25. Ripolone M, Ronchi D, Violano R, et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol. 2015;72:666‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stam M, Wadman RI, Bartels B, et al. A continuous repetitive task to detect fatigability in spinal muscular atrophy. Orphanet J Rare Dis. 2018;13:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spinraza [Summary of product characteristics] . https://www.ema.europa.eu/en/documents/product‐information/spinraza‐epar‐product‐information_en.pdf. Accessed May 1, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information