

Abstract
Oxidative chemical vapour deposition of (5,15‐diphenylporphyrinato)nickel(II) (NiDPP) with iron(III) chloride as oxidant yielded a conjugated poly(metalloporphyrin) as a highly coloured thin film, which is potentially useful for optoelectronic applications. This study clarified the reactive sites of the porphyrin monomer NiDPP by HRMS, UV/Vis/NIR spectroscopy, cyclic voltammetry and EPR spectroscopy in combination with quantum chemical calculations. Unsubstituted meso positions are essential for successful polymerisation, as demonstrated by varying the porphyrin meso substituent pattern from di‐ to tri‐ and tetraphenyl substitution. DFT calculations support the proposed radical oxidative coupling mechanism and explain the regioselectivity of the C−C coupling processes. Depositing the conjugated polymer on glass slides and on thermoplastic transparent polyethylene naphthalate demonstrated the suitability of the porphyrin material for flexible optoelectronic devices.
Keywords: chemical vapor deposition, nickel, polymerization, porphyrins, thin films
Introduction
Porphyrins and porphyrinic compounds experience widespread and growing interest due to their remarkable electronic and optoelectronic properties,1 which enable applications in photovoltaics,2 electro‐ and photocatalysis3, 4 and chemical sensing.5, 6 Particular interest focuses on directly fused metalloporphyrins, which have an extended π system, fascinating optical properties such as near‐infrared (NIR) absorption,7 two‐photon absorption,8 nonlinear optical properties9 and enhanced electrocatalytic activity.10 Although the current synthetic routes to directly fused metalloporphyrins consist of treating a metalloporphyrin having free meso and β positions (Scheme 1) with a suitable oxidant in solution, the poor solubility of metalloporphyrin monomers, oligomers and polymers requires the introduction of solubilising pendant groups to achieve high chain lengths.11
Scheme 1.
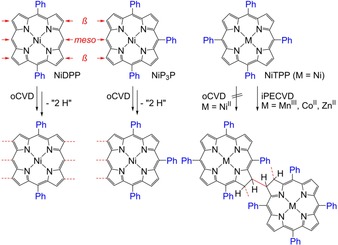
Molecular structures of the porphyrins investigated in this work. NiDPP can form linear polymers with up to three linkages between monomers. NiP3P should merely form dimers and MTPP should not readily polymerise in an oCVD process, but can form sterically congested β–β‐linked polychlorins by iPECVD.
The best results are obtained by dehydrogenative coupling of zinc(II) porphyrins with silver(I) salts followed by a second oxidation step to obtain β–β/meso–meso/β–β triply fused metalloporphyrins.7 Inspired by these seminal studies, alternatives for the formation of directly fused metalloporphyrins have been developed.11, 12, 13, 14, 15, 16, 17 However, fused metalloporphyrins are generally poorly soluble and infusible, and hence straightforward integration into devices is prevented.18, 19 On the other hand, introduction of solubilising agents on the porphyrins hinders the π–π interactions that are required for electronically conducting materials.1, 20
Chemical vapour deposition (CVD) processes enable simultaneous synthesis and deposition of a plethora of thin‐film compositions directly from the vapour phase. In particular, CVD circumvents the challenges related to the poor solubility of the starting monomers and resulting polymers, since no solvents are required in this process.21 Recent ground‐breaking studies also demonstrated successful free‐radical polymerisation and oxidative polymerisation of porphyrins in CVD processes (Scheme 1).20, 22
Initiated plasma‐enhanced CVD (iPECVD) of free‐base, manganese(III), cobalt(II) and zinc(II) 5,10,15,20‐tetraphenylporphyrins22 yielded polymeric films by free‐radical polymerisation of one of the exo‐pyrrole double bonds of the monomers (Scheme 1).23 The propagation reaction occurs at the β position of the porphyrin (22 π electrons) and leads to a reduced porphyrin (chlorin with 20 π electrons). In these polychlorin thin films the monomers are covalently connected through β–β linkages, yet π conjugation is absent (Scheme 1).22
On the other hand, the oxidative chemical vapour deposition (oCVD) reaction of (5,15‐diphenylporphyrinato)nickel(II) (NiDPP, Scheme 1) with FeCl3 furnished π‐conjugated poly(NiDPP) thin films by dehydrogenative C−C coupling. These films show a remarkably high conductivity of 3.6×10−2 S cm−1.20 Moreover, patterned conductive poly(NiDPP) thin films were readily formed even on printer paper owing to the potential of oCVD to form conjugated polymer thin films directly from the vapour phase without the need for solvents.20 In addition, the oxidative coupling of the NiDPP monomer in oCVD yielded NIR‐absorbing coatings consistent with the formation of directly fused metalloporphyrins.
Although thin film preparation by oCVD is rather straightforward, mechanistic details and the side reactions occurring in the oCVD process are not yet fully understood. Herein, we report insights into the dehydrogenative fusion sites for di‐, tri‐ and tetraphenyl meso‐substituted nickel(II) porphyrins (Scheme 1). Laser desorption ionisation (LDI) HRMS and UV/Vis/NIR spectroscopy were used to probe the direct fusion of di‐ and triphenyl‐substituted nickel(II) porphyrins to form conjugated oligomers and polymers. Characterisation of the thin films was complemented by studies on the monomers and monomer radical cations as the active species by cyclic voltammetry (CV), EPR spectroscopy and DFT calculations to provide insights into the initial steps in oCVD of nickel(II) porphyrins.
Results and Discussion
Preparation of thin films by oCVD and film characterisation
Nickel(II) 5,15‐diphenylporphyrins form linear polymers both in solution and in oCVD‐based approaches with suitable oxidants (Scheme 2).18, 20 Nickel(II) as central metal ion is inert towards demetallation by acids, which can be produced during the coupling reactions. Consequently, all oCVD processes in this study were performed with the respective stable nickel(II) porphyrins.24 FeCl3 is quite volatile25 and its oxidation potential is sufficiently high to make it an ideal oxidant for oCVD processes20, 26 and for the dehydrogenative coupling of porphyrins in solution‐based approaches.14
Scheme 2.
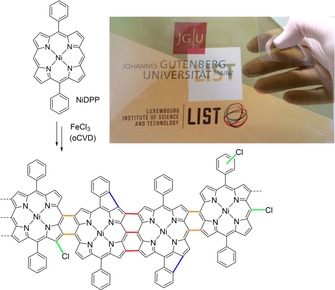
The oCVD reaction of NiDPP yields a conjugated fused metalloporphyrin polymer with meso–β/β–meso (orange bonds) or β–β/meso–meso/β‐β (red bonds) links between the monomer units. Potential side reactions, namely, cyclisation between a phenyl substituent and the porphyrin macrocycle (blue bonds) and chlorination of the porphyrin (green bonds), are indicated. The location of cyclisation and chlorination is not clear and merely representative. The inset shows an optical image of a bent transparent PEN foil coated by the greenish oCVD NiDPP film (top area of the foil) and the orange reference NiDPP film (bottom area of the foil).
With the aim of identifying the sites involved in the oxidative coupling of 5,15‐diphenyl‐substituted porphyrins during the oCVD process, we selectively blocked the remaining meso sites by phenyl substituents and investigated the oCVD reaction of tri‐ and tetra‐meso‐substituted nickel(II) 5,10,15‐triphenylporphyrin (NiP3P) and nickel(II) 5,10,15,20‐tetraphenylporphyrin (NiTPP), respectively (Scheme 1). NiTPP should be unable to form linkages at its meso positions. On the other hand, NiP3P should mainly form dimers and trimers through its single free meso position (Scheme 1).
The oCVD reaction of NiDPP and NiP3P with FeCl3 as oxidant under the reported conditions (see Experimental Section)20 yielded greenish coatings on glass and on polyethylene naphthalate (PEN) foils, in contrast to the reddish colour of the respective reference NiDPP and NiP3P coatings formed in the absence of oxidant (Scheme 2; Supporting Information, Figure S1). On the other hand, the oCVD reaction of NiTPP with FeCl3 has a far less pronounced impact on the colour of the coatings, which only turned from reddish for the reference NiTPP coating to orange for the oCVD NiTPP coating (Supporting Information, Figure S1 c).
All films produced by oCVD covered the substrates homogeneously with thicknesses determined by profilometry of 200, 120 and about 10 nm for NiDPP, NiP3P and NiTPP, respectively. The decreasing film thickness in this series suggests a relation between the number of phenyl rings and the evaporation rate of the phenyl‐substituted porphyrins in the oCVD process.
UV/Vis/NIR absorption spectroscopy revealed broader absorption of the oCVD coatings compared to the reference coatings obtained by sublimation of the respective porphyrin. The absorption of the oCVD NiP3P and NiDPP coatings even tails significantly into the NIR region (Figure 1 a and b). In particular, the oCVD NiDPP coating appreciably absorbs up to 2000 nm (Figure 1 a). According to reports of Osuka et al.,7, 18 these observations suggest the formation of oligomers with higher conjugation lengths.
Figure 1.
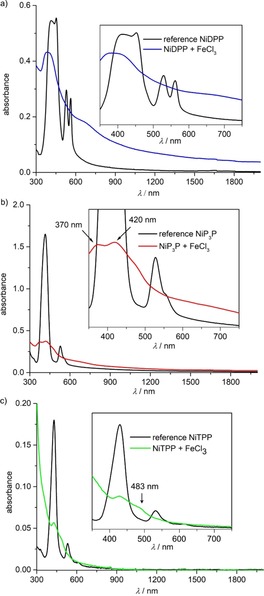
UV/Vis/NIR absorption spectra of the oCVD coatings grown from a) NiDPP (blue line), b) NiP3P (red line) and c) NiTPP (green line) on glass slides. UV/Vis/NIR absorption spectra of the respective sublimed porphyrins are provided for comparison (black lines).
In addition, the oCVD NiDPP coating is almost insoluble in many common nonpolar, polar aprotic and protic organic solvents (e.g., chloroform, dichloromethane, acetone, methanol, hexafluoro‐2‐propanol), whereas the oCVD NiP3P and NiTPP coatings are partly and fully soluble in these solvents, respectively (Supporting Information, Figure S2). Clearly, the solubility of the coating decreases on reducing the number of phenyl rings attached to the porphyrins from four to two. This is consistent with the proposed polymerisation through the meso positions (meso–β, meso–meso), which is impossible for NiTPP, while NiP3P may yield less soluble dimers and possibly short oligomers. On the other hand, NiDPP essentially forms insoluble coatings. As expected, all reference coatings formed by sublimation, that is, porphyrin monomers, are completely soluble in the aforementioned solvents.
UV/Vis/NIR spectroscopic analysis of the acetone‐ and dichloromethane‐soluble phases of all oCVD coatings revealed broadened and bathochromically shifted Soret and Q bands (Supporting Information, Figure S3 and Table S1) together with unconsumed FeCl3 showing an absorption band at 350 nm in acetone. The characteristic changes of the Soret bands of the soluble, low molecular weight fractions of the NiP3P and NiTPP oCVD coatings are very similar to those observed for the NiDPP films. We had attributed these absorption changes to dehydrogenative intramolecular cyclisation reactions between the phenyl rings and the β position of the porphyrin and/or chlorination of the porphyrins (Scheme 2).20 Both reactions modify the porphyrin symmetry and the HOMO–LUMO energy gap, which results in red shifts and different numbers of Q bands compared with the respective starting materials.27, 28, 29
These two side reactions are particularly noticeable for the NiTPP oCVD coating, which shows a strong decrease and bathochromic shift of the Soret and Q bands (Figure 1 c). UV/Vis/NIR spectroscopic analysis of the dichloromethane phase of this fully soluble coating revealed a broad absorption band with a pronounced maximum at 429 nm and weaker maxima at 462 and 514 nm (Supporting Information, Figure S3 c and f). The two weaker maxima agree very well with literature reports for singly and doubly ring fused ZnTPP in DMF (471 and 532 nm, respectively).28b The bathochromic shift of the main band to 429 nm and the broadening are attributed to chlorination of the porphyrin.27
Atmospheric‐pressure LDI‐HRMS of all oCVD coatings (Figure 2) confirmed that, similar to solution‐based approaches, a free meso position is required to promote the oxidative polymerisation of the porphyrin through oCVD.14, 30 Indeed, the mass spectrum of the oCVD NiTPP coating solely exhibits signals related to NiTPP monomers (Figure 2 a), while peaks corresponding to oligomers are additionally observed for the oCVD NiP3P and NiDPP coatings (Figure 2 b and c). Besides signals of the expected NiP3P dimers, weak signals of NiP3P oligomers are detected as well (Figure 2 b). This is in contrast to the reaction of NiP3P and iron(III) salts in solution, which gives mainly doubly and triply linked dimers in a combined yield of 92 %.14 On the other hand, DFT calculations had suggested the possibility that in a CVD process tetraphenylporphyrins can undergo a free‐radical polymerisation reaction through exo‐pyrrole double bonds leading to a sterically congested β–β linked‐staircase structure.22 During the oCVD process discussed here, NiP3P could possibly exhibit similar reactivity to form short oligomers through β–meso or β–β linkages at the less hindered side of the porphyrin (Figure 2 b, inset). Clearly, linkages involving the β positions are possible in CVD processes, yet they appear to be insignificant in solution processes involving NiP3P. In such congested β–meso or β–β connections, the almost perpendicular arrangement of the monomeric units hampers π extension and conjugation compared with doubly or triply linked NiDPP. Less conjugation also explains the lower NIR absorbance of the oCVD NiP3P coating compared to the oCVD NiDPP coating (Figure 1 a and b). Further investigations on this intriguing reactivity pattern are ongoing but lie outside the scope of the present study.
Figure 2.
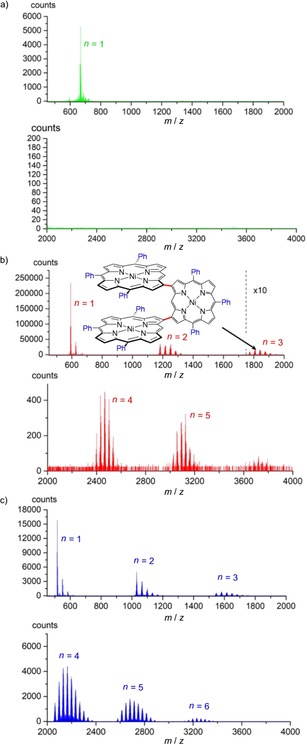
LDI‐HRMS analysis of a) oCVD NiTPP, b) oCVD NiP3P and c) oCVD NiDPP coatings. Peak distributions related to the chlorination of the porphyrin are present in all mass spectra. The spectra show the formation of oligomers for NiP3P and NiDPP (n>1). The m/z 500–2000 and 2000–4000 ranges were recorded in separate analyses and thus a meaningful comparison of intensities is not possible. b) The inset shows a conceivable structure of a β–β‐linked NiP3P trimer.
Similar to previously reported data for NiDPP,20 the LDI‐HR mass spectra of oCVD NiP3P and NiTPP coatings show signals related to the loss of an even number of hydrogen atoms (−2n H) for the monomer (Figure 3). Interestingly, the maximum number of 2 H pairs eliminated from the porphyrin monomers is proportional to the number of phenyl substituents, that is, 4 H for NiDPP, 6 H for NiP3P and 8 H for NiTPP. This observation strongly supports the previously suggested intramolecular cyclisation between the phenyl substituent and the β position of the porphyrin (Scheme 2).
Figure 3.
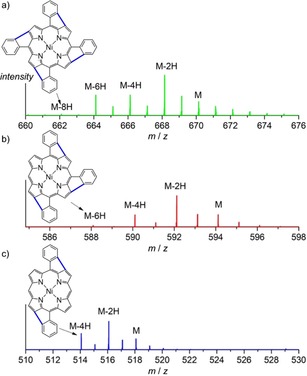
LDI‐HR mass spectra of a) oCVD NiTPP, b) oCVD NiP3P and c) oCVD NiDPP in the monomer range. The spectra show the loss of hydrogen atoms (M−2n H) attributed to the intramolecular cyclisation between the phenyl ring and the macrocycle. The coupling is drawn at arbitrary positions.
As already observed for oCVD NiDPP, the mass spectra of oCVD NiP3P and NiTPP coatings show multiple chlorine‐atom incorporation leading to several sets of peak clusters separated by 35 mass units (Figure 2). The mass spectra did not allow any preferential reactive site on the porphyrin for chlorination (Scheme 2) to be determined. This chlorination is likely due to a reaction between the porphyrin and iron(III) chloride or Cl2 produced from FeCl3 during sublimation.27, 31, 32 Chlorinated by‐products are also commonly observed in reactions of aromatic substrates and oxidizing metal halides.33
Mechanistic aspects of oCVD of nickel porphyrins
To gain a deeper understanding of the NiDPP/oxidant reactivity, electrochemical experiments were conducted (Figure 4). The cyclic voltammogram of NiDPP in CH2Cl2/[nBu4N][PF6] shows quasireversible waves at peak potentials of E p=0.57, 0.95 and 1.10 V versus the ferrocene/ferrocenium couple, assigned to the first, second and third oxidation of NiDPP, respectively. The first oxidation of NiP3P has been reported at a very similar potential (E p≈0.55 V vs. ferrocene/ferrocenium).14 The quasireversibility suggests a follow‐up reaction after the initial one‐electron oxidation.
Figure 4.
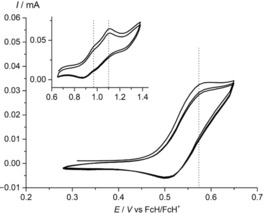
Cyclic voltammograms of NiDPP in CH2Cl2/[nBu4N][PF6] solution.
The oxidation potential of iron(III) triflate is high enough to promote the oxidative coupling of NiP3P in CH2Cl2/CH3NO2 solution yielding triply and doubly linked dimers.14 Furthermore, conjugated polyporphyrins have an even lower oxidation potential than their parent monomers, which favours extension of polymeric chains over initiation of new ones.18 Similarly, FeCl3 promotes oxidative polymerisation of NiDPP and NiP3P in oCVD, as sublimation of FeCl3(s) most probably yields FeCl3(g), Fe2Cl6(g) and Cl2(g) among other oxidizing/chlorinating species under the oCVD conditions.34 We suggest that the formation of strongly oxidising species such as Cl2 could be involved in the intramolecular cyclisation of the porphyrin. Indeed, it has been shown previously that a second oxidation of the porphyrin is needed to achieve electrochemical extension of the π system.28b, 28c
In all cases, the initial step of the oxidative polymerisation is most probably the one‐electron oxidation of the nickel(II) porphyrins to [NiDPP].+, [NiP3P].+ and [NiTPP].+, respectively. In coordinating solvents or in the presence of coordinating ligands, [NiTPP].+ is best described as a nickel(III) porphyrin, whereas a description as a nickel(II) porphyrin π cation radical is more appropriate in the absence of axial coordination.35 To simulate the oCVD conditions, NiDPP (1 mm in CH2Cl2) was oxidised with one equivalent of WCl6 36 under inert atmosphere and in the absence of coordinating ligands (apart from potentially dissociated chloride) in an EPR tube. The solution was rapidly frozen by immersing the EPR tube in liquid nitrogen and examined by X‐band EPR spectroscopy. The corresponding EPR spectrum at 77 K (Figure 5 a) shows a weakly anisotropic resonance at g av=2.004 [NiDPP].+. No hyperfine coupling to 14N nuclei was resolved. For [Ni(TPP)].+, g av=2.005 (gx=2.012, gy≈2.004, gz≈2.002) has been reported, consistent with a nickel(II) porphyrin radical cation. On the other hand, nickel(III) porphyrins show highly anisotropic EPR patterns, for example, gx=2.328, gy=2.305 and gz=2.093 for [NiIII(TPP)(THF)2].+.35 Consequently, the electronic structure of [NiDPP].+ is best described as nickel(II) porphyrin radical cation under these conditions. Upon warming the solution to room temperature, the isotropic EPR spectra evolved over time towards an isotropic value of g iso=2.007 suggesting an ongoing chemical reaction of the radical cations (Figure 5 a). Persistent radical cations of aromatic molecules had been observed previously, for example, by using MoCl5 or FeCl3 with suitable aromatic substrates.37, 38
Figure 5.
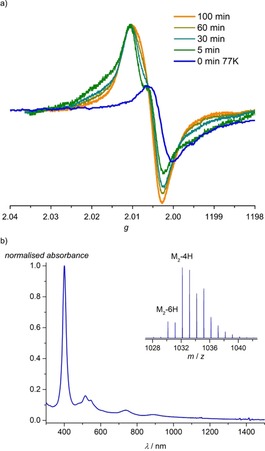
a) EPR spectrum of NiDPP/WCl6 solution in CH2Cl2 rapidly frozen at 77 K (orange trace) and evolution of the EPR spectrum (normalised) upon thawing the solution and warming to room temperature over time. b) UV/Vis/NIR and MALDI‐HR mass spectrum of the final NiDPP/WCl6 solution (inset).
UV/Vis/NIR spectroscopic analysis of the solution at the end of the EPR measurements (Figure 5 b) showed new weak bands at 478, 615, 736 and 888 nm, which are consistent with porphyrin coupling reactions. LDI‐HR mass spectra confirmed such coupling reactions. The mass spectrum in the m/z region of dimers shows a main peak at 1032.176 amu for [C64H36N8Ni2]+, attributed to a doubly linked bis‐porphyrin, and a five times less intense signal at 1030.160 amu for [C64H34N8Ni2]+ (inset of Figure 5 b). The latter peak corresponds to the loss of 2 H from the dimer, although it remains speculative whether a further porphyrin–porphyrin linkage or a phenyl cyclisation has occurred (Scheme 2). Clearly, WCl6 promotes the dimerisation/oligomerisation of NiDPP in CH2Cl2 solution. Furthermore, the evolution of the EPR spectra demonstrates a radical chain growth mechanism with the EPR pattern of porphyrin oligomers varying with the oligomer length.
All obtained EPR data fit to essentially ligand‐centred radicals without significant nickel contributions and are similar to literature data of [NiTPP].+.39, 40 This electronic description of [NiDPP].+ should apply for [NiP3P].+ as well, both for the solution (in the absence of axial ligands) and for the gas phase.
DFT calculations with B3LYP, TPSSH and PBE0 functionals converged to weakly distorted nickel(III) valence isomers as ground states, which are not observed under our conditions in the EPR spectra. Ambiguities and difficulties in assigning the correct valence isomeric descriptions have been noted before for [NiTPP].+.35, 39, 41 DFT calculations at the RIJCOSX‐BP86‐D3‐ZORA/def2‐TZVP level of theory [with and without solvent modelling by CPCM(CH2Cl2)] correctly describe [NiDPP].+ and [NiP3P].+ as π radical cations with saddle‐shaped macrocycles (Figure 6).42
Figure 6.
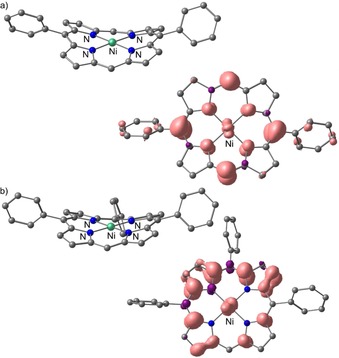
DFT calculated optimised geometries (approximately side view) and the respective spin densities (isosurface value 0.003 a.u.; approximately top view) of a) [NiDPP].+ and b) [NiP3P].+ radical cations (side view). Hydrogen atoms omitted for clarity.
The actual symmetry of the spin density in the π radical (a1u or a2u in the idealised D 4h symmetry point group) is often ambiguous as well due to static and/or dynamic pseudo‐Jahn–Teller effects which mix these states by appropriate vibrational modes.42 According to the DFT calculations, [NiDPP].+ features spin density at the meso positions (approximate a2u symmetry), and [NiP3P].+ shows spin density at the pyrrole rings and nodal planes at the meso positions (approximate a1u symmetry) at this level of theory (Figure 6). In fact, both descriptions may contribute to the true electronic structure of the π radical cations due to vibrationally induced mixing of the states. Considering the Jahn–Teller bistability of nickel porphyrin radical cations, spin density could be present at both meso and β positions of the radical cations and affect also the distribution of electrophilic sites within the macrocycle. Consequently, both sites are amenable to nucleophilic attack, that is, by a neutral nickel porphyrin with its meso positions having large orbital coefficients in the HOMO (a2u symmetry; Supporting Information, Figure S5). Hence, both initial connections, β–meso and meso–meso, can in principle form. On oxidation with tris(4‐bromophenyl)aminium hexachloroantimonate, NiDPP forms meso–β links in CHCl3 (among other products),17 while NiP3P is reported to form both meso–β and meso–meso links.14, 17 In fact, the solvent/oxidant combination influences the meso–β/meso–meso link ratio.14, 17 We suggest that oxidant and environment may influence the actual spin density of the radical cations [NiDPP].+ and [NiP3P].+ and consequently the regioselectivity of the initial C−C bond‐forming reaction. The electron‐deficient pyrrole rings of the radical cations might also be amenable to nucleophilic attack by the phenyl substituents leading to the intramolecular cyclisation. Such intramolecular dehydrogenative coupling reactions might be preferred under oCVD conditions, that is, in the gas phase, and by reactions of less‐mobile porphyrins on the substrate surface.
Beyond the polymerization process, the observed diverse reactivity of nickel porphyrins in oCVD processes with FeCl3 includes chlorination by highly reactive oxidants such as Cl2 in the gas phase, formation of β–β linkages to form oligomers from NiP3P and intramolecular cyclisation enabled by high dilution (gas phase), low mobility (surface) and the presence of strong oxidants (Cl2).
Conclusion
The oCVD reaction of nickel(II) porphyrins with FeCl3 requires free meso positions to form conjugated polyporphyrins. The first step in the polymerisation reaction is the one‐electron oxidation of nickel(II) porphyrins to the respective π radical cations coordinated to nickel(II). In the gas phase and in non‐coordinating solvents, the nickel(III) porphyrin isomer is less favourable. The pseudo‐Jahn–Teller distortion of the π radical cations mixes states with different symmetry (a1u and a2u), and this allows for nucleophilic attack of the electron‐rich meso position of neutral nickel(II) porphyrins at both the β and meso positions of the radical cations. This leads to both meso–β and meso–meso linkages in the growing polymer and gives doubly and triply linked units after further dehydrogenative coupling.
Compared to conventional solution‐based methods, which mainly yield doubly and triply linked oligoporphyrins, the oCVD process of nickel(II) porphyrins and FeCl3 shows an even broader reactivity pattern, including polymer formation, chlorination, formation of single β–β linkages and intramolecular cyclisation. The diverse reactivity in the oCVD process is explained by the presence of chlorinating agents, the low porphyrin concentration in the gas phase and the low mobility on the substrate surface.
In addition to glass or silicon wafers as substrates, the reported oCVD process is also compatible with transparent, thermally stable43 plastic foils (PEN). This could lead to the design of a new family of flexible optoelectronic devices thanks to the combination of the optical, electric and mechanical properties of the fused porphyrin thin films. Finally, the oxidant WCl6 34, 36, 44 can also be used in the oxidative coupling of nickel porphyrins in solution and possibly also in oCVD processes.
Experimental Section
Materials
(5,15‐Diphenylporphyrinato)nickel(II) (NiDPP) and (5,10,15,20‐tetraphenylporphyrinato)nickel(II) (NiTPP) were prepared by metallation of 5,15‐diphenylporphyrin (PorphyChem, 98 %) (H2DPP) and 5,10,15,20‐tetraphenylporphyrin (PorphyChem, 98 %), (H2TPP), respectively, with Ni(OAc)2 ⋅4 H2O.45 (5,10,15‐Triphenylporphyrinato)nickel(II) (NiP3P) was obtained from PorphyChem and used without further purification (98 %). Iron(III) chloride (97 %; Sigma‐Aldrich) and tungsten hexachloride (99.9 %; ABCR) were used without further purification. Dichloromethane for EPR measurements was dried with CaH2 and distilled prior to use.
oCVD experiments
oCVD experiments were performed in a custom‐built oCVD reactor as described elsewhere.20 The deposition experiments were carried out in argon (Air Liquide, 99.999 %) at a pressure of 10−3 mbar. The evaporators, located at the bottom of the reaction chamber, were loaded with 10 mg of porphyrins and 150 mg of FeCl3 and heated to 250 and 170 °C, respectively. The substrate holder, oriented face‐down, was maintained at 130 °C. The deposition time was 30 min in all experiments. Microscope glass slides, silicon wafers and 125 μm PEN43 foils (DuPont Teijin Films) were used as substrates.
Spectroscopy and film characterization
The optical absorbance of the films was measured in the range of 250–2000 nm with a UV/Vis/NIR spectrophotometer (PerkinElmer, Lambda 950) with a 150 mm‐diameter integrating sphere. The absorption spectra were recorded directly on the glass substrates before and after rinsing the glass with dichloromethane and acetone. UV/Vis/NIR spectra of the soluble fractions of the coating in acetone or CH2Cl2 were measured in quartz cuvettes of 3.5 mL and 1 cm path length. The thin films thicknesses were measured using a KLA‐Tencor P‐17 Stylus profiler. Cyclic voltammetric measurements were carried out with a BioLogic SP‐50 voltammetric potentiostat 1 mm in CH2Cl2 containing 0.1 m [nBu4N][PF6] as supporting electrolyte with a platinum working electrode, a platinum wire as counter electrode and a 0.01 m Ag/AgNO3 reference electrode. Cyclic voltammograms were recorded at 100 mV s−1 scan rate. Ferrocene was employed as an internal reference redox system. X‐band CW EPR spectra were recorded with a Magnettech MS 300 spectrometer and a Hewlett Packard 5340A frequency counter at a microwave frequency of 9.39 GHz in CH2Cl2 solution (298 and 77 K). Mn2+ in ZnS was used as external standard (g=2.118, 2.066, 2.027, 1.986, 1.946, 1.906). Atmospheric‐pressure LDI‐HRMS was employed for characterisation of the coatings. HRMS analyses were performed with an LTQ/Orbitrap Elite Hybrid Linear Ion Trap‐Orbitrap Mass Spectrometer from Thermo Scientific (San Jose, CA) coupled with an AP‐LDI (ng) UHR source from MassTech Inc (Columbia, MA) with a 355 nm Nd:YAG laser. The thin films were directly probed without any matrix deposition by the laser following a spiral motion during 30 s per sample. An in‐source decay (ISD) of 70 V was applied to the samples in order to prevent any formation of non‐covalent porphyrin clusters that could interfere with the distribution of the oligomers. A maximum injection time of 800 ms and a resolving power of 240 000 at m/z 400 in the normal mass range (m/z 300–2000) and the high mass range (m/z 1800–4000) were employed for the HRMS analyses. The solution prepared for EPR analysis was studied by HR‐MALDI mass spectrometry by using α‐cyano‐4‐hydroxycinnamic acid as matrix.
Preparation of EPR samples
In an EPR tube, 0.5 mL of a 10−3 m CH2Cl2 solution of NiDPP was treated with 0.7 mL of a 10−3 m CH2Cl2 solution of WCl6 under inert atmosphere. The solution was rapidly frozen by immersing the tube in liquid nitrogen. X‐band EPR: g av=2.004 (77 K); g iso=2.007 (298 K); MALDI‐HRMS (α‐cyano‐4‐hydroxycinnamic acid): m/z=518.103 [M].+, 1030.160 [M 2−6 H].+, 1032.176 [M 2−4 H].+, 1542.217 [M 3−12 H].+, 1544.233 [M 3−10 H].+, 1546.252 [M 3−8 H].+; UV/Vis/NIR: λ max=478, 615, 736, 888 nm.
DFT calculations
DFT calculations were performed with the ORCA program package (Version 4.0.1).46 All calculations were carried out by using the BP8647, 48 functional in combination with the D349, 50 dispersion correction and the RIJCOX51, 52 approximation. A conductor‐like screening solvation model (CPCM) modelling CH2Cl2 was used for the calculations unless noted otherwise.53 Relativistic effects were included with the zeroth‐order regular approximation (ZORA) level.54 Geometry optimisations were performed by means of Ahlrichs’ triple zeta valence basis set def2‐TZVP55, 56, 57 and Weigend's auxiliary basis set.58 The optimised geometries were confirmed to be local minima on the respective potential energy surface by numerical frequency analyses that showed the absence of negative frequencies. Explicit counterions and/or solvent molecules were not taken into account.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge the financial support of the Luxembourg National Research Fund (http://fnr.lu) through the POLYPORPH project (C15/MS/10340560/POLYPORH/Boscher). D. El Assad from LIST is acknowledged for insightful discussions and acquisition of the mass spectra. Parts of this research were conducted using the supercomputer MOGON and advisory services offered by Johannes Gutenberg University of Mainz (http://www.hpc.uni‐mainz.de), which is a member of the AHRP and the Gauss Alliance e.V..
G. Bengasi, K. Baba, O. Back, G. Frache, K. Heinze, N. D. Boscher, Chem. Eur. J. 2019, 25, 8313.
Contributor Information
Prof. Dr. Katja Heinze, Email: katja.heinze@uni-mainz.de.
Dr. Nicolas D. Boscher, Email: nicolas.boscher@list.lu.
References
- 1. Kesters J., Verstappen P., Kelchtermans M., Lutsen L., Vanderzande D., Maes W., Adv. Energy Mater. 2015, 5, 1500218. [Google Scholar]
- 2. Mathew S., Yella A., Gao P., Humphry-Baker R., Curchod B. F. E., Ashari-Astani N., Tavernelli I., Rothlisberger U., Nazeeruddin M. K., Grätzel M., Nat. Chem. 2014, 6, 242–247. [DOI] [PubMed] [Google Scholar]
- 3. Lin S., Diercks C. S., Zhang Y.-B., Kornienko N., Nichols E. M., Zhao Y., Paris A. R., Kim D., Yang P., Yaghi O. M., Chang C. J., Science 2015, 349, 1208–1213. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Zhang W., Lai W., Cao R., Chem. Rev. 2017, 117, 3717–3797; [DOI] [PubMed] [Google Scholar]
- 4b. Liu Y., Han Y., Zhang Z., Zhang W., Lai W., Wang Y., Cao R., Chem. Sci. 2019, 10, 2613–2622; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Wadsworth B. L., Khusnutdinova D., Moore G. F., J. Mater. Chem. A 2018, 6, 21654–21665; [Google Scholar]
- 4d. Rybicka-Jasińska K., Shan W., Zawada K., Kadish K. M., Gryko D., J. Am. Chem. Soc. 2016, 138, 15451–15458. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Ding Y., Zhu W.-H., Xie Y., Chem. Rev. 2017, 117, 2203–2256; [DOI] [PubMed] [Google Scholar]
- 5b. Rakow N. A., Suslick K. S., Nature 2000, 406, 710–713; [DOI] [PubMed] [Google Scholar]
- 5c. Heier P., Boscher N. D., Choquet P., Heinze K., Inorg. Chem. 2014, 53, 11086–11095. [DOI] [PubMed] [Google Scholar]
- 6. Singh S., Aggarwal A., Bhupathiraju N. V. S. D. K., Arianna G., Tiwari K., Drain C. M., Chem. Rev. 2015, 115, 10261–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsuda A., Osuka A., Science 2001, 293, 79–82. [DOI] [PubMed] [Google Scholar]
- 8. Pawlicki M., Collins H. A., Denning R. G., Anderson H. L., Angew. Chem. Int. Ed. 2009, 48, 3244–3266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3292–3316. [Google Scholar]
- 9. Senge M. O., Fazekas M., Notaras E. G. A., Blau W. J., Zawadzka M., Locos O. B., Ni Mhuircheartaigh E. M., Adv. Mater. 2007, 19, 2737–2774. [Google Scholar]
- 10. Khusnutdinova D., Wadsworth B. L., Flores M., Beiler A. M., Cruz E. A. R., Zenkov Y., Moore G. F., ACS Catal. 2018, 8, 9888–9898. [Google Scholar]
- 11. Yoshida N., Aratani N., Osuka A., Chem. Commun. 2000, 197–198. [Google Scholar]
- 12. Brennan B. J., Arero J., Liddell P. A., Moore T. A., Moore A. L., Gust D., J. Porphyrins Phthalocyanines 2013, 17, 247–251. [Google Scholar]
- 13. Sahoo A. K., Nakamura Y., Aratani N., Kim K. S., Noh S. B., Shinokubo H., Kim D., Osuka A., Org. Lett. 2006, 8, 4141–4144. [DOI] [PubMed] [Google Scholar]
- 14. Feng C.-M., Zhu Y.-Z., Zhang S.-C., Zang Y., Zheng J.-Y., Org. Biomol. Chem. 2015, 13, 2566–2569. [DOI] [PubMed] [Google Scholar]
- 15. Jin L. M., Chen L., Yin J. J., Guo C. C., Chen Q. Y., Eur. J. Org. Chem. 2005, 3994–4001. [Google Scholar]
- 16. Sugiura K., Matsumoto T., Ohkouchi S., Naitoh Y., Kawai T., Takai Y., Ushiroda K., Sakata Y., Chem. Commun. 1999, 1957–1958. [Google Scholar]
- 17. Tsuda A., Nakano A., Furuta H., Yamochi H., Osuka A., Angew. Chem. Int. Ed. 2000, 39, 558–561; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 572–575. [Google Scholar]
- 18. Tsuda A., Nakamura Y., Osuka A., Chem. Commun. 2003, 1096–1097. [DOI] [PubMed] [Google Scholar]
- 19. Brennan B. J., Kenney M. J., Liddell P. A., Cherry B. R., Li J., Moore A. L., Moore T. A., Gust D., Chem. Commun. 2011, 47, 10034–10036. [DOI] [PubMed] [Google Scholar]
- 20. Bengasi G., Baba K., Frache G., Desport J., Gratia P., Heinze K., Boscher N. D., Angew. Chem. Int. Ed. 2019, 58, 2103–2108; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2125–2130. [Google Scholar]
- 21. Wang M., Wang X., Moni P., Liu A., Kim D. H., Jo W. J., Sojoudi H., Gleason K. K., Adv. Mater. 2017, 29, 1604606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M., Boscher N. D., Heinze K., Gleason K. K., Adv. Funct. Mater. 2017, 27, 1606652. [Google Scholar]
- 23. Boscher N. D., Minghui W., Perrotta A., Heinze K., Creatore M., Gleason K. K., Adv. Mater. 2016, 28, 7479–7485. [DOI] [PubMed] [Google Scholar]
- 24. Kadish K. M., Smith K. M., Guillard R., The Porphyrin Handbook: Inorganic, Organometallic and Coordination Chemistry, Vol. 3, Academic Press, London, 2000. [Google Scholar]
- 25. CRC Handbook of Chemistry and Physics, 92nd ed. (Ed.: W. M. Haynes), CRC, Boca Raton, 2011. [Google Scholar]
- 26. Gharahcheshmeh M. H., Gleason K. K., Adv. Mater. Interfaces 2019, 6, 1801564. [Google Scholar]
- 27. Wijesekera T., Matsumoto A., Dolphin D., Lexa D., Angew. Chem. Int. Ed. Engl. 1990, 29, 1028–1030; [Google Scholar]; Angew. Chem. 1990, 102, 1073–1074. [Google Scholar]
- 28.
- 28a. Ishizuka T., Saegusa Y., Shiota Y., Ohtake K., Yoshizawa K., Kojima T., Chem. Commun. 2013, 49, 5939–5941; [DOI] [PubMed] [Google Scholar]
- 28b. Chen P., Fang Y., Kadish K. M., Lewtak J. P., Koszelewski D., Janiga A., Gryko D. T., Inorg. Chem. 2013, 52, 9532–9538; [DOI] [PubMed] [Google Scholar]
- 28c. Fang Y., Koszelewski D., Kadish K. M., Gryko D. T., Chem. Commun. 2014, 50, 8864–8867. [DOI] [PubMed] [Google Scholar]
- 29. Saegusa Y., Ishizuka T., Komamura K., Shimizu S., Kotani H., Kobayashi N., Kojima T., Phys. Chem. Chem. Phys. 2015, 17, 15001–15011. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka T., Osuka A., Chem. Soc. Rev. 2015, 44, 943–969. [DOI] [PubMed] [Google Scholar]
- 31. Wilson L. E., Gregory N. W., J. Phys. Chem. 1958, 62, 433–437. [Google Scholar]
- 32. Lewtak J. P., Gryko D., Bao D., Sebai E., Vakuliuk O., Ścigaj M., Gryko D. T., Org. Biomol. Chem. 2011, 9, 8178–8181. [DOI] [PubMed] [Google Scholar]
- 33. Schubert M., Leppin J., Wehming K., Schollmeyer D., Heinze K., Waldvogel S. R., Angew. Chem. Int. Ed. 2014, 53, 2494–2497; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2527–2530. [Google Scholar]
- 34. M. W. Chase, Jr. , Davies C. A., J. R. Downey, Jr. , Frurip D. J., McDonald R. A., Syverud A. N., J. Phys. Chem. Ref. Data 1985, 14, 903. [Google Scholar]
- 35. Seth J., Palaniappan V., Bocian D. F., Inorg. Chem. 1995, 34, 2201–2206. [Google Scholar]
- 36. Herndon J. W., Jung M. E., Encyclopedia of Reagents for Organic Synthesis, Wiley, Hoboken, 2007. [Google Scholar]
- 37. Leppin J., Schubert M., Waldvogel S. R., Heinze K., Chem. Eur. J. 2015, 21, 4229–4232. [DOI] [PubMed] [Google Scholar]
- 38. Horibe T., Ohmura S., Ishihara K., J. Am. Chem. Soc. 2019, 141, 1877–1881. [DOI] [PubMed] [Google Scholar]
- 39. Dolphin D., Niem T., Felton R. H., Fujita I., J. Am. Chem. Soc. 1975, 97, 5288–5290. [DOI] [PubMed] [Google Scholar]
- 40. Chatterjee D., Balasubramanian E., J. Coord. Chem. 1999, 46, 467–470. [Google Scholar]
- 41. Wolberg A., Manassen J., Inorg. Chem. 1970, 9, 2365–2367. [Google Scholar]
- 42. Czernuszewicz R. S., Macor K. A., Li X. Y., Kincaid J. R., Spiro T. G., J. Am. Chem. Soc. 1989, 111, 3860–3869. [Google Scholar]
- 43. Lillwitz L. D., Appl. Catal. A 2001, 221, 337–358. [Google Scholar]
- 44. Bortoluzzi M., Marchetti F., Pampaloni G., Pinzino C., Zacchini S., Inorg. Chem. 2016, 55, 887–893. [DOI] [PubMed] [Google Scholar]
- 45. Yao S. A., Hansen C. B., Berry J. F., Polyhedron 2013, 58, 2–6. [Google Scholar]
- 46. Neese F., Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- 47. Becke A. D., Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- 48. Perdew J. P., Phys. Rev. Β 1986, 33, 8822–8824. [DOI] [PubMed] [Google Scholar]
- 49. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 50. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 51. Neese F., Wennmohs F., Hansen A., Becker U., Chem. Phys. 2009, 356, 98–109. [Google Scholar]
- 52. Izsák R., Neese F., J. Chem. Phys. 2011, 135, 144105. [DOI] [PubMed] [Google Scholar]
- 53. Barone V., Cossi M., J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- 54. Pantazis D. A., Chen X.-Y., Landis C. R., Neese F., J. Chem. Theory Comput. 2008, 4, 908–919. [DOI] [PubMed] [Google Scholar]
- 55. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297. [DOI] [PubMed] [Google Scholar]
- 56. Schäfer A., Horn H., Ahlrichs R., J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- 57. Schäfer A., Huber C., Ahlrichs R., J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
- 58. Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary