

Abstract
Objective
Systemic sclerosis (SSc) is a heterogeneous connective tissue disease that is typically subdivided into limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc) depending on the extent of skin involvement. This subclassification may not capture the entire variability of clinical phenotypes. The European Scleroderma Trials and Research (EUSTAR) database includes data on a prospective cohort of SSc patients from 122 European referral centers. This study was undertaken to perform a cluster analysis of EUSTAR data to distinguish and characterize homogeneous phenotypes without any a priori assumptions, and to examine survival among the clusters obtained.
Methods
A total of 11,318 patients were registered in the EUSTAR database, and 6,927 were included in the study. Twenty‐four clinical and serologic variables were used for clustering.
Results
Clustering analyses provided a first delineation of 2 clusters showing moderate stability. In an exploratory attempt, we further characterized 6 homogeneous groups that differed with regard to their clinical features, autoantibody profile, and mortality. Some groups resembled usual dcSSc or lcSSc prototypes, but others exhibited unique features, such as a majority of lcSSc patients with a high rate of visceral damage and antitopoisomerase antibodies. Prognosis varied among groups and the presence of organ damage markedly impacted survival regardless of cutaneous involvement.
Conclusion
Our findings suggest that restricting subsets of SSc patients to only those based on cutaneous involvement may not capture the complete heterogeneity of the disease. Organ damage and antibody profile should be taken into consideration when individuating homogeneous groups of patients with a distinct prognosis.
INTRODUCTION
Systemic sclerosis (SSc) is a chronic disease that affects connective tissue and is characterized by vascular damage, autoimmunity, and fibrosis. The European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) have recently developed new classification criteria for SSc 1. To date, the subclassification of SSc patients mainly relies on the cutaneous involvement subsets proposed by LeRoy et al in 1988 2, 3, 4. It separates patients into 2 main groups: diffuse cutaneous SSc (dcSSc) associated with early skin changes affecting the trunk and proximal limbs, and limited cutaneous SSc (lcSSc), in which skin fibrosis is limited to the hands, face, feet, and forearms. Organ damage can vary between the 2 subsets, with an early and significant incidence of organ damage (lung fibrosis, gastrointestinal [GI] involvement, heart disease, and renal crisis) in dcSSc and pulmonary hypertension (PH) in lcSSc 4. The 2 subsets also differ in autoantibody profile, with a high prevalence (70–80%) of anticentromere antibodies (ACAs) in lcSSc, and a predominant presence of antibodies against topoisomerase I (anti–topo I) in dcSSc (30%) compared to lcSSc in the study by LeRoy et al 4. In addition, mortality is higher in patients with dcSSc than in patients with lcSSc 5, 6. Overall, previous studies suggest that lcSSc and dcSSc are 2 clearly differentiated phenotypes with regard to clinical characteristics, serologic profiles, and prognosis 7.
Yet, past and recent studies of large cohorts have challenged this distinction by highlighting an often‐neglected heterogeneity among clinical subsets 8, 9, 10, 11, 12, as suggested by, for example, lcSSc patients with anti–topo I antibodies and severe interstitial lung disease (ILD). One method of dealing with heterogeneity is to conduct a cluster analysis in order to organize data from a heterogeneous population into a fairly small number of homogeneous groups. Cluster analysis has been applied to various conditions, such as gout 13, chronic heart failure 14, asthma 15, mixed connective tissue diseases 16, and antineutrophil cytoplasmic antibody–associated vasculitis 17. Cluster analyses have also been carried out in 2 SSc studies, to our knowledge 18, 19. One of them included patients from the EULAR European Scleroderma Trials and Research (EUSTAR) cohort but was centered on capillaroscopy patterns 18. Another recent study took into account a limited number of cluster variables and a limited number of patients 19. The aim of this study was to distinguish and characterize homogeneous groups of SSc patients using cluster analysis within the large EUSTAR cohort, and analyze survival between the clusters obtained.
PATIENTS AND METHODS
Patient population
SSc patients were included in the prospective, open, multinational SSc EUSTAR cohort beginning in June 2004 20, 21, 22. For the present study, the EUSTAR database was locked in April 2014. Eligible patients were age ≥18 years, fulfilled the ACR criteria for SSc 23, and had a calculable SSc disease duration, i.e., a date of disease onset (defined as the onset of the first non–Raynaud's phenomenon symptom) and at least one date of study visit.
All patients agreed to participate in the EUSTAR cohort by signing informed consent forms approved by the local ethics committees. The study was conducted in accordance with the principles of the Declaration of Helsinki, local laws, and Guidelines for Good Clinical Practice 21, 22. See Appendix A for a list of the EUSTAR Collaborators.
Definition and selection of variables
The EUSTAR database contains data on demographic characteristics, disease features, organ damage, laboratory parameters, capillaroscopy, echocardiography, pulmonary function tests (PFTs), and medication. In order to harmonize clinical practices and ensure reliable evaluation of parameters among centers, EUSTAR arranges regular training courses and edits SSc management guidelines 24, 25.
Autoantibodies were identified and characterized according to the local center's guidelines 21, 22. Clustering variables were selected in order to ensure a global phenotype of SSc patients by considering clinical relevance and representativeness of disease features, eliminating redundant variables providing analogous information, and dismissing variables with a high rate of missing values. We retained 24 variables, including symptoms or organ involvement observed at least once among visits (Raynaud's phenomenon, esophageal, stomach, and intestinal symptoms, digital ulcers, joint synovitis, joint contractures, tendon friction rubs, muscle weakness, muscle atrophy, arterial hypertension, palpitations, and renal crisis), laboratory values (creatine kinase elevation, proteinuria, antinuclear antibody, ACA, and anti–topo I antibody positivity), results of other tests (restrictive defect on PFTs, lung fibrosis on plain radiography, conduction blocks, abnormal diastolic function, suspected PH on cardiac echography), and the peak modified Rodnan skin thickness score (MRSS) observed during follow‐up (Table 1 and Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract). Each variable included for symptoms or organ involvement, laboratory values, and results of other tests was considered positive for a specific patient if “yes” was recorded at least once for that variable at any of the visits included.
Table 1.
Characteristics of the EUSTAR patients analyzed and not analyzed and characteristics of the patients in the present study by cutaneous subseta
| EUSTAR population | Study population | |||||
|---|---|---|---|---|---|---|
| Patients analyzed (n = 6,927) | Patients not analyzed (n = 1,505) | P b | dcSSc | lcSSc | P b | |
| % of patients | – | – | – | 42 | 58 | – |
| Demographic characteristics | ||||||
| Sex, female | 86 (6,924) | 83 (1,505) | <0.001 | 80 | 91 | <0.001 |
| Ethnicity | <0.001 | <0.001 | ||||
| White | 95 (3,973) | 87 (1,176) | 92 | 97 | ||
| Asian | 3 (3,973) | 11 (1,176) | 5 | 2 | ||
| Black | 2 (3,973) | 2 (1,176) | 3 | 1 | ||
| Age, mean ± SD years (n) | 58.7 ± 13.2 (6,927) | 56.3 ± 13.9 (1,505) | <0.001 | 55.6 ± 13.0 | 60.9 ± 13.0 | <0.001 |
| Age at first non–Raynaud's phenomenon symptom, mean ± SD years (n) | 47.3 ± 13.3 (6,927) | 47.6 ± 14.1 (1,505) | 0.474 | 45.6 ± 13.2 | 48.5 ± 13.3 | <0.001 |
| Disease duration, mean ± SD years (n)c | 11.4 ± 8.1 (6,927) | 8.7 ± 8.1 (1,505) | <0.001 | 10.0 ± 7.4 | 12.4 ± 8.5 | <0.001 |
| Time from onset of Raynaud's phenomenon to first non–Raynaud's phenomenon symptom, mean ± SD years (n) | 3.9 ± 8.0 (5,868) | 3.4 ± 8.1 (1,351) | <0.001 | 2.0 ± 5.6 | 5.2 ± 9.2 | <0.001 |
| Time from first non–Raynaud's phenomenon symptom to EUSTAR enrollment, mean ± SD years (n) | 9.4 ± 7.8 (4,875) | 7.8 ± 7.8 (1,271) | <0.001 | 8.0 ± 7.3 | 10.3 ± 8.1 | <0.001 |
| Time from EUSTAR enrollment to last visit, mean ± SD years (n) | 2.6 ± 2.5 (4,875) | 0.8 ± 1.7 (1,271) | <0.001 | 2.7 ± 2.6 | 2.5 ± 2.5 | 0.031 |
| Body mass index, mean ± SD kg/m2 (n) | 23.6 ± 4.3 (2,483) | 24.4 ± 4.8 (889) | <0.001 | 22.9 ± 4.0 | 24.1 ± 4.4 | <0.001 |
| SSc characteristics | ||||||
| Autoantibody status | ||||||
| Antinuclear antibody positived | 96 (6,927) | 94 (1,412) | <0.001 | 97 | 96 | 0.400 |
| Anticentromere antibody positived | 37 (6,927) | 36 (1,264) | 0.751 | 14 | 54 | <0.001 |
| Anti–topoisomerase I antibody positived | 39 (6,927) | 36 (1,270) | 0.028 | 61 | 23 | <0.001 |
| Anti–U1 RNP antibody positive | 5 (4,054) | 7 (807) | 0.006 | 5 | 5 | 0.770 |
| Anti‐PM/Scl antibody positive | 3 (3,335) | 4 (648) | 0.278 | 5 | 2 | <0.001 |
| Anti–RNA polymerase III antibody positive | 4 (3,163) | 6 (563) | 0.025 | 6 | 3 | <0.001 |
| Cutaneous involvement | ||||||
| dcSSc | 42 (6,913) | 38 (1,437) | 0.011 | – | – | – |
| Peak MRSS value, mean ± SD (n)d | 12.0 ± 9.2 (6,927) | 10.9 ± 9.7 (1,170) | <0.001 | 18.3 ± 9.8 | 7.5 ± 5.2 | <0.001 |
| Gastrointestinal involvemente | ||||||
| Esophageal symptomsd | 81 (6,927) | 69 (1,498) | <0.001 | 84 | 79 | <0.001 |
| Stomach symptomsd | 42 (6,927) | 27 (1,491) | <0.001 | 47 | 38 | <0.001 |
| Intestinal symptomsd | 43 (6,927) | 33 (1,497) | <0.001 | 44 | 42 | 0.027 |
| Joint involvement | ||||||
| Joint contracturesd | 48 (6,927) | 35 (1,492) | <0.001 | 64 | 36 | <0.001 |
| Joint synovitisd | 26 (6,927) | 18 (1,496) | <0.001 | 32 | 22 | <0.001 |
| Tendon friction rubsd | 17 (6,927) | 8 (1,477) | <0.001 | 28 | 9 | <0.001 |
| Vascular involvement | ||||||
| Raynaud's phenomenond | 98 (6,927) | 97 (1,500) | <0.001 | 98 | 98 | 0.340 |
| History of or current digital ulcersd | 49 (6,927) | 35 (1,491) | <0.001 | 58 | 42 | <0.001 |
| Muscular involvement | ||||||
| Muscle weaknessd | 39 (6,927) | 24 (1,488) | <0.001 | 47 | 33 | <0.001 |
| Muscle atrophyd | 22 (6,927) | 12 (1,484) | <0.001 | 30 | 16 | <0.001 |
| CK elevationd | 13 (6,927) | 13 (1,231) | 0.711 | 18 | 9 | <0.001 |
| Cardiac involvement | ||||||
| Systemic arterial hypertensiond | 34 (6,927) | 27 (1,492) | <0.001 | 33 | 35 | 0.150 |
| Palpitationsd | 39 (6,927) | 26 (1,483) | <0.001 | 41 | 38 | 0.014 |
| Conduction blocksd | 22 (6,927) | 14 (1,152) | <0.001 | 24 | 20 | <0.001 |
| LVEF <50% | 5 (4,239) | 5 (879) | 0.799 | 6 | 4 | <0.001 |
| Abnormal diastolic functiond | 33 (6,927) | 22 (1,116) | <0.001 | 34 | 33 | 0.588 |
| Pericardial effusion | 11 (4,442) | 8 (920) | 0.042 | 13 | 9 | <0.001 |
| Pulmonary hypertension | ||||||
| Pulmonary hypertension on echocardiographyd | 31 (6,927) | 22 (1,173) | <0.001 | 33 | 29 | <0.001 |
| Systolic PAP measured by echocardiography, mean ± SD mm Hg (n) | 34.5 ± 15.3 (3,983) | 34.2 ± 15.1 (727) | 0.041 | 34.8 ± 16.4 | 34.2 ± 14.5 | 0.013 |
| Interstitial lung disease | ||||||
| Lung fibrosis on plain radiographyd | 49 (6,927) | 39 (1,033) | <0.001 | 63 | 39 | <0.001 |
| Lung fibrosis on HRCT | 57 (3,424) | 53 (816) | 0.023 | 68 | 48 | <0.001 |
| Restrictive defect on PFTsd | 43 (6,927) | 33 (1,083) | <0.001 | 57 | 32 | <0.001 |
| FVC, mean ± SD % predicted (n) | 89.3 ± 21.7 (4,349) | 90.0 ± 21.8 (903) | 0.437 | 81.4 ± 21.1 | 94.9 ± 20.3 | <0.001 |
| Dlco, mean ± SD % predicted (n) | 61.8 ± 20.1 (6,196) | 66.1 ± 21.1 (1,026) | <0.001 | 57.4 ± 19.9 | 64.9 ± 19.7 | <0.001 |
| 6‐minute walking distance, mean ± SD meters (n) | 392 ± 134 (1,179) | 411 ± 145 (338) | 0.007 | 394 ± 137 | 391 ± 131 | 0.872 |
| Renal involvement | ||||||
| History of renal crisisd | 3 (6,927) | 3 (1,497) | 0.626 | 5 | 2 | <0.001 |
| Proteinuriad | 12 (6,927) | 10 (1,308) | 0.082 | 15 | 9 | <0.001 |
| Blood tests | ||||||
| CRP elevation | 36 (4,736) | 31 (1,100) | <0.001 | 44 | 30 | <0.001 |
| Hypocomplementemia | 11 (4,469) | 10 (860) | 0.409 | 12 | 11 | 0.504 |
| Treatment | ||||||
| Past or current steroids | 43 (4,647) | 38 (1,081) | 0.006 | 55 | 34 | <0.001 |
| Prednisone, mean ± SD mg/day (n) | 4.4 ± 7.5 (4,644) | 5.1 ± 9.7 (1,080) | 0.081 | 6.0 ± 8.7 | 3.3 ± 6.1 | <0.001 |
| Past or current immunosuppressive drugs | 42 (4,631) | 44 (1,085) | 0.162 | 60 | 28 | <0.001 |
Except where indicated otherwise, values are the percent (number with data available). EUSTAR = European Scleroderma Trials and Research; dcSSc = diffuse cutaneous systemic sclerosis; lcSSc = limited cutaneous systemic sclerosis; MRSS = modified Rodnan skin thickness score; CK = creatine kinase; LVEF = left ventricular ejection fraction; PAP = pulmonary artery pressure; HRCT = high‐resolution computed tomography; PFTs = pulmonary function tests; FVC = forced vital capacity; DLco = diffusing capacity for carbon monoxide; CRP = C‐reactive protein.
By Student's t‐test for continuous variables and Fisher's exact test for categorical variables.
Time between the first non–Raynaud's phenomenon symptom and the last visit.
Clustering variables.
Esophageal symptoms included dysphagia and/or reflux, stomach symptoms included early satiety and/or vomiting, and intestinal symptoms included diarrhea, bloating, and/or constipation.
Statistical analysis
Cluster analysis
Cluster analysis determines the distances between individuals using the combined values of their measured features to obtain groups of individuals who have a greater resemblance to each other than to those in the other groups. Cluster analysis was carried out by ascendant hierarchical clustering of the 24 selected variables using Ward's minimum variance method. Results were graphically represented in a dendrogram. We estimated the number of clusters using the visual distance criterion of the horizontal intersection at the highest dissimilarity level on the dendrogram (i.e., where the vertical branches were the longest). In an exploratory approach, we increased the number of clusters considered in the suboptimal visual distance criterion by cutting the dendrogram horizontally at the second highest level of dissimilarity 26.
Evaluation of clusterwise stability and reproducibility is a major issue in cluster analysis 27. To assess stability and reproducibility, we conducted 100 iterations of the clustering process (with the number of clusters in the primary analysis) in randomly selected subsets of up to 50% of the original data set, and estimated the clusterwise stability by computing the Jaccard coefficient (which is a measure of similarity between data sets) between every cluster of the primary analysis and the most comparable cluster retrieved in each iteration 27. A Jaccard similarity index of ≤0.5 indicates a weakly stable and reproducible cluster 28.
The main cluster analysis was carried out in patients without missing data for the 24 selected variables. In order to estimate the impact of late complications on the cluster analysis, we performed a sensitivity analysis by selecting patients with a disease duration of >10 years (adequate time for the occurrence of organ damage). In order to study the possible impact of rare antibodies on the clustering process, we performed a second sensitivity analysis by adding in the clustering variables anti–RNA polymerase III, anti‐PM/Scl, and anti–U1 RNP antibodies. Finally, a third sensitivity analysis was conducted to evaluate the potential survival bias, and was restricted to patients with a disease duration at the enrollment visit of <5 years. The descriptive words used to refer to disease features or severity in the Results section (low/mild/moderate/severe) were not used during the clustering process but were used to describe and interpret the groups of patients in accordance with established practice 13, 14.
Survival analysis
Survival was assessed using disease duration (the time from disease onset to the most recent date data were obtained). We found that a high percentage (52%) of patients were lost to follow‐up (i.e., data last obtained prior to January 2012), which was responsible for a significant overestimation of survival. Because we could not update data with actual vital status, we chose to exclude those patients from the survival analysis. A sensitivity analysis that included those patients was therefore performed. We also performed a sensitivity analysis using onset of Raynaud's phenomenon as the definition of disease onset.
Survival rates were examined using several Cox proportional hazards models: unadjusted, adjusted for age at disease onset, adjusted for age at disease onset and sex, and adjusted for age at disease onset, sex, and immunosuppressive treatment. The proportional hazards assumption for Cox regression models was assessed by the graphical study of Schonfeld's residues, and the log linearity assumption for quantitative predictors was assessed using cubic spline functions. Finally, we calculated the C‐index for each Cox regression model (i.e., the estimation of the probability of concordance, which is equivalent to the area under the receiver operating characteristic curve for logistic regression models). Statistical analyses were carried out using the “survival” and “fastcluster” packages in R software, version 2.14 29. P values less than 0.05 were considered significant.
RESULTS
Patient characteristics
A total of 11,318 patients (from 122 centers) were registered in the EUSTAR database as of April 2014, and 34,066 visits were recorded. Of these patients, 2,886 were excluded and 1,505 were not analyzed (due to ≥1 missing value for the variables used for clustering). Therefore 6,927 patients (from 120 centers) were incorporated in the cluster analysis (Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract). Compared to patients who were not included in the analysis, patients who were included were slightly older (mean ± SD age 58.7 ± 13.2 versus 56.3 ± 13.9 years; P < 0.001), had a longer disease duration (mean ± SD 11.4 ± 8.1 versus 8.7 ± 8.1 years; P < 0.001), had a higher rate of dcSSc (42% versus 38%, P = 0.011), and had generally more severe disease as indicated by proportions of organ damage (Table 1). The median number of visits per patient was 3 (interquartile range 4).
Of the patients included, 42% had dcSSc and 58% had lcSSc. Patients with dcSSc were significantly younger than those with lcSSc, and had more severe disease. Of the patients with dcSSc, 14% had ACAs and 61% had anti–topo I antibodies, and of the patients with lcSSc, 54% had ACAs and 23% had anti–topo I antibodies (Table 1).
Primary cluster analysis
Clustering of individuals on the basis of the 24 selected variables yielded an optimal number of 2 clusters: cluster A and cluster B (Figure 1A). Jaccard indexes showed moderate stability: 0.64 for cluster A and 0.66 for cluster B. The characteristics of the 2 clusters are summarized in Table 2, Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract), and Figures 1B and 2. Contingency tables (Supplementary Tables 2 and 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract) show the proportions of patients with ACAs and anti–topo I antibodies in the different subsets of SSc according to skin involvement (lcSSc or dcSSc).
Figure 1.
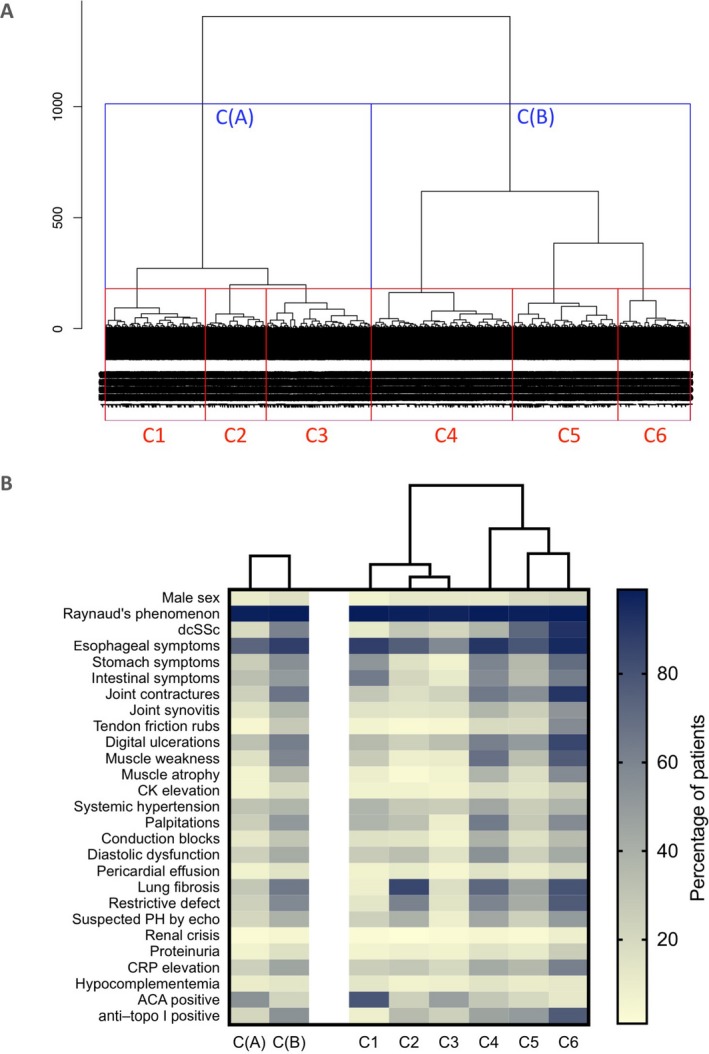
A, Dendrogram of the 6,927 patients with systemic sclerosis (SSc) included in the cluster analysis. The length of the vertical lines represents the degree of similarity between patients. Patients were divided into 2 clusters (cluster A and B) and into 6 clusters (clusters 1–6). B, Heatmap showing the clinical characteristics in each cluster. dcSSc = diffuse cutaneous SSc; CK = creatine kinase; PH = pulmonary hypertension; CRP = C‐reactive protein; ACA = anticentromere antibody; anti–topo I = anti–topoisomerase I.
Table 2.
Characteristics of the patients in the 2 and 6 clusters found in the cluster analysis (n = 6,927)a
| 2 clusters | 6 clusters | |||||||
|---|---|---|---|---|---|---|---|---|
| Cluster A | Cluster B | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | |
| Jaccard index | 0.64 | 0.66 | 0.39 | 0.32 | 0.57 | 0.38 | 0.68 | 0.43 |
| No. of patients | 3,149 | 3,778 | 1,186 | 720 | 1,243 | 1,673 | 1,249 | 856 |
| Demographic characteristics | ||||||||
| Sex, female | 90 | 84 | 94 | 88 | 88 | 88 | 81 | 79 |
| Ethnicity | ||||||||
| White | 94 | 96 | 97 | 88 | 94 | 96 | 94 | 96 |
| Asian | 5 | 2 | 2 | 10 | 4 | 2 | 3 | 2 |
| Black | 2 | 2 | 1 | 2 | 2 | 2 | 3 | 2 |
| Age, mean ± SD years | 59.2 ± 13.3 | 58.2 ± 13.2 | 61.3 ± 12.9 | 60 ± 12.8 | 56.6 ± 13.5 | 61.2 ± 12.6 | 55.8 ± 13.2 | 55.9 ± 13.2 |
| Age at first non‐Raynaud's symptom, mean ± SD years | 47.9 ± 13.3 | 46.7 ± 13.3 | 48.9 ± 13.1 | 48.3 ± 12.8 | 46.7 ± 13.6 | 48.1 ± 13.1 | 46 ± 13.4 | 45.1 ± 13.4 |
| Disease duration, mean ± SD yearsb | 11.3 ± 8.2 | 11.5 ± 8.1 | 12.4 ± 8.1 | 11.8 ± 8.3 | 9.9 ± 7.9 | 13.2 ± 8.4 | 9.8 ± 7.6 | 10.8 ± 7.5 |
| Time from onset of Raynaud's phenomenon to first non–Raynaud's phenomenon symptom, mean ± SD years | 4.8 ± 8.7 | 3.1 ± 7.3 | 5.4 ± 8.7 | 4.4 ± 9.1 | 4.4 ± 8.5 | 3.9 ± 8.2 | 2.8 ± 6.6 | 2.2 ± 6.1 |
| Time from first non–Raynaud's phenomenon symptom to EUSTAR enrollment, mean ± SD years | 9.4 ± 7.9 | 9.3 ± 7.8 | 10.3 ± 7.9 | 9.8 ± 8.2 | 8.2 ± 7.4 | 10.5 ± 8.1 | 8.1 ± 7.4 | 8.6 ± 7.4 |
| Time from EUSTAR enrollment to last visit, mean ± SD years | 2.2 ± 2.3 | 2.8 ± 2.6 | 2.5 ± 2.3 | 2.3 ± 2.5 | 1.8 ± 2.2 | 3 ± 2.7 | 2.4 ± 2.5 | 2.9 ± 2.5 |
| Body mass index, mean ± SD kg/m2 | 24.1 ± 4.3 | 23.2 ± 4.2 | 24.3 ± 4.4 | 24.5 ± 4.6 | 23.6 ± 4 | 23.6 ± 4.4 | 23.3 ± 3.9 | 22.1 ± 4.2 |
| SSc characteristics | ||||||||
| Autoantibody status | ||||||||
| Antinuclear antibody positivec | 96 | 97 | 98 | 94 | 95 | 97 | 95 | 98 |
| Anticentromere antibody positivec | 54 | 22 | 79 | 24 | 48 | 29 | 20 | 12 |
| Anti–topoisomerase I antibody positivec | 21 | 54 | 8 | 35 | 24 | 46 | 50 | 77 |
| Anti–U1 RNP antibody positive | 5 | 5 | 3 | 8 | 5 | 7 | 3 | 4 |
| Anti‐PM/Scl antibody positive | 2 | 4 | 1 | 3 | 1 | 4 | 4 | 6 |
| Anti–RNA polymerase III antibody positive | 3 | 5 | 2 | 3 | 4 | 3 | 6 | 6 |
| Cutaneous involvement | ||||||||
| dcSSc | 19 | 61 | 11 | 29 | 21 | 37 | 72 | 92 |
| Peak MRSS, mean ± SDc | 6.6 ± 4.3 | 16.5 ± 9.8 | 6.6 ± 4.2 | 7.2 ± 4.6 | 6.3 ± 4.1 | 9.2 ± 5.3 | 19 ± 6.7 | 27.2 ± 8.7 |
| Gastrointestinal involvementd | ||||||||
| Esophageal symptomsc | 73 | 88 | 88 | 76 | 58 | 91 | 79 | 95 |
| Stomach symptomsc | 26 | 55 | 52 | 16 | 7 | 60 | 36 | 70 |
| Intestinal symptomsc | 33 | 50 | 64 | 21 | 11 | 57 | 34 | 63 |
| Joint involvement | ||||||||
| Joint contracturesc | 24 | 67 | 29 | 17 | 23 | 65 | 55 | 91 |
| Joint synovitisc | 14 | 37 | 15 | 13 | 15 | 37 | 25 | 53 |
| Tendon friction rubsc | 4 | 28 | 6 | 3 | 4 | 19 | 19 | 57 |
| Vascular involvement | ||||||||
| Raynaud's phenomenonc | 98 | 99 | 99 | 98 | 97 | 99 | 98 | 99 |
| History of or current digital ulcersc | 32 | 63 | 35 | 24 | 33 | 62 | 50 | 85 |
| Muscular involvement | ||||||||
| Muscle weaknessc | 16 | 59 | 27 | 8 | 10 | 69 | 33 | 77 |
| Muscle atrophyc | 6 | 35 | 9 | 3 | 6 | 38 | 17 | 57 |
| CK elevationc | 6 | 18 | 7 | 7 | 5 | 17 | 13 | 26 |
| Cardiac involvement | ||||||||
| Systemic arterial hypertensionc | 31 | 37 | 38 | 28 | 26 | 44 | 26 | 38 |
| Palpitationsc | 25 | 51 | 38 | 32 | 9 | 64 | 28 | 57 |
| Conduction blocksc | 12 | 30 | 16 | 14 | 6 | 39 | 16 | 34 |
| LVEF <50% | 3 | 7 | 3 | 3 | 2 | 6 | 5 | 10 |
| Abnormal diastolic functionc | 24 | 42 | 27 | 33 | 15 | 54 | 24 | 43 |
| Pericardial effusion | 7 | 14 | 7 | 11 | 4 | 15 | 9 | 18 |
| Pulmonary hypertension | ||||||||
| Pulmonary hypertension on echocardiographyc | 21 | 39 | 24 | 39 | 8 | 44 | 24 | 50 |
| Systolic PAP measured by echocardiography, mean ± SD mm Hg | 32.5 ± 13.7 | 36 ± 16.2 | 33 ± 14.3 | 36.7 ± 14.1 | 29.4 ± 12 | 37.2 ± 14.6 | 32.4 ± 12 | 38.1 ± 22.1 |
| Interstitial lung disease | ||||||||
| Lung fibrosis on plain radiographyc | 29 | 65 | 8 | 85 | 17 | 72 | 46 | 80 |
| Lung fibrosis on HRCT | 38 | 70 | 22 | 78 | 29 | 73 | 56 | 82 |
| Restrictive defect on PFTsc | 24 | 58 | 13 | 61 | 14 | 60 | 42 | 77 |
| FVC, mean ± SD % predicted | 97.8 ± 19.3 | 82.7 ± 21.1 | 101.2 ± 17.4 | 86.7 ± 21.9 | 99.9 ± 17.7 | 84.4 ± 20.8 | 87.5 ± 19.8 | 72.8 ± 20.3 |
| Dlco, mean ± SD % predicted | 68 ± 18.9 | 56.6 ± 19.7 | 69.8 ± 17.2 | 57.7 ± 19.3 | 72.3 ± 18 | 55.2 ± 18.8 | 62.5 ± 20.3 | 50.6 ± 18.1 |
| 6‐minute walking distance, mean ± SD meters | 411 ± 129 | 381 ± 136 | 400 ± 135 | 405 ± 130 | 427 ± 121 | 366 ± 133 | 418 ± 130 | 362 ± 138 |
| Renal involvement | ||||||||
| History of renal crisisc | 2 | 4 | 2 | 1 | 2 | 4 | 3 | 8 |
| Proteinuriac | 7 | 16 | 6 | 8 | 7 | 15 | 11 | 26 |
| Blood tests | ||||||||
| CRP elevation | 24 | 45 | 25 | 29 | 20 | 43 | 36 | 62 |
| Hypocomplementemia | 10 | 13 | 13 | 7 | 8 | 14 | 10 | 12 |
| Treatment | ||||||||
| Past or current steroids | 27 | 55 | 22 | 45 | 24 | 57 | 44 | 65 |
| Prednisone, mean ± SD mg/day | 2.8 ± 6.4 | 5.7 ± 7.9 | 2 ± 4.9 | 5.5 ± 9.3 | 2.3 ± 5.6 | 5.6 ± 7.6 | 4.6 ± 7.6 | 7.3 ± 8.8 |
| Past or current immunosuppressive drugs | 27 | 54 | 17 | 44 | 27 | 48 | 54 | 66 |
| Mortality | ||||||||
| Number of deaths per 1,000 patient‐years | 10.3 | 22.6 | 7.5 | 17.3 | 9.7 | 19.1 | 20.8 | 31.9 |
Except where indicated otherwise, values are the percent of patients. See Table 1 for definitions.
Time between the first non–Raynaud's phenomenon symptom and the last visit.
Clustering variables.
Esophageal symptoms included dysphagia and/or reflux, stomach symptoms included early satiety and/or vomiting, and intestinal symptoms included diarrhea, bloating, and/or constipation.
Figure 2.
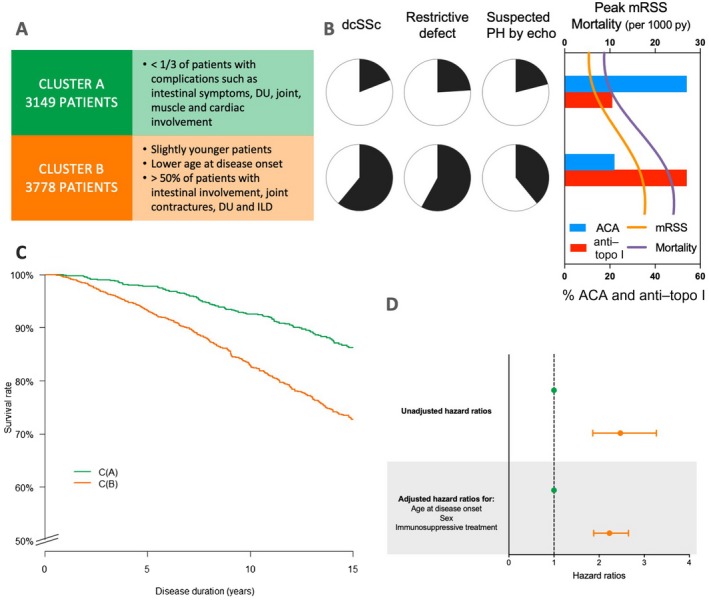
A, Main characteristics of the 2 clusters (cluster A and cluster B) of patients with systemic sclerosis (SSc). B, Left, Proportions of each cluster with the main clinical characteristics of diffuse cutaneous SSc (dcSSc), restrictive defect, and suspected pulmonary hypertension (PH) on echocardiography (echo). Right, Peak modified Rodnan skin thickness score (MRSS), mortality (per 1,000 patient‐years [py]), and percentages of patients with anticentromere antibodies (ACAs) and anti–topoisomerase I (anti–topo I) antibodies in each cluster. C, Kaplan‐Meier survival curves for the 2 clusters. D, Forest plot showing mortality hazard ratios and 95% confidence intervals for the 2 clusters. Broken line shows the hazard ratio for the reference group. Green symbols represent cluster A; orange symbols represent cluster B. DU = digital ulcer; ILD = interstitial lung disease.
Cluster A (n = 3,149; 45.5%)
Cluster A contained principally patients with lcSSc (81%). Less than a third of the patients in this cluster had severe organ damage (digital ulcers, intestinal symptoms, or muscle, joint, cardiac, or lung involvement). ACAs were present in 54% of the patients, and anti–topo I antibodies were present in 21%.
Cluster B (n = 3,778; 54.5%)
Patients in cluster B were a little younger than those in cluster A, with a younger age at disease onset. In cluster B, 61% of the patients had dcSSc. A majority of the patients presented with digital ulcers, joint contractures, intestinal involvement, and ILD. The autoantibody profile was the opposite of that seen in cluster A; 54% of the patients were positive for anti–topo I antibodies and 22% were positive for ACAs.
Exploratory cluster analysis
In an exploratory attempt to decipher the heterogeneity of the disease, we then increased the number of clusters. Graphical observation of the dendrogram determined that a suboptimal number of clusters was 6 (Figure 1A). As a consequence, we observed a decrease in Jaccard coefficients (ranging from 0.32 to 0.68). The characteristics of clusters 1–6 are summarized in Table 2, Figure 1B, and Figure 3.
Figure 3.
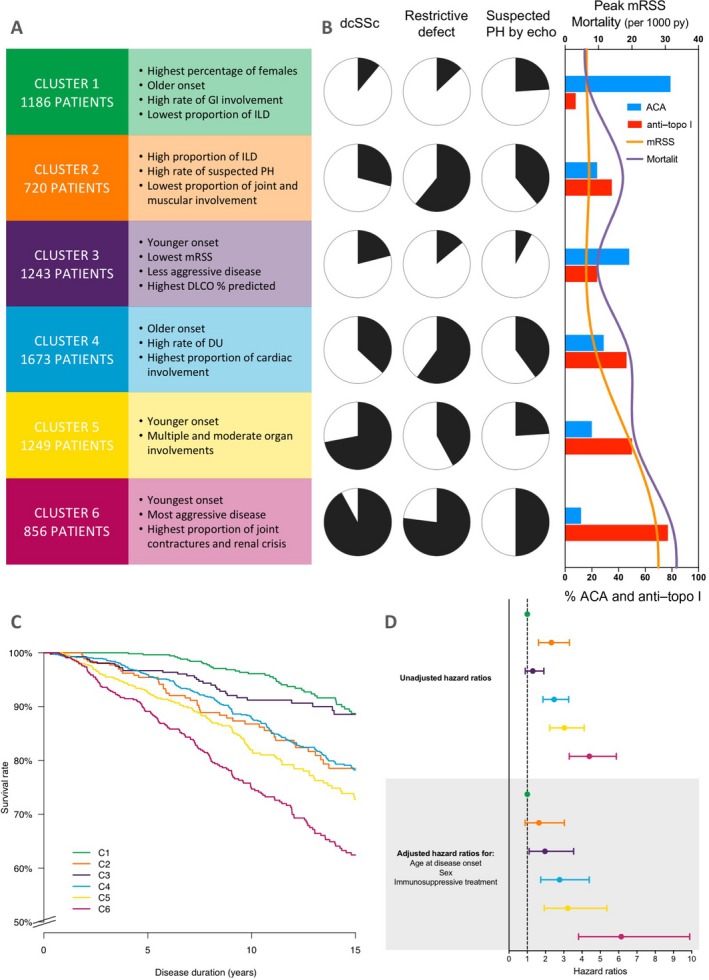
A, Main characteristics of the 6 clusters (clusters 1–6) of patients with systemic sclerosis (SSc). B, Left, Proportions of each cluster with the main clinical characteristics of diffuse cutaneous SSc (dcSSc), restrictive defect, and suspected pulmonary hypertension (PH) on echocardiography (echo). Right, Peak modified Rodnan skin thickness score (MRSS), mortality (per 1,000 patient‐years [py]), and percentages of patients with anticentromere antibodies (ACAs) and anti–topoisomerase I (anti–topo I) antibodies in each cluster. C, Kaplan‐Meier survival curves for the 6 clusters. D, Forest plot showing mortality hazard ratios and 95% confidence intervals for the 6 clusters. Broken line shows the hazard ratio for the reference group. Colors represent the different clusters as indicated in C. GI = gastrointestinal; ILD = interstitial lung disease; DL co = diffusing capacity for carbon monoxide; DU = digital ulcer.
Cluster 1 (n = 1,186; 17%)
A majority of the patients in cluster 1 (89%) had lcSSc, and most were female. They were older at disease onset, had a high prevalence of GI involvement, and had a low proportion of patients with ILD. Most of the patients in cluster 1 (79%) were ACA positive.
Cluster 2 (n = 720; 10%)
Cluster 2 was composed mainly of lcSSc patients (71%), with increased frequencies of suspected PH by echocardiography (39%), ILD (85%), and restrictive defect (61%). Anti–topo I antibodies were present in 35% of the patients, and ACAs were present in 24%.
Cluster 3 (n = 1,243; 18%)
Cluster 3 included mainly patients with lcSSc (79%) characterized by low prevalence of GI involvement and ILD. ACAs were twice as frequent as anti–topo I antibodies (48% versus 24%, respectively).
Cluster 4 (n = 1,673; 24%)
Patients in cluster 4 were mainly lcSSc patients (63%) with severe disease as demonstrated by high proportions of cardiac and lung, muscular, joint, and GI involvement and digital ulcers. Anti–topo I antibodies were present in 46% of the patients and ACAs in 29%.
Cluster 5 (n = 1,249; 18%)
Cluster 5 consisted mainly of patients with dcSSc (72%), with a notable proportion of male patients (19%), and GI, joint, and cardiac disease and moderate lung involvement. Half of the patients in cluster 5 were anti–topo I antibody positive and 20% were ACA positive.
Cluster 6 (n = 856; 12%)
Cluster 6 was characterized by the highest proportion of patients with dcSSc (92%) and men (21%), the highest mean peak MRSS (27.2), and severe disease as shown by high frequencies of GI, joint, muscular, renal, lung, and cardiac disease. Anti–topo I antibodies were present in 77% of the patients and ACAs in 12% of the patients.
Sensitivity cluster analyses
Three sensitivity cluster analyses were conducted. The first included only patients with a disease duration of >10 years (Supplementary Table 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract), the second included anti–U1 RNP, anti–RNA polymerase III, and anti‐PM/Scl antibodies as clustering variables (Supplementary Table 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract), and the third included only patients with a disease duration of <5 years at the enrollment visit (Supplementary Table 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract). Results of the sensitivity analyses were similar to those of the main cluster analysis.
Survival analyses
Kaplan‐Meier curves are shown in Figures 2 and 3 and Supplementary Figures 3 and 4 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract). Survival rates are presented in Supplementary Table 7 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract), and the results of Cox regression analyses are shown in Table 3.
Table 3.
Cox regression analysesa
| Univariable analysis (n = 3,352) | Multivariable analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Adjusted for age at disease onset(n = 3,352) | Adjusted for age at disease onset and sex (n = 3,352) | Adjusted for age at disease onset, sex, and immunosuppressive treatment (n = 2,887) | ||||||
| HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | |
| Cutaneous involvement | ||||||||
| lcSSc | Reference | Reference | Reference | Reference | ||||
| dcSSc | 1.90 (1.64–2.19) | <0.001 | 2.39 (2.07–2.77) | <0.001 | 2.14 (1.85–2.48) | <0.001 | 2.03 (1.61–2.56) | <0.001 |
| C‐indexb | 0.60 ± 0.01 | 0.73 ± 0.01 | 0.75 ± 0.01 | 0.78 ± 0.02 | ||||
| 2 clusters | ||||||||
| Cluster A | Reference | Reference | Reference | Reference | ||||
| Cluster B | 2.23 (1.88–2.65) | <0.001 | 2.40 (2.02–2.85) | <0.001 | 2.26 (1.91–2.69) | <0.001 | 2.47 (1.86–3.27) | <0.001 |
| C‐indexb | 0.59 ± 0.01 | 0.72 ± 0.01 | 0.74 ± 0.01 | 0.78 ± 0.02 | ||||
| 6 clusters | ||||||||
| Cluster 1 | Reference | Reference | Reference | Reference | ||||
| Cluster 2 | 2.32 (1.62–3.31) | <0.001 | 2.10 (1.46–3.00) | <0.001 | 1.97 (1.38–2.82) | <0.001 | 1.64 (0.88–3.03) | 0.119 |
| Cluster 3 | 1.30 (0.89–1.91) | 0.172 | 1.63 (1.11–2.38) | 0.012 | 1.62 (1.11–2.37) | 0.013 | 1.97 (1.10–3.54) | 0.023 |
| Cluster 4 | 2.47 (1.86–3.27) | <0.001 | 2.49 (1.88–3.30) | <0.001 | 2.40 (1.81–3.19) | <0.001 | 2.77 (1.74–4.39) | <0.001 |
| Cluster 5 | 3.03 (2.23–4.11) | <0.001 | 3.77 (2.77–5.12) | <0.001 | 3.37 (2.47–4.58) | <0.001 | 3.22 (1.93–5.36) | <0.001 |
| Cluster 6 | 4.40 (3.30–5.87) | <0.001 | 5.85 (4.38–7.81) | <0.001 | 5.20 (3.89–6.95) | <0.001 | 6.14 (3.81–9.89) | <0.001 |
| C‐indexb | 0.63 ± 0.01 | 0.75 ± 0.01 | 0.76 ± 0.01 | 0.79 ± 0.02 | ||||
Disease onset was defined as the first non–Raynaud's phenomenon symptom (see Supplementary Table 8, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract, for sensitivity analysis using the onset of Raynaud's phenomenon as the definition of disease onset). HR = hazard ratio; 95% CI = 95% confidence interval; lcSSc = limited cutaneous systemic sclerosis; dcSSc = diffuse cutaneous systemic sclerosis.
The C‐index was calculated for each Cox regression model, and corresponds to the estimation of the probability of concordance, equivalent to the area under the receiver operating characteristic curve for logistic regression models. A value of 1 indicates perfect agreement and 0.5 indicates an agreement that is no better than chance. Values for the C‐index are the mean ± SEM.
The risk of death was increased for patients with dcSSc compared to patients with lcSSc, with a hazard ratio (HR) of 2.03 (95% confidence interval [95% CI] 1.61–2.56) in the most‐adjusted model. An increased risk of death was also present in cluster B compared to cluster A (HR 2.47 [95% CI 1.86–3.27]). When analyzing 6 clusters, we noticed a continuous increasing mortality from cluster 1 to cluster 6 in the most‐adjusted model. The risk of death had a magnitude superior to those noted in the 2 previous analyses (i.e., HR 6.14 [95% CI 3.81–9.89] for cluster 6 compared to cluster 1). C‐indexes were similar for the most‐adjusted models: lcSSc versus dcSSc, cluster A versus cluster B, and for the 6 clusters (mean ± SEM 0.78 ± 0.02, 0.78 ± 0.02, and 0.79 ± 0.02, respectively).
The sensitivity analysis taking into account patients who were lost to follow‐up yielded comparable HRs when we examined survival in clusters A and B and clusters 1–6 (data not shown). We also performed a sensitivity analysis using the onset of Raynaud's phenomenon as the date of disease onset (Supplementary Table 8, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40906/abstract), which yielded similar results, albeit the number of patients with available data was lower.
DISCUSSION
This study aimed to distinguish homogeneous groups in a substantial population of ~7,000 SSc patients using a cluster analysis. The study had 2 main findings. First, the optimal clustering divided patients into 2 distinct groups according to their clinical and serologic features and disease severity and prognosis; these 2 categories partially overlapped with the classifications dcSSc and lcSSc. Second, an exploratory analysis yielded 6 homogeneous subsets of individuals that broadly differed with regard to clinical features, autoantibody profiles, and survival.
The fact that 2 clusters were found could be considered a validation of the expected dichotomy between dcSSc and lcSSc. However, 19% of the patients in cluster A had dcSSc and 21% had anti–topo I antibodies. In cluster B, 39% of the patients had lcSSc and 22% had ACAs. No clear parallels between the severity of organ damage and the cutaneous extent of SSc were observed. This finding is consistent with the results of recent studies. For example, Nihtyanova et al demonstrated that the presence of significant organ involvement was a strong predictor of prognosis, in both lcSSc and dcSSc, in a study of nearly 400 consecutive patients followed up for up to 15 years. Notably, survival curves were close for the 2 cutaneous subsets when organ damage was present 30. Taken together, these results suggest that, while there is consensus on the relevance and practicality of subdividing SSc into lcSSc and dcSSc 31, this binary classification may be too restrictive as a separation within a continuous spectrum of varying severity primarily driven by organ damage and subsequent prognosis 12.
In an exploratory attempt to study the heterogeneity of SSc more in depth, we found 6 additional clusters. Some of the 6 clusters obtained were expected, since they were consistent with the historical descriptions of lcSSc and dcSSc. Indeed, cluster 1 included patients with the classic presentation of lcSSc, i.e., older female patients with a low rate of severe organ damage, a high frequency of ACA positivity, and a generally favorable prognosis. Cluster 6 resembled the classic description of dcSSc, with a high rate of male patients, the highest frequency of anti–topo I antibody–positive patients, and a high rate of severe organ damage and poor prognosis. Intriguingly, we observed clusters of patients that seemed to be grouped together based on characteristics other than the degree of skin involvement. Cluster 2 was composed principally of patients with lcSSc but with a rather high frequency of anti–topo I antibody–positivity and high rates of ILD and suspected PH. Of note, the prognosis for patients in cluster 2 was significantly worse than that for patients in cluster 1. Similarly, cluster 4 consisted of predominantly patients with lcSSc, often with visceral complication. Cluster 5 comprised, for the most part, patients with dcSSc, but we noted lower frequencies of ILD and suspected PH in this group than in clusters 2, 4, or 6. These findings indicate that subclassifications established solely on the extent of skin involvement might not be entirely representative of the severity of organ damage and prognosis.
Furthermore, this work highlighted some groups of patients in which the classic relationships between lcSSc and ACAs and between dcSSc and anti–topo I antibodies were not obvious. For example, in cluster 2, 71% of the patients were classified as having lcSSc, although 85% had lung fibrosis. Moreover, we found a relatively small proportion of ACA‐positive patients (24%) and a notable rate of anti–topo I antibody positivity (35%), which was unexpected in a group in which the majority of the patients had lcSSc. The prognosis for the patients in this group was worse than that for the patients in cluster 1, which included mainly patients with lcSSc and few with organ damage, which supports the findings of Nihtyanova et al 30. Likewise, a Canadian Scleroderma Research Group study examined the clinical features and mortality of anti–topo I antibody–positive lcSSc and ACA‐positive dcSSc patients. The autoantibody profile seemed to be more strongly associated with demographic characteristics and visceral damage than with the skin subgroup. Mortality was related to both skin and serologic profile 9. Kranenburg et al also demonstrated that lcSSc patients who were positive for anti–topo I antibodies contrasted with lcSSc patients who were negative for anti–topo I antibodies and dcSSc patients who were positive for anti–topo I antibodies in terms of survival and organ involvement 32. Taken together, those studies suggest that subclassification combining antibody profile and skin involvement might predict clinical outcomes more accurately than skin or serologic features alone 9, 32.
The heterogeneity of SSc has been discussed over a long period, and many studies were published both before and after the work of LeRoy et al describing the limited and diffuse subsets 2, 3, 4, 33, 34. The significance of serologic profile has also been highlighted by Patterson et al, who characterized 5 groups of patients with homogeneous clinical and organ involvement 11, 12. Significant efforts to classify patients into subsets on the basis of common clinical phenotypes, rather than through a predetermined decision process, have proposed to classify individuals using changes in MRSS over time 34, 35, changes in the forced vital capacity percent predicted value 36, 37, or gene expression patterns in the skin 38, 39. Each of these attempts has resulted in a small number of subsets that define the range of phenotypes captured by the stratification characteristics 12. There is growing interest in a new subclassification of SSc that combines patterns of underlying pathogenesis, organ damage, and prognosis in order to personalize disease management and ameliorate outcomes 12, 31.
This study has strengths and limitations. The principal strengths are the number of patients included in this large, prospective, multicenter cohort, and the lack of any a priori assumptions. The main weakness is that several clinically relevant variables were lacking or were disregarded due to the proportion of missing data being too high (e.g., autoantibodies other than ACAs/anti–topo I antibodies, extent of ILD on high‐resolution computed tomography [HRCT] scan, detailed skin involvement, and overlap syndromes). In addition, 1,505 of 8,432 patients were excluded from the cluster analysis because of missing data for any of the selected clustering variables. Since those excluded patients had slightly less severe disease than the included ones, it could affect the extrapolation of our results. Imputation of missing data by model‐based clustering was not performed because we could not assume that these data were missing at random 40, 41. Moreover, several definitions of variables lacked precision (e.g., ILD was defined as lung fibrosis on radiography whereas HRCT scan is now widely used, and PH was defined as suspicion on echocardiography without invasive confirmation).
We also acknowledge that a thorough analysis of treatment regimens was not possible due to missing data. Nevertheless, for a majority of the patients we were able to determine whether or not they had been taking an immunosuppressive drug. To account for the potential effect of these drugs on survival, survival analyses were adjusted for immunosuppressive treatment. A potentially important bias is the influence of disease duration on the clustering process, since the frequency of organ damage tends to increase as the disorder progresses. Also, disease duration at the enrollment visit was relatively long, raising the possibility that study results were influenced by survival bias. Yet, the sensitivity analyses that included only patients with a long disease duration and those that included only patients with a short disease duration yielded similar results.
Another limitation is that a significant number of patients were excluded from the survival analysis because of loss to follow‐up. Nevertheless, this exclusion did not alter the survival differences between clusters in a sensitivity analysis. The primary aim of our study was not to assess the prognosis factors for survival in SSc, but to decipher the heterogeneity of SSc by a cluster analysis and describe the survival rate in the clusters obtained, allowing us to validate this approach post hoc. In studies assessing the prognosis factors of survival, baseline data are most often used. In our study, we had to include follow‐up data in order to identify the occurrence of organ involvement. Therefore, we considered an organ complication to be present if the corresponding variable was described as “positive” at least once among all the visits included for a specific patient. We did not describe the progression of organ involvement in the whole population or in the different clusters because the limited number of follow‐up visits precluded us from performing a precise temporal description. In the end, the weak reproducibility of the exploratory analysis with 6 clusters precludes translating these results to a new subclassification (e.g., to allocate an individual to a designated group on the basis of their features). Moreover, previous studies have shown differences between distinct geographical cohorts 42. Of note, 95% of the patients included in this study were white. It is likely that inclusion of a higher proportion of Asian or black patients could have modified the results.
In conclusion, this study shows that SSc is a very heterogeneous condition. While there is consensus regarding the relevance and practicality of the subclassification of SSc into lcSSc and dcSSc, this binary system might omit a wider spectrum of clinical phenotypes characterized not only by skin involvement but also by organ damage, serologic profile, and subsequent prognosis. There is an increasing demand for a future SSc classification that combines these different patterns, in order to personalize approaches to diagnosis and clinical management.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Sobanski had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Sobanski, Giovannelli, Allanore, Launay, Hachulla.
Acquisition of data
Sobanski, Giovannelli, Allanore, Riemekasten, Airò, Vettori, Cozzi, Distler, Matucci‐Cerinic, Denton, Launay, Hachulla.
Analysis and interpretation of data
Sobanski, Giovannelli, Allanore, Distler, Matucci‐Cerinic, Denton, Launay, Hachulla.
Supporting information
APPENDIX A: THE EUSTAR COLLABORATORS
EUSTAR Collaborators (numerical order of centres): (1) Marco Matucci Cerinic, Serena Guiducci, Department of Medicine, Section of Rheumatology, University of Florence, Italy; (2) Ulrich Walker, Diego Kyburz, Department of Rheumatology, University Hospital Basel, Switzerland; (4) Giovanni Lapadula, Florenzo Iannone, Rheumatology Unit‐DiMIMP, School of Medicine University of Bari, Italy; (6) Oliver Distler, Britta Maurer, Suzana Jordan, Department of Rheumatology, University Hospital Zürich, Switzerland; (7) Radim Becvar, Institute of Rheumatology, 1st Medical School, Charles University, Prague, Czech Republic; (8) Stanislaw Sierakowsky, Otylia Kowal Bielecka, Department of Rheumatology and Internal Medicine, Medical University of Bialystok, Bialystok, Poland; (11) Maurizio Cutolo, Alberto Sulli, Research Laboratory and Division of Rheumatology, Department of Internal Medicine, University of Genova, Italy; (13) Gabriele Valentini, Giovanna Cuomo, Serena Vettori, Department of Clinical and Experimental Medicine ‘F‐Magrassi’ II Policlinico, Unit of Rheumatology, Naples, Italy; (15) Elise Siegert, Department of Rheumatology, Charité University Hospital, Berlin, German Rheumatism Research Centre Berlin (DRFZ), a Leibniz Institute, Germany; (16) Simona Rednic, Ileana Nicoara, Department of Rheumatology, University of Medicine and Pharmacy ‘Iuliu Hatieganu’ Cluj, Cluj‐Napoca, Romania; (17) André Kahan, Yannick Allanore, Department of Rheumatology, University Paris Descartes and Cochin Hospital, Paris, France; (18) Panayiotis Vlachoyiannopoulos, Department of Pathophysiology, Medical School, National University of Athens, Greece; (19) Carlo Montecucco, Roberto Caporali, Unita’ Operativa e Cattedra di Reumatologia, IRCCS Policlinico S Matteo, Pavia, Italy; (20) Jiri Stork, Department of Dermatology the 1st faculty of Medicine, Charles University, Prague, Czech Republic; (21) Murat Inanc, Istanbul Medical Faculty, Department of Internal Medicine, Division of Rheumatology, Istanbul, Turkey; (23) Patricia E. Carreira, Division of Rheumatology, Hospital 12 de Octubre, Madrid, Spain; (24) Srdan Novak, Department of Rheumatology and Clinical Immunology, Internal Medicine, KBC Rijeka, Croatia; (25) László Czirják, Cecilia Varju, Department of Immunology and Rheumatology, Faculty of Medicine, University of Pécs, Hungary; (28) Carlo Chizzolini, Department of Immunology and Allergy, University Hospital, Geneva, Switzerland; (29) Eugene J. Kucharz, Anna Kotulska, Magdalena Kopec‐Medrek, Malgorzata Widuchowska, Department of Internal Medicine and Rheumatology, Medical University of Silesia, Katowice, Poland; (31) Franco Cozzi, Rheumatology Unit, Department of Clinical and Experimental Medicine, University of Padova, Italy; (32) Blaz Rozman, University Medical Center Ljublijana, Division of Internal Medicine, Department of Rheumatology, Ljubliana, Slovenia; (33) Carmel Mallia, Bernard Coleiro, ‘Stella Maris’, Balzan, Malta; (34) Armando Gabrielli, Dipartimento di Scienze Cliniche e Molecolari, Clinica Medica, Università Politecnica delle Marche, Ancona, Italy; (35) Dominique Farge, Chen Wu, Zora Marjanovic, Helene Faivre, Darin Hij, Roza Dhamadi, Department of Internal Medicine, Hospital Saint‐Louis, Paris, France; (38) Paolo Airò, Spedali Civili di Brescia, Servizio di Reumatologia Allergologia e Immunologia Clinica, Brescia, Italy; (40) Roger Hesselstrand, Frank Wollheim, Dirk M Wuttge, Kristofer Andréasson, Department of Rheumatology, Lund University, Lund, Sweden; (41) Duska Martinovic, Department of Internal Medicine, Clinical Hospital of Split, Croatia; (42) Alexandra Balbir‐Gurman, Yolanda Braun‐Moscovici, B. Shine Rheumatology Unit, Rambam Health Care Campus, Rappaport Faculty of Medicine, Technion, Haifa, Israel; (43) Francesco Trotta, Andrea Lo Monaco, Department of Clinical and Experimental Medicine, Rheumatology Unit, University of Ferrara, Italy; (44) Nicolas Hunzelmann, Department of Dermatology, University Hospital Cologne, Germany; (49) Raffaele Pellerito, Ospedale Mauriziano, Centro di Reumatologia, Torino, Italy; (50) Lisa Maria Bambara, Paola Caramaschi, Unità di Reumatologia, AOUI, Verona Italy; (51) Jadranka Morovic‐Vergles, Division of Clinical Immunology and Rheumatology Department of Internal Medicine, Dubrava University Hospital, Zagreb, Croatia; (52) Carol Black, Christopher Denton, Centre for Rheumatology, Royal Free and University College London Medical School, Royal Free Campus, United Kingdom; (55) Nemanja Damjanov, Institute of Rheumatology, Belgrade, Serbia and Montenegro; (56) Jörg Henes, Medizinische Universitätsklinik, Abt. II (Onkologie, Hämatologie, Rheumatologie, Immunologie, Pulmonologie), Tübingen, Germany; (57) Vera Ortiz Santamaria, Rheumatology Granollers General Hospital, Barcelona, Spain; (58) Stefan Heitmann, Department of Rheumatology, Marienhospital Stuttgart, Germany; (59) Dorota Krasowska, Department of Dermatology, Medical University of Lublin, Poland; (60) Matthias Seidel, Medizinische Universitäts‐Poliklinik, Department of Rheumatology, Bonn, Germany; (61) Paul Hasler, Rheumaklinik und Institut für Physikalische Medizin und Rehabilitation, Kantonsspital Aarau, Switzerland; (63) Harald Burkhardt, Andrea Himsel, Klinikum der Johann Wolfgang Goethe Universität, Medizinische Klinik III, Rheumatologische Ambulanz, Frankfurt am Main, Germany; (66) Gianluigi Bajocchi, Arcispedale Santa Maria Nuova, Dipartimento Area Medica I, U.O. di Reumatologia, Padiglione Spallanzani, Reggio Emilia, Italy; (68) Maria João Salvador, José Antonio Pereira Da Silva, Rheumatology Department, Hospitais da Universidade, Coimbra, Portugal; (73) Bojana Stamenkovic, Aleksandra Stankovic, Institute for Prevention, Treatment and Rehabilitation of Rheumatic and Cardiovascular Diseases, Niska Banja, Serbia and Montenegro; (74) Carlo Francesco Selmi, Maria De Santis, Bianca Marasini, Division of Rheumatology and Clinical Immunology Humanitas Clinical and Research Center, BIOMETRA Department, University of Milan, Italy; (77) Mohammed Tikly, Rheumatology Unit, Department of Medicine Chris Hani Haragwanath, Hospital and University of the Witwatersrand, Johannesburg, South Africa; (78) Lidia P. Ananieva, Lev N. Denisov, Institute of Rheumatology, Russian Academy of Medical Science, Moscow, Russia; (81) Ulf Müller‐Ladner, Marc Frerix, Ingo Tarner, Justus‐Liebig University Giessen, Department of Rheumatology and Clinical Immunology, Kerckhoff‐Klinik Bad Nauheim, Germany; (83) Raffaella Scorza, U.O. Immunologia Clinica, Centro di Riferimento per le Malattie Autoimmuni Sistemiche, Milano, Italy; (85) Francesco Puppo, Clinica di Medicina Interna ad orientamento immunologico‐Università di Genova, IRCCS Azienda Ospedaliero‐Universitaria, Università San Martino, Genova, Italy; (86) Merete Engelhart, Gitte Strauss, Henrik Nielsen, Kirsten Damgaard, Department of Rheumatology, University Hospital of Gentofte, Hellerup, Denmark; (87) Gabriella Szücs, Szilvia Szamosi, Third Department of Medicine, Rheumatology Division, University of Debrecen, Hungary; (91) Antonio Zea Mendoza, Carlos de la Puente, Walter Alberto Sifuentes Giraldo, Servicio de Reumatología, Hospital Ramon Y Cajal, Madrid, Spain; (92) Øyvind Midtvedt, Silje Reiseter, Torhild Garen, Department of Rheumatology, Rikshospitalet University Hospital, Oslo, Norway; (93) Eric Hachulla, David Launay, Department of Internal Medicine, Hôpital Claude Huriez, Lille, France; (94) Guido Valesini, Valeria Riccieri, Department of Internal Medicine and Medical Specialities, ‘Sapienza’ University of Rome, Italy; (96) Ruxandra Maria Ionescu, Daniela Opris, Laura Groseanu, Department of Rheumatology, St. Mary Hospital, Carol Davila, University of Medicine and Pharmacy, Bucharest, Romania; (99) Fredrick M. Wigley, Johns Hopkins University Division of Rheumatology, Johns Hopkins School of Medicine, Baltimore, USA; (100) Roxana Sfrent Cornateanu, Razvan Ionitescu, Ana Maria Gherghe, Alina Soare, Marilena Gorga, Mihai Bojinca, Carina Mihai, Mihaela Milicescu, Department of Internal Medicine and Rheumatology, Cantacuzino Hospital, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania; (102) Cord Sunderkötter, Annegret Kuhn, Department of Dermatology, University of Münster, Germany; (104) Nora Sandorfi, Thomas Jefferson University, Philadelphia, PA, USA; (106) Georg Schett, Jörg HW Distler, Christian Beyer, Department of Internal Medicine 3, University Hospital Erlangen, Germany; (110) Pierluigi Meroni, Francesca Ingegnoli, Division of Rheumatology, Istituto Gaetano Pini, Department of Clinical Sciences and Community Health, University of Milano, Milano, Italy; (112) Luc Mouthon, Department of Internal Medicine, Hôpital Cochin, Paris, France; (113) Filip De Keyser, Vanessa Smith, University of Ghent, Department of Rheumatology, Gent, Belgium; (115) Francesco Paolo Cantatore, Ada Corrado, U.O. Reumatologia‐Università degli Studi di Foggia, Ospedale ‘Col. D'Avanzo’, Foggia, Italy; (116) Susanne Ullman, Line Iversen, University Hospital of Copenhagen, Department of Dermatology D‐40, HS‐Bispebjerg Hospital, Copenhagen, Denmark; (117) Carlos Alberto von Mühlen, Jussara Marilu Bohn, Lilian Scussel Lonzetti, Rheuma Clinic, Porto Alegre, Brazil; (118) Maria Rosa Pozzi, Dipartimento di Medicina, Ospedale San Gerardo, Monza, Italy; (119) Kilian Eyerich, Rüdiger Hein, Elisabeth Knott, Department of Dermatology and Allergy of the TU Munich, Germany; (120) Piotr Wiland, Magdalena Szmyrka‐Kaczmarek, Renata Sokolik, Ewa Morgiel, Marta Madej, Department of Rheumatology and Internal Diseases, Wroclaw University of Medicine, Wroclaw, Poland; (122) Frédéric A. Houssiau, Université Catholique de Louvain, Bruxelles, Belgium; (123) Juan Jose Alegre‐Sancho, Hospital Universitario Dr Peset, Valencia, Spain; (124) Brigitte Krummel‐Lorenz, Petra Saar, Endokrinologikum Frankfurt, Germany; (125) Martin Aringer, Claudia Günther, Division of Rheumatology, Department of Medicine III/Department of Dermatology, University Medical Center Carl Gustav Carus, Technical University of Dresden, Germany; (126) Rene Westhovens, Ellen de Langhe, Jan Lenaerts, Catholic University of Leuven, Department of Rheumatology, Leuven, Belgium; (128) Branimir Anic, Marko Baresic, Miroslav Mayer, University Hospital Centre Zagreb, Division of Clinical Immunology and Rheumatology, Department of Medicine, Zagreb, Croatia; (130) Maria Üprus, Kati Otsa, East‐Tallin Central Hospital, Department of Rheumatology, Tallin, Estonia; (133) Sule Yavuz, University of Marmara, Dept. of Rheumatology, Istanbul, Turkey; (134) Brigitte Granel, Service de Médecine Interne, Hôpital Nord de Marseille, Marseille, France; (135) Sebastião Cezar Radominski, Carolina de Souza Müller, Valderílio Feijó Azevedo, Hospital de Clinicas da Universidade Federal do Parana, Curitiba – Parana, Brazil; (136) Sergio Jimenez, Joanna Busquets, Thomas Jefferson Scleroderma Center, Division of Rheumatology and Jefferson Institute of Molecular Medicine, Philadelphia PA, USA; (137) Svetlana Agachi, Liliana Groppa, Lealea Chiaburu, Eugen Russu, Sergei Popa, Municipal Centres of Research in Scleroderma, Hospital ‘Sacred Trinity’, Department of Rheumatology/Department of Rheumatology, Republican Clinical Hospital, Chisinau, Republic of Moldova; (138) Thierry Zenone, Department of Medicine, Unit of Internal Medicine, Valence, France; (140) Margarita Pileckyte, Kaunas University of Medicine Hospital, Department of Rheumatology, Lithuania; (141) Simon Stebbings, John Highton, Dunedin School of Medicine, Dunedin, New Zealand; (142) Alessandro Mathieu, Alessandra Vacca, II Chair of Rheumatology, University of Cagliari‐Policlinico Universitario, Monserrato (CA), Italy; (145) Percival D. Sampaio‐Barros, Natalino H. Yoshinari, Roberta G. Marangoni, Patrícia Martin, Luiza Fuocco, University of São Paulo, Rheumatology Division, Faculdade de Medicina de Universidade de São Paulo, São Paulo SP, Brasil; (147) Lisa Stamp, Peter Chapman, John O'Donnell, Department of Medicine, University of Otago, Christchurch, New Zealand; (148) Kamal Solanki, Alan Doube, Waikato University Hospital, Rheumatology Unit, Hamilton City, New Zealand; (149) Douglas Veale, Marie O'Rourke, Department of Rheumatology, Bone and Joint Unit, St. Vincent's University Hospital, Dublin, Ireland; (152) Esthela Loyo, Reumatologia e Inmunologia Clinica, Hospital Regional Universitario Jose Ma Cabral y Baez, Clinica Corominas, Santiago, Dominican Republic; (154) Mengtao Li, Department of Rheumatology, Peking Union Medical College Hospital (West Campus), Chinese Academy of Medical Sciences, Beijing, China; (155) Walid Ahmed Abdel Atty Mohamed, Alexandria University, Unit of Rheumatology, Alexandria, Egypt; (158) Edoardo Rosato, Antonio Amoroso, Antonietta Gigante, Centro per la Sclerosi Sistemica ‐ Dipartimento di Medicina Clinica, Università La Sapienza, Policlinico Umberto I, Roma, Italy; (159) Fahrettin Oksel, Figen Yargucu, Ege University, Faculty of Medicine, Dept. of Internal Medicine, Division of Rheumatology, Izmir, Turkey; (160) Cristina‐Mihaela Tanaseanu, Monica Popescu, Alina Dumitrascu, Isabela Tiglea, Clinical Emergency Hospital St. Pantelimon, Bucharest, Romania; (161) Rosario Foti, U.O. di Reumatologia, A.O.U. Policlinico Vittorio Emanuele, Catania, Italy; (162) Rodica Chirieac, Codrina Ancuta, Division of Rheumatology and Rehabilitation GR.T.Popa, Center for Biomedical Research, European Center for Translational Research, “GR.T.Popa” University of Medicine and Pharmacy, Rehabilitation Hospital, Iasi, Romania; (163) Daniel E. Furst, Division of Rheumatology, Department of Medicine University of California at Los Angeles, Los Angeles, CA, USA; (164) Peter Villiger, Sabine Adler, Department of Rheumatology and Clinical Immunology/Allergology, Inselspital, University of Bern, Switzerland; (165) Jacob van Laar, James Cook University, Hospital Marton Road, Middlesbrough, United Kingdom; (167) Cristiane Kayser, Andrade Luis Eduardo C., Universidade Federal de São Paulo, Disciplina de Reumatologia, São Paulo, SP, Brasil; (168) Nihal Fathi, Manal Hassanien, Assiut and Sohage University Hospital, Rheumatology Department, Assiut, Egypt; (169) Paloma García de la Peña Lefebvre, Silvia Rodriguez Rubio, Marta Valero Exposito, Hospital Universitario Madrid Norte Sanchinarro, Madrid, Spain; (172) Jean Sibilia, Emmanuel Chatelus, Jacques Eric Gottenberg, Hélène Chifflot, University Hospital of Strasbourg, Department of Rheumatology, Hôpital de Hautepierre, Service de Rhumatologie, Strasbourg Cedex, France; (173) Ira Litinsky, Department of Rheumatology, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; (175) Paul Emery, Maya Buch, Francesco Del Galdo, Scleroderma Programme, Institute of Molecular Medicine, Division of Musculoskeletal Diseases, University of Leeds, Leeds, United Kingdom; (176) Algirdas Venalis, Irena Butrimiene, Paulius Venalis, Rita Rugiene, Diana Karpec, State Research Institute for Innovative Medicine, Vilnius University, Vilnius, Lithuania; (177) Lesley Ann Saketkoo, Joseph A. Lasky, Tulane University Lung Center, Tulane/University Medical Center Scleroderma and Sarcoidosis Patient Care and Research Center, New Orleans, USA; (178) Eduardo Kerzberg, Fabiana Montoya, Vanesa Cosentino, Osteoarticular Diseases and Osteoporosis Centre, Pharmacology and Clinical Pharmacological Research Centre, School of medicine, University of Buenos Aires, Rheumatology and Collagenopathies Department, Ramos Mejía Hospital, Buenos Aires, Argentina; (182) Massimiliano Limonta, Antonio Luca Brucato, Elide Lupi, USSD Reumatologia, Ospetali Riuniti di Bergamo, Italy; (183) Itzhak Rosner, Michael Rozenbaum, Gleb Slobodin, Nina Boulman, Doron Rimar, Rheumatology Unit Bnai Zion Medical Center, Haifa, Israel; (184) Maura Couto, Unidade de Reumatologia de Viseu, Centro Hospitalar Tondela‐Viseu (Unidade de Reumatologia), Viseu, Portugal; (185) François Spertini, Camillo Ribi, Guillaume Buss, Department of Rheumatology, Clinical Immunology and Allergy, Lausanne, Switzerland; (187) Sarah Kahl, Universitätsklinikum Schleswig‐Holstein, Campus Lübeck, Innere Medizin/Rheumatologie/Immunologie, Rheumaklinik Bad Bramstedt, Bad Bramstedt, Germany; (188) Vivien M. Hsu, Fei Chen, Deborah McCloskey, Halina Malveaux, UMDNJ ‐ Scleroderma Program, Clinical Research Center ‐ Robert Wood Johnson Medical School, Acute Care Building, New Brunswick NJ, USA; (189) Jean Louis Pasquali, Thierry Martin, Audrey Gorse, Aurélien Guffroy, Vincent Poindron, Clinical Immunology and Internal Medicine, National Referral Center for Systemic Autoimmune Diseases, Strasbourg, France.
1Vincent Sobanski, MD, PhD, David Launay, MD, PhD, Eric Hachulla, MD, PhD: Univ. Lille, INSERM U995 LIRIC, CHU Lille, and Referral Center for Rare Systemic Autoimmune Diseases North and North‐West of France, Lille, France; 2Jonathan Giovannelli, MD, PhD: Univ. Lille, INSERM U995 LIRIC, and CHU Lille, Lille, France; 3Yannick Allanore, MD, PhD: Hôpital Cochin, AP‐HP, and Université Paris Descartes, Paris, France; 4Gabriela Riemekasten, MD, PhD: University Clinic Schleswig‐Holstein, University of Lübeck, Lübeck, Germany; 5Paolo Airò, MD, ASST: Spedali Civili di Brescia, Brescia, Italy; 6Serena Vettori, MD, PhD: Second University of Naples, Naples, Italy; 7Franco Cozzi, MD, PhD: University of Padua, Padua, Italy; 8Oliver Distler, MD, PhD: University Hospital Zurich, Zurich, Switzerland; 9Marco Matucci‐Cerinic, MD, PhD: Azienda Ospedaliero‐Universitaria Careggi, University of Florence, Florence, Italy; 10Christopher Denton, MD, PhD: Royal Free Hospital, University College London, London, UK.
Drs. Sobanski and Giovannelli contributed equally to this work. Drs. Launay and Hachulla contributed equally to this work.
Dr. Sobanski has received consulting fees and speaking fees from Grifols (less than $10,000) and has received research support from Actelion, Grifols, GlaxoSmithKline, Octapharma, Pfizer, and Shire. Dr. Distler has received consulting fees and speaking fees from Actelion, Bayer, Biogen Idec, Boehringer Ingelheim, ChemomAb, EspeRare Foundation, Genentech, GlaxoSmithKline, Inventiva, Eli Lilly, Medac, MedImmune, Mitsubishi Tanabe Pharma, Pharmacyclics, Novartis, Pfizer, Sanofi, Sinoxa, UCB, and Roche (less than $10,000 each); has received research support from Actelion, Bayer, Boehringer Ingelheim, Mitsubishi Tanabe Pharma, and Roche; and holds a patent for mir‐29 for the treatment of systemic sclerosis. Dr. Denton has received consulting fees and speaking fees from Actelion, Boehringer Ingelheim, Bayer, GlaxoSmithKline, Inventiva, Roche, and Sanofi‐Aventis (less than $10,000 each) and has received research support from Bayer, CSL Behring, GlaxoSmithKline, Inventiva, and Roche. Dr. Launay has received research support from Actelion, GlaxoSmithKline, Octapharma, Pfizer, and Shire. No other disclosures relevant to this article were reported.
Data are available from the European Scleroderma Trials and Research database upon request.
Contributor Information
David Launay, Email: david.launay@univ-lille.fr.
the EUSTAR Collaborators:
Serena Guiducci, Ulrich Walker, Diego Kyburz, Giovanni Lapadula, Florenzo Iannone, Britta Maurer, Suzana Jordan, Radim Becvar, Stanislaw Sierakowsky, Otylia Kowal Bielecka, Maurizio Cutolo, Alberto Sulli, Gabriele Valentini, Giovanna Cuomo, Elise Siegert, Simona Rednic, Ileana Nicoara, André Kahan, Panayiotis Vlachoyiannopoulos, Carlo Montecucco, Roberto Caporali, Jiri Stork, Murat Inanc, Patricia E. Carreira, Srdan Novak, László Czirják, Cecilia Varju, Carlo Chizzolini, Eugene J. Kucharz, Anna Kotulska, Magdalena Kopec‐Medrek, Malgorzata Widuchowska, Blaz Rozman, Carmel Mallia, Bernard Coleiro, Armando Gabrielli, Dominique Farge, Chen Wu, Zora Marjanovic, Helene Faivre, Darin Hij, Roza Dhamadi, Paolo Airò, Roger Hesselstrand, Frank Wollheim, Dirk M Wuttge, Kristofer Andréasson, Duska Martinovic, Alexandra Balbir‐Gurman, Yolanda Braun‐Moscovici, Francesco Trotta, Andrea Lo Monaco, Nicolas Hunzelmann, Raffaele Pellerito, Ospedale Mauriziano, Lisa Maria Bambara, Paola Caramaschi, Jadranka Morovic‐Vergles, Carol Black, Nemanja Damjanov, Jörg Henes, Vera Ortiz Santamaria, Stefan Heitmann, Dorota Krasowska, Matthias Seidel, Paul Hasler, Harald Burkhardt, Andrea Himsel, Gianluigi Bajocchi, Arcispedale Santa Maria Nuova, Maria João Salvador, José Antonio Pereira Da Silva, Bojana Stamenkovic, Aleksandra Stankovic, Carlo Francesco Selmi, Maria De Santis, Bianca Marasini, Mohammed Tikly, Lidia P. Ananieva, Lev N. Denisov, Ulf Müller‐Ladner, Marc Frerix, Ingo Tarner, Raffaella Scorza, Francesco Puppo, Merete Engelhart, Gitte Strauss, Henrik Nielsen, Kirsten Damgaard, Gabriella Szücs, Szilvia Szamosi, Antonio Zea Mendoza, Carlos de la Puente, Walter Alberto Sifuentes Giraldo, Øyvind Midtvedt, Silje Reiseter, Torhild Garen, Guido Valesini, Valeria Riccieri, Ruxandra Maria Ionescu, Daniela Opris, Laura Groseanu, Fredrick M. Wigley, Roxana Sfrent Cornateanu, Razvan Ionitescu, Ana Maria Gherghe, Alina Soare, Marilena Gorga, Mihai Bojinca, Carina Mihai, Mihaela Milicescu, Cord Sunderkötter, Annegret Kuhn, Nora Sandorfi, Georg Schett, Jörg HW Distler, Christian Beyer, Pierluigi Meroni, Francesca Ingegnoli, Luc Mouthon, Filip De Keyser, Vanessa Smith, Francesco Paolo Cantatore, Ada Corrado, Susanne Ullman, Line Iversen, Carlos Alberto von Mühlen, Jussara Marilu Bohn, Lilian Scussel Lonzetti, Maria Rosa Pozzi, Kilian Eyerich, Rüdiger Hein, Elisabeth Knott, Piotr Wiland, Magdalena Szmyrka‐Kaczmarek, Renata Sokolik, Ewa Morgiel, Marta Madej, Frédéric A. Houssiau, Juan Jose Alegre‐Sancho, Brigitte Krummel‐Lorenz, Petra Saar, Martin Aringer, Claudia Günther, Rene Westhovens, Ellen de Langhe, Jan Lenaerts, Branimir Anic, Marko Baresic, Miroslav Mayer, Maria Üprus, Kati Otsa, Sule Yavuz, Brigitte Granel, Sebastião Cezar Radominski, Carolina de Souza Müller, Valderílio Feijó Azevedo, Sergio Jimenez, Joanna Busquets, Svetlana Agachi, Liliana Groppa, Lealea Chiaburu, Eugen Russu, Sergei Popa, Thierry Zenone, Margarita Pileckyte, Simon Stebbings, John Highton, Alessandro Mathieu, Alessandra Vacca, Percival D. Sampaio‐Barros, Natalino H. Yoshinari, Roberta G. Marangoni, Patrícia Martin, Luiza Fuocco, Lisa Stamp, Peter Chapman, John O'Donnell, Kamal Solanki, Alan Doube, Douglas Veale, Marie O'Rourke, Esthela Loyo, Mengtao Li, Walid Ahmed Abdel Atty Mohamed, Edoardo Rosato, Antonio Amoroso, Antonietta Gigante, Fahrettin Oksel, Figen Yargucu, Cristina‐Mihaela Tanaseanu, Monica Popescu, Alina Dumitrascu, Isabela Tiglea, Rosario Foti, Rodica Chirieac, Codrina Ancuta, Daniel E. Furst, Peter Villiger, Sabine Adler, Jacob van Laar, Cristiane Kayser, Andrade Luis Eduardo C., Nihal Fathi, Manal Hassanien, Paloma García de la Peña Lefebvre, Silvia Rodriguez Rubio, Marta Valero Exposito, Jean Sibilia, Emmanuel Chatelus, Jacques Eric Gottenberg, Hélène Chifflot, Ira Litinsky, Paul Emery, Maya Buch, Francesco Del Galdo, Algirdas Venalis, Irena Butrimiene, Paulius Venalis, Rita Rugiene, Diana Karpec, Lesley Ann Saketkoo, Joseph A. Lasky, Eduardo Kerzberg, Fabiana Montoya, Vanesa Cosentino, Massimiliano Limonta, Antonio Luca Brucato, Elide Lupi, Itzhak Rosner, Michael Rozenbaum, Gleb Slobodin, Nina Boulman, Doron Rimar, Maura Couto, François Spertini, Camillo Ribi, Guillaume Buss, Sarah Kahl, Vivien M. Hsu, Fei Chen, Deborah McCloskey, Halina Malveaux, Jean Louis Pasquali, Thierry Martin, Audrey Gorse, Aurélien Guffroy, and Vincent Poindron
References
- 1. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson SR, Soowamber ML, Fransen J, Khanna D, Van Den Hoogen F, Baron M, et al. There is a need for new systemic sclerosis subset criteria: a content analytic approach. Scand J Rheumatol 2018;47:62–70. [DOI] [PubMed] [Google Scholar]
- 3. Wollheim FA. Classification of systemic sclerosis: visions and reality. Rheumatology (Oxford) 2005;44:1212–6. [DOI] [PubMed] [Google Scholar]
- 4. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988;15:202–5. [PubMed] [Google Scholar]
- 5. Komócsi A, Vorobcsuk A, Faludi R, Pintér T, Lenkey Z, Költo G, et al. The impact of cardiopulmonary manifestations on the mortality of SSc: a systematic review and meta‐analysis of observational studies. Rheumatology (Oxford) 2012;51:1027–36. [DOI] [PubMed] [Google Scholar]
- 6. Tyndall AJ, Bannert B, Vonk M, Airò P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010;69:1809–15. [DOI] [PubMed] [Google Scholar]
- 7. Medsger TA Jr. Natural history of systemic sclerosis and the assessment of disease activity, severity, functional status, and psychologic well‐being. Rheum Dis Clin N Am 2003;29:255–73. [DOI] [PubMed] [Google Scholar]
- 8. Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of pulmonary complications and long‐term survival in systemic sclerosis. Arthritis Rheumatol 2014;66:1625–35. [DOI] [PubMed] [Google Scholar]
- 9. Srivastava N, Hudson M, Tatibouet S, Wang M, Baron M, Fritzler MJ, et al. Thinking outside the box: the associations with cutaneous involvement and autoantibody status in systemic sclerosis are not always what we expect. Semin Arthritis Rheum 2015;45:184–9. [DOI] [PubMed] [Google Scholar]
- 10. Sobanski V, Dauchet L, Lefèvre G, Lambert M, Morell‐Dubois S, Sy T, et al. Prevalence of anti–RNA polymerase III antibodies in systemic sclerosis: new data from a French cohort and a systematic review and meta‐analysis. Arthritis Rheumatol 2014;66:407–17. [DOI] [PubMed] [Google Scholar]
- 11. Patterson KA, Roberts‐Thomson PJ, Lester S, Tan JA, Hakendorf P, Rischmueller M, et al. Interpretation of an extended autoantibody profile in a well‐characterized Australian systemic sclerosis (scleroderma) cohort using principal components analysis. Arthritis Rheumatol 2015;67:3234–44. [DOI] [PubMed] [Google Scholar]
- 12. Ligon CB, Wigley FM. Scleroderma: bringing a disease from black‐and‐white into technicolor [editorial]. Arthritis Rheumatol 2015;67:3101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richette P, Clerson P, Périssin L, Flipo RM, Bardin T. Revisiting comorbidities in gout: a cluster analysis. Ann Rheum Dis 2015;74:142–7. [DOI] [PubMed] [Google Scholar]
- 14. Ahmad T, Pencina MJ, Schulte PJ, O'Brien E, Whellan DJ, Piña IL, et al. Clinical implications of chronic heart failure phenotypes defined by cluster analysis. J Am Coll Cardiol 2014;64:1765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourdin A, Molinari N, Vachier I, Varrin M, Marin G, Gamez AS, et al. Prognostic value of cluster analysis of severe asthma phenotypes. J Allergy and Clin Immunol 2014;134:1043–50. [DOI] [PubMed] [Google Scholar]
- 16. Szodoray P, Hajas A, Kardos L, Dezso B, Soos G, Zold E, et al. Distinct phenotypes in mixed connective tissue disease: subgroups and survival. Lupus 2012;21:1412–22. [DOI] [PubMed] [Google Scholar]
- 17. Mahr A, Katsahian S, Varet H, Guillevin L, Hagen EC, Höglund P, et al. Revisiting the classification of clinical phenotypes of anti‐neutrophil cytoplasmic antibody‐associated vasculitis: a cluster analysis. Ann Rheum Dis 2013;72:1003–10. [DOI] [PubMed] [Google Scholar]
- 18. Ingegnoli F, Ardoino I, Boracchi P, Cutolo M, EUSTAR co‐authors . Nailfold capillaroscopy in systemic sclerosis: data from the EULAR Scleroderma Trials and Research (EUSTAR) database. Microvasc Res 2013;89:122–8. [DOI] [PubMed] [Google Scholar]
- 19. Leclair V, Hudson M, Proudman SM, Stevens WM, Fritzler MJ, Wang M, et al. Subsets in systemic sclerosis: one size does not fit all. J Scleroderma Relat Disord 2016;1:298–306. [Google Scholar]
- 20. Tyndall A, Mueller‐Ladner U, Matucci‐Cerinic M. Systemic sclerosis in Europe: first report from the EULAR Scleroderma Trials and Research (EUSTAR) group database. Ann Rheum Dis 2005;64:1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walker UA, Tyndall A, Czirják L, Denton C, Farge‐Bancel D, Kowal‐Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2007;66:754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meier FMP, Frommer KW, Dinser R, Walker UA, Czirjak L, Denton CP, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2012;71:1355–60. [DOI] [PubMed] [Google Scholar]
- 23. Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic, Therapeutic Criteria Committee . Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 1980;23:581–90. [DOI] [PubMed] [Google Scholar]
- 24. Park JW, Ahn GY, Kim JW, Park ES, Kang JH, Chang SH, et al. Impact of EUSTAR standardized training on accuracy of modified Rodnan skin score in patients with systemic sclerosis. Int J Rheum Dis 2019;22:96–102. [DOI] [PubMed] [Google Scholar]
- 25. Kowal‐Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017;76:1327–39. [DOI] [PubMed] [Google Scholar]
- 26. Han L, Benseler SM, Tyrrell PN. Cluster and multiple correspondence analyses in rheumatology: paths to uncovering relationships in a sea of data. Rheum Dis Clin North Am 2018;44:349–60. [DOI] [PubMed] [Google Scholar]
- 27. Hennig C. Cluster‐wise assessment of cluster stability. Comput Stat Data Anal 2007;52:258–71. [Google Scholar]
- 28. Hennig C. Dissolution point and isolation robustness: robustness criteria for general cluster analysis methods. J Multivar Anal 2008;99:1154–76. [Google Scholar]
- 29. R Core Team . R: a language and environment for statistical computing Vienna, Austria: R Foundation for Statistical Computing; 2011. URL: https://www.R-project.org. [Google Scholar]
- 30. Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of pulmonary complications and long‐term survival in systemic sclerosis. Arthritis Rheumatol 2014;66:1625–35. [DOI] [PubMed] [Google Scholar]
- 31. Varga J, Hinchcliff M. Systemic sclerosis: beyond limited and diffuse subsets? Nat Rev Rheumatol 2014;10:200–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kranenburg P, van den Hombergh WM, Knaapen‐Hans HK, van den Hoogen FH, Fransen J, Vonk MC. Survival and organ involvement in patients with limited cutaneous systemic sclerosis and anti‐topoisomerase‐I antibodies: determined by skin subtype or auto‐antibody subtype? A long‐term follow‐up study. Rheumatology (Oxford) 2016;55:2001–8. [DOI] [PubMed] [Google Scholar]
- 33. Giordano M, Valentini G, Migliaresi S, Picillo U, Vatti M. Different antibody patterns and different prognoses in patients with scleroderma with various extent of skin sclerosis. J Rheumatol 1986;13:911–6. [PubMed] [Google Scholar]
- 34. Perera A, Fertig N, Lucas M, Rodriguez‐Reyna TS, Hu P, Steen VD, et al. Clinical subsets, skin thickness progression rate, and serum antibody levels in systemic sclerosis patients with anti–topoisomerase I antibody. Arthritis Rheum 2007;56:2740–6. [DOI] [PubMed] [Google Scholar]
- 35. Shand L, Lunt M, Nihtyanova S, Hoseini M, Silman A, Black CM, et al. Relationship between change in skin score and disease outcome in diffuse cutaneous systemic sclerosis: application of a latent linear trajectory model. Arthritis Rheum 2007;56:2422–31. [DOI] [PubMed] [Google Scholar]
- 36. Man A, Davidyock T, Ferguson LT, Ieong M, Zhang Y, Simms RW. Changes in forced vital capacity over time in systemic sclerosis: application of group‐based trajectory modelling. Rheumatology (Oxford) 2015;54:1464–71. [DOI] [PubMed] [Google Scholar]
- 37. Schulam P, Wigley F, Saria S. Clustering longitudinal clinical marker trajectories from electronic health data: applications to phenotyping and endotype discovery. Association for the Advancement of Artificial Intelligence. 2015. URL: https://www.aaai.org/ocs/index.php/AAAI/AAAI15/paper/download/10015/9966.
- 38. Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol 2012;132:1363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 2013;133:1979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Little RJ, Rubin DB. Statistical analysis with missing data. 2nd ed Hoboken (NJ): Wiley; 2002. [Google Scholar]
- 41. Storlie CB, Myers SM, Katusic SK, Weaver AL, Voigt RG, Croarkin PE, et al. Clustering and variable selection in the presence of mixed variable types and missing data. Stat Med 2018. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walker UA, Tyndall A, Czirják L, Denton CP, Farge‐Bancel D, Kowal‐Bielecka O, et al. Geographical variation of disease manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research (EUSTAR) group database. Ann Rheum Dis 2009;68:856–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials