

Abstract
The endosomal system plays an essential role in cell homeostasis by controlling cellular signaling, nutrient sensing, cell polarity and cell migration. However, its place in the regulation of tissue, organ and whole body physiology is less well understood. Recent studies have revealed an important role for the endosomal system in regulating glucose and lipid homeostasis, with implications for metabolic disorders such as type 2 diabetes, hypercholesterolemia and non‐alcoholic fatty liver disease. By taking insights from in vitro studies of endocytosis and exploring their effects on metabolism, we can begin to connect the fields of endosomal transport and metabolic homeostasis. In this review, we explore current understanding of how the endosomal system influences the systemic regulation of glucose and lipid metabolism in mice and humans. We highlight exciting new insights that help translate findings from single cells to a wider physiological level and open up new directions for endosomal research.
Keywords: diabetes, endocytosis, fatty liver disease, glucose and lipid metabolism, metabolic signaling, nutrient transport
1. INTRODUCTION
The role of the endosomal system in maintaining cellular homeostasis has been studied for decades and has provided us with a deep appreciation for its complexity, specificity and functionality.1, 2, 3, 4 This homeostasis is mediated by a highly interconnected network of compartments that regulate cellular signaling, nutrient sensing and transport, cell polarity and cell migration. The dynamic interaction between the endosomal compartments enables the amount and spatial distribution of more than 5000 integral membrane proteins to be controlled.5, 6 This function is accomplished by numerous endosomal multi‐protein complexes that regulate the amount of protein on the plasma membrane depending on the physiological condition by sending them to lysosomes for degradation or routing them back to the plasma membrane for recycling. Many elegant studies have unraveled the functions of the endosomal network within cellular systems. However, we have very limited knowledge on how these functions relate to the role of cells in tissues, organs and whole organism physiology.
Interestingly, recent findings have begun to support a role for the endosomal system in metabolic homeostasis. For instance, reduction of the endo‐lysosomal compartments in the liver through knockdown of the small GTPase Rab5 was sufficient to induce a severe metabolic phenotype,7, 8 normally only achieved through interference with metabolically relevant genes. This rapid and profound effect of Rab5 knockdown places the endosomal system into the regulatory network of glucose and lipid metabolism. Although these results might have been surprising, data in humans with metabolic diseases already show that mutations in endosomal genes are associated with metabolic pathologies, such as type 2 diabetes, hypercholesterolemia and non‐alcoholic fatty liver disease (NAFLD).9, 10, 11, 12, 13, 14, 15, 16, 17 These recent findings highlight a wider function of the endosomal system at an organism level and provide us with a possibility to connect and translate our fundamental knowledge on endocytosis to metabolic (patho‐) physiologies. In light of the increasing pandemics of type 2 diabetes and obesity, which will collectively affect 2 billion people worldwide by 2040 (WHO), this research could also reveal potential new targets for therapeutic interventions.
A key feature of type 2 diabetes is the dysregulation of metabolic homeostasis. This is characterized by an imbalance between nutrient intake, storage and consumption. Under physiological conditions, metabolically active organs, such as muscle, fat, liver, pancreas and brain store nutrients upon food intake by turning on anabolic processes, such as glycogen and fatty acid synthesis and glycolysis, and by repressing catabolic processes, such as gluconeogenesis (glucose production), glycogenolysis and fatty acid oxidation (Figure 1). When nutrient availabilities are low during fasting, these responses are inverted to ensure sufficient energy supplies for the body (Figure 2). The switch between fasting and feeding is mainly regulated by the hormones glucagon and insulin, where insulin is responsible for anabolic processes. This metabolic regulation is perturbed in type 2 diabetes, where constant food intake and endogenous energy production lead to a permanent increase in blood glucose levels (hyperglycemia) and insulin resistance. Thus, the maintenance of metabolic homeostasis requires proper sensing of insulin and glucagon signals and differential interpretation of those signals in different tissues. For instance, insulin induces glucose uptake in muscle and fat, but represses glucose production in liver (see below). In addition, the expression of nutrient transporters on the plasma membrane to mediate the uptake of glucose, low density lipoprotein‐cholesterol (LDL‐C), free fatty acids, etc. needs to be modulated in target tissues depending on nutrient availabilities. This implies a requirement for fine‐tuned regulation of transporter recycling vs degradation. Plasma membrane plasticity is essential for internalization of receptors but also for secretion of insulin granules, for instance, in the pancreas. These different requirements for signal transduction, nutrient transport, receptor degradation and induced recycling are all hallmarks of the endosomal system.
Figure 1.
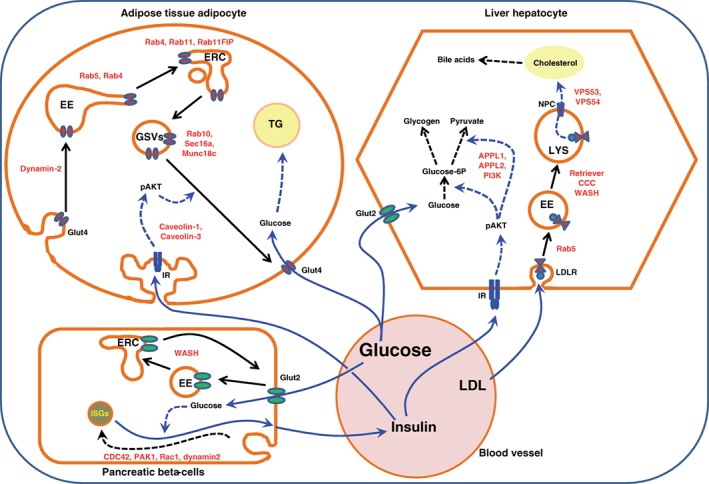
Involvement of endocytic components in systemic metabolism in the postprandial (feeding) period. After a meal, circulating blood glucose levels rise, leading to a massive entry of glucose into pancreatic β‐cells via the glucose transporter GLUT2, which causes the release of insulin into the blood stream. The cell surface amounts of GLUT2 are maintained constant via recycling of GLUT2. Adipocytes, the main cell type of adipose tissue, sense the increase in circulating insulin levels resulting in an induction of p‐AKT‐dependent signaling cascades that favor the fusion of GLUT4 storage vesicles with the plasma membrane, increasing the surface expression of GLUT4. When GLUT4 is translocated at the cell surface, glucose will enter into adipocytes and be stored as triglycerides within the lipid droplets. Concomitantly, insulin acts on the main liver cells, hepatocytes, by inducing a cascade of signaling events downstream of the insulin receptor. These signals will restructure the genomic program of the hepatocytes towards anabolic processes, either storing glucose that enters the cells in a concentration‐dependent manner via GLUT2 into glycogen, or utilizing the glucose for glycolysis. In addition to controlling glucose levels, hepatocytes also participate in the regulation of cholesterol levels. To achieve this function, circulating low‐density lipoprotein cholesterol (LDL‐C) is taken up by the hepatocytes and sent to lysosomes, where the digested cholesterol will be extracted via the Niemann‐Pick proteins into the cytosol. Cytosolic cholesterol can then be used either for incorporation into membranes or for bile acid production. Examples of endocytic components that play a role in these metabolic processes are labeled in red. EE, early endosomes; ERC, endosomal recycling compartment; GSVs, GLUT4 storage vesicles; ISGs, insulin secretory vesicles; LYS, lysosome; TG, triglycerides; IR, insulin receptor; LDLR, LDL receptor; NPC, Niemann‐Pick protein
Figure 2.
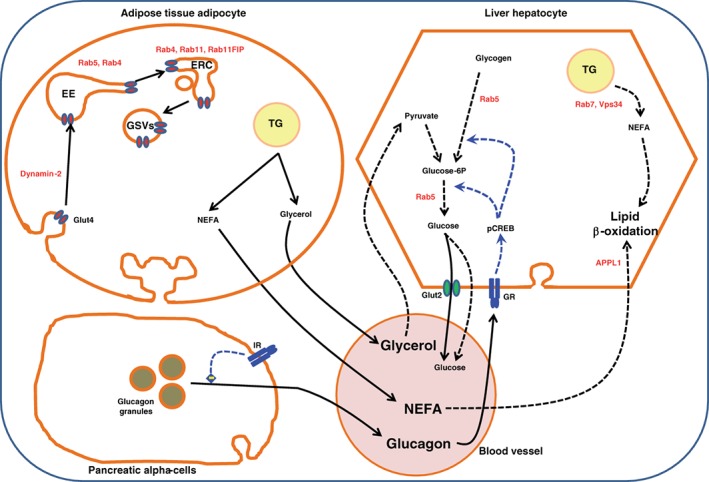
Involvement of endocytic components in systemic metabolism during the fasting period. During fasting, blood glucose levels drop, leading to a reduction in insulin secretion by pancreatic β‐cells. Instead glucagon is secreted from pancreatic α‐cells, which mainly acts on hepatocytes and induces a signaling cascade that shifts the genomic program towards catabolism, producing glucose either by the breakdown of glycogen or de novo synthesis from pyruvate through gluconeogenesis. Hepatocytes will also produce energy by breaking down their triglyceride stores into non‐esterified fatty acids and burning them via β‐oxidation. Concomitantly, in the adipocytes, the drop in insulin will stop GLUT4 translocation, which will be removed from the plasma membrane through endocytosis and stored in GLUT4 storage vesicles, leading to the arrest of glucose uptake by the adipocyte. This is also accompanied by an activation of triglyceride lipolysis releasing glycerol and NEFA into the blood. Glycerol will be used by hepatocytes to generate pyruvate essential for gluconeogenesis, and NEFA will be burned via β‐oxidation. Examples of endocytic components that play a role in these metabolic processes are labeled in red. EE, early endosomes; ERC, endosomal recycling compartment; GSVs, GLUT4 storage vesicles; ISGs, insulin secretory vesicles; LYS, lysosome; TG, triglycerides; GR, glucagon receptor; IR, insulin receptor; NEFA, non‐esterified fatty acids
This review will highlight how different levels of endosomal regulation can be integrated into our understanding of metabolic homeostasis and discuss components of the endocytic system that regulate systemic glucose and lipid metabolism, based mainly on in vivo studies in mice and humans. We will not address amino acid metabolism and lysosomal nutrient sensing via mechanistic Target of Rapamycin, as this has been discussed extensively elsewhere.18, 19, 20 Instead, we will focus on the parts of the endosomal system that contribute to metabolic control and potentially participate in the development of insulin resistance, hyperglycemia and steatosis (lipid accumulation within liver and muscle), leading to type 2 diabetes.
2. REGULATION OF GLUCOSE AND LIPID METABOLISM THROUGH THE ENDOSOMAL SYSTEM
Glucose and lipid homeostasis are achieved by a balance between uptake of these nutrients, storage as glycogen or lipid droplets, and mobilization of those nutrient stores for energy production. The endosomal system participates in glucose and lipid uptake in the postprandial phase by regulating the expression of nutrient transporters and receptors on the cell surface. This can be achieved by enhanced recycling or reduced internalization. Upon surface appearance, nutrients can either bind to their receptors and be internalized, as exemplified by the LDL receptor (LDLR), or be actively transported into the cell, as in the case of fatty acids and glucose.
2.1. Regulation of glucose uptake and storage
Glucose uptake is induced upon food intake, when nutrients are enriched and blood glucose concentrations rise (Figure 1). This induces the secretion of insulin from the β‐cells of the pancreas, which leads to the uptake of glucose into peripheral tissues, such as adipose tissue and muscle, causing blood glucose levels to drop. Internalized glucose is used for the build up of glycogen in muscle or is metabolized through glycolysis to be incorporated into fatty acids for storage in fat. The uptake of glucose is mediated by the insulin‐dependent recruitment of the glucose transporter, GLUT4 from intracellular storage vesicles (GSVs).21
2.2. Glucose transport through GLUT4
The regulation of GLUT4 trafficking is one of the best examples of how endosomal transport participates in metabolic control. Many detailed mechanistic studies describing the intracellular transport steps of GLUT4 have been performed in cell lines, and these have been discussed in many elegant reviews.21, 22, 23, 24, 25 In this review, we will focus instead on in vivo studies, where components of the endosomal system have been shown to affect GLUT4 trafficking in tissues with immediate impact on systemic metabolism, and which are affected in metabolic disorders in humans.
As a transporter that cycles between intracellular compartments and the plasma membrane, GLUT4 traffics through all stages of the endosomal system, hence it is not surprising that many endosomal proteins have been shown to affect the behavior of GLUT4. Starting at the plasma membrane, dynamin 2 was shown already in the late 1990s to be required for GLUT4 internalization in primary rat adipocytes,26 which is consistent with its role in endocytic vesicle scission. Mutations in DNM2 have been linked to centronuclear myopathy (CNM), a diabetic late complication involving impaired actin‐dependent trafficking in human muscle cells.9 Interestingly, biopsies of patients with CNM reveal aberrant perinuclear accumulation of GLUT4, underscoring the importance of dynamin 2 in internalization and actin remodeling even in humans.9 In diabetes‐induced nephropathy, induction of actin‐dependent dynamin oligomerization has been shown to improve renal health in models of transient and chronic kidney diseases, which is relevant for diabetes‐induced renal dysfunction.27 Whether this is mediated through the spatiotemporal reorganization of cell surface molecules upon dynamin activation or alterations in signaling properties in diverse models of transient kidney diseases remains to be elucidated.
Upon internalization, GLUT4 traffics through the early endosomes, where the requirement for early endosomal Rabs, such as Rab4 and Rab5, has been shown in 3T3‐L1 adipocytes in vitro.28, 29, 30, 31, 32, 33 Here, Huang et al32 raised the interesting idea that insulin inhibits Rab5 activity and the interaction of dynein with microtubules, and that these effects may inhibit the rate of GLUT4 internalization, resulting in enhanced surface expression of GLUT4. Whether this mechanism affects the presence of GLUT4 at the cell surface in adipose tissue and muscle in mice in vivo needs to be determined. Whole‐body knockout mice for Rab4a and Rab4b display a reduction in circulating triglycerides and an increase in their total body fat mass, respectively, indicating a function of Rab4 in lipid metabolism (IMPC). The phenotype of Rab4b knockout mice could be in part due to the key role of Rab4b in adipose tissue T cells, as was recently demonstrated.34 After passing through early endosomes, GLUT4 traffics through the endosomal recycling compartment, involving Rab11. Although Rab11 has been shown to be required for proper GLUT4 trafficking in adipocytes in vitro,35 its function in metabolic regulation in vivo remains unclear. Whole‐body knockout mice indicate an essential role for Rab11a in development, whereas Rab11b knockout mice have no phenotype (IMPC). Only knockout mice of a Rab11 interactor, Rab11‐FIP3, exhibit decreased body fat accumulations (IMPC). However, how this connects to GLUT4 trafficking and glucose homeostasis still needs to be elucidated.
An interesting feature of the recycling endosomes is to sequester cargo into a specialized recycling compartment, which is utilized in many tissues to specifically initiate the delivery of these cargoes to the plasma membrane in response to stimuli. This feature is used in GLUT4 trafficking for targeting of GLUT4 to the cell surface in adipocytes and muscle cells. Interestingly, in addition to GLUT4, the delivery of aquaporins to the basal membrane in the kidney36 or the transport of bile acid transporters to the apical bile canaliculi in hepatocytes37 is mediated via a similar storage compartment, highlighting a common concept of cargo sequestration and recruitment in physiology.
GLUT4 is retained in a specialized intracellular compartment and is targeted to the plasma membrane via insulin‐regulated vesicular trafficking.21, 23 Insulin induces an inactivation of the Rab GTPase activating proteins (GAPs) necessary for GLUT4 translocation, namely TBC1D4/AS160 and TBC1D1. This is achieved by an AKT‐dependent phosphorylation of these Rab‐GAPs, leading to a loss of their inhibitory role on the downstream Rabs necessary for GLUT4 trafficking to the cell surface. The Rabs for GLUT4 include Rab8a, Rab13 and Rab14 in muscle cells38, 39, 40 and Rab10 and Rab35 in adipocytes.41, 42, 43 While many elegant studies have unraveled the function of these Rabs and other interacting proteins in cell lines (for detailed review see Reference 21), only Rab10's function has been elucidated in mice. Adipose‐specific knockout of Rab10 causes a 50% reduction in insulin‐stimulated glucose uptake and GLUT4 translocation to the plasma membrane in fat, leading to a near complete failure of insulin to suppress hepatic glucose production without significant inhibition of muscle glucose uptake.44 These data indicate that the amount of glucose uptake in adipocytes is a measure of whole‐body glucose homeostasis that affects liver insulin sensitivity. Interestingly, Rab10 has been shown to interact with Sec16a, which is important for the biogenesis of GLUT4 transport vesicles at the trans Golgi network (TGN), suggesting a cooperative action of GLUT4 vesicle recycling and accelerated biogenesis to maintain the elevated cell‐surface expression of GLUT4 under insulin stimulation.45 It will be interesting to see the role Rab8a and Rab13 play in muscle tissue in vivo and how interfering with their function affects GLUT4 trafficking and whole body glucose homeostasis.
The fusion of GLUT4 containing vesicles (GSVs) with the plasma membrane is an essential aspect in GLUT4 translocation upon insulin stimulation and has been extensively reviewed.46 This process has been shown to require Munc18c in 3T3‐L1 adipocytes.47 Interestingly, heterozygous Munc18c knockout mice show a decrease in GLUT4 translocation in skeletal muscle and alterations of glucose‐stimulated insulin secretion in pancreatic islets. This leads to severe glucose intolerance upon feeding a high fat diet, suggesting its importance not only for regulation of GLUT4 traffic but also other processes in β‐cells.48
Altogether, these data support an essential function of the specialized recycling compartment in regulating GLUT4 distribution and glucose transport that is essential in whole body glucose homeostasis. In fact, it has been shown that defective GLUT4 translocation is a strong feature of insulin resistance and type 2 diabetes in humans.12, 13, 14 In addition, variants in the clathrin heavy chain CHC22 in humans are associated with altered GLUT4 trafficking and correlate with impaired GLUT4 translocation observed in type 2 diabetes.15 These data underscore the important contribution of the endosomal system in proper GLUT4 trafficking, where many components have been studied in cell lines. As GLUT4 traffics through many stages of the endosomal network throughout its cellular cycles, it is not surprising that many components affect GLUT4 behavior. However, it would be interesting to see whether these components are altered in diabetic conditions and how trafficking of GLUT4 might change in diabetes. Trafficking studies in isolated primary adipocytes from diabetic mice might give answers in this direction.
2.3. Glucose transport through GLUT1 and GLUT2
Other glucose transporters, such as GLUT1 and GLUT2 are not insulin‐dependent. GLUT1 is expressed in all cell types and is responsible for steady‐state uptake of glucose. Interestingly, depletion of the retromer complex, which is involved in the retrieval of cargoes back to the TGN,5, 6, 49, 50 was shown to enhance degradation of GLUT1 and reduce cellular glucose uptake.51, 52, 53, 54 The retromer complex is composed of a VPS complex (Vps35, Vps29 and Vps26), which engages cargoes, and a sorting nexin‐Bin/amphiphysin/Rvs (SNX‐BAR) complex (SNX1, SNX2, SNX3, SNX5, SNX6 and SNX27), which bends the membrane (for review see References 6, 55). The whole‐body knockout mice of the core component of the retromer complex, Vps35, are embryonically lethal,56 but whole‐body heterozygous depletion is not associated with a specific phenotype (IMPC). Consistent with these observations, knockout of the cargo adapter proteins SNX3 and SNX27 are homozygous lethal or sub‐lethal (IMPC), suggesting that the retromer complex is essential for cellular homeostasis.
GLUT2 is expressed on β‐cells and in the liver. It has not been shown to be sequestered into storage vesicles or to be recruited upon stimulation. In the liver, GLUT2 is a bi‐directional transporter that internalizes and secretes glucose depending on its intracellular and extracellular concentrations.57 In the postprandial state, glucose is taken up into the liver and used mainly for glycogen production (Figure 1). This glycogen can then be used for maintaining blood glucose levels under starvation. The reduction of blood glucose concentrations induces the release of glucagon from the α‐cells of the pancreas, causing the breakdown of glycogen and induction of glucose production through gluconeogenesis in the liver, resulting in normalization of blood glucose concentrations (Figure 2).
In rodent β‐cells, GLUT2 is the principal glucose transporter, while in human β‐cells both GLUT1 and GLUT2 are involved in glucose transport.58, 59 Interestingly, GLUT2/SLC2A2 variants confer type 2 diabetes risk, indicating that GLUT1 and GLUT2 function is not completely redundant.58 Importantly, extra‐ and intra‐cellular glucose concentrations equilibrate quickly in pancreatic islets, suggesting that glucose transport is not the rate‐limiting step in β‐cell glucose metabolism. Instead, β‐glucokinase (GCK/hexokinase 4) activity is crucial to keep glucose inside the cell and to initiate glycolysis.60 Glucose‐dependent mitochondrial ATP production in turn initiates insulin secretion by closing ATP‐dependent K+ channels and subsequent opening of voltage‐gated Ca2+ channels.61
The trafficking of GLUT2 was found to be controlled by the Wiskott‐Aldrich syndrome protein and SCAR homolog (WASH) complex in mouse pancreatic β‐cells. Indeed, pancreas‐specific deletion of the WASH component WASHC1 causes intracellular accumulation of GLUT2 and decreased GLUT2 protein levels leading to impaired blood glucose clearance and reduced insulin secretion.62 Because the WASH complex, composed of WASHC1, WASHC2, WASHC3, WASHC4 and WASHC5, is an important player in the retrieval of cargoes from the endosomes (for review see Reference 6), it is likely that WASH complex depletion enhances protein degradation upon reduced protein retrieval. Whether the retrieval of cargo through the WASH complex is sensitive to extracellular metabolic cues remains to be determined. However, it is important to realize that glucose uptake via GLUT2 in β‐cells seems to be mainly observed in rodents, whereas humans internalize glucose also through GLUT1,58, 59 making the direct translation of these trafficking studies to the human situation difficult.
An important part of glucose uptake is the adequate release of insulin from β‐cells. This is mediated in a bi‐phasic mode, where pre‐docked and ready to fuse insulin secretory granules are secreted in the first phase of glucose‐induced insulin secretion happening in seconds. The second phase insulin secretion requires re‐internalization of the machinery necessary for insulin granule production and trafficking, where endocytosis plays an important role. Here, rearrangements of the actin cytoskeleton are needed for proper insulin secretion. Components of the endosomal machinery, such as Cdc42 have been implicated in this process. Knockdown of Cdc42 in isolated mouse islets results in the selective loss of the second phase insulin release, potentially causing less insulin secretion, which could lead to an increase in blood glucose levels.63 In fact, islets from donors with type 2 diabetes have profound defects in glucose‐stimulated Cdc42‐PAK1 activation and insulin secretion, which might contribute to the failure of the β‐cells to secrete insulin in diabetes.64 Here, it is suggested that glucose induces the translocation of Cdc42 to the plasma membrane, which activates the Cdc42‐PAK1‐RAC1 pathway, leading to cytoskeletal rearrangements and enhanced fusion of insulin granules with the plasma membrane.64 In addition, β‐cell‐specific inducible DMN2 knockout mice show a requirement for dynamin 2 in glucose tolerance and proper glucose‐induced second phase insulin secretion in mice.65 Interestingly, the decrease in insulin secretion is due to a disorganization and enhancement of the actin cytoskeletal network,65 which underscores a function for dynamin 2 in plasma membrane actin remodeling that is not directly related to its role in vesicle scission.
2.4. Regulation of LDL‐C uptake
The uptake of LDL‐C by the liver is crucial for lipid homeostasis. In hepatocytes, internalized cholesterol esters in LDL are hydrolyzed to free cholesterol in late endosomes/lysosomes. Subsequently, through the Nieman‐Pick Type‐C proteins, cholesterol is further transported to other cellular compartments and either used as a component of intracellular membranes66 or metabolized into bile acids and released into the bile canaliculi.67 Through this, the liver can actively reduce whole‐body cholesterol levels via LDL‐C endocytosis. A common feature of type 2 diabetes is hypercholesterolemia and fatty liver disease, which can develop through reduced uptake of LDL‐C or enhanced secretion of very low density lipoprotein from the liver. Thus, by altering LDLR trafficking, endocytosis participates actively in LDL‐C clearance. While it was thought that the recycling of receptors, including the LDLR, is a relatively passive process following bulk flow,68 recent evidence has clearly illustrated that these trafficking processes are spatially and temporally regulated by a large number of multi‐protein complexes.5, 6, 49, 50, 69
Underlining the pivotal role of endocytosis on LDLR trafficking, we previously demonstrated that the depletion of Rab5 in mouse liver in vivo leads to a decrease in LDL‐C uptake due to failure of LDLR internalization.7, 8 In this study, we took advantage of a hepatocyte‐specific delivery system, where siRNAs encapsulated into lipid nanoparticles are exclusively taken up by the liver,70 inducing a tissue‐specific knockdown of Rab5 in adult mice. The defects in LDL‐C uptake were sufficient to induce hypercholesterolemia and hyperlipidemia in the blood of these animals. Remarkably, depletion of Rab5 was sufficient to abolish the biogenesis of early endosomes causing a reduction of the entire degradative pathway,7 making the dissection of the metabolic phenotypes and underlying molecular mechanisms challenging.
2.5. The role of the retriever‐CCC‐WASH complex in controlling lipid homeostasis
The retriever‐CCC‐WASH complex participates in cargo sorting6 downstream of Rab5 by recruiting F‐actin onto endosomes via the WASH complex, as described above, recognizing cargoes via the CCC complex (COMMD, CCDC22 and CCDC93) and sorting them via the retriever complex (DSCR3, C16orf62 and Vps29). Interestingly, this large complex was also found to be implicated in lipid homeostasis. Indeed, patients with X‐linked intellectual disability involving a CCDC22 p.T17A or CCDC22 p.Y557C mutation were found to have an increase in total plasma cholesterol and LDL‐C levels.71 Moreover, liver‐specific COMMD1 knockout mice show elevated plasma LDL‐C and exhibit a mislocalization of LDLR associated with decreased LDL‐C uptake in the liver.71 COMMD1 is the founding member of a family of 10 proteins expressing the Copper Metabolism MURR1 Domain (COMMD) at their carboxy terminus. All are able to engage the CCC complex to fulfill their discrete functions, including cell proliferation, copper homeostasis and regulation of NF‐κB and sodium channels activities.72, 73, 74, 75 Strikingly, the increase in plasma cholesterol in liver‐specific COMMD1, COMMD6 or COMMD9 knockout mice causes an exacerbation of atherosclerosis, highlighting a collective function of COMMDs and CCC components in modulating plasma lipid levels in humans.10
Considering the large number of nutrient transporters and receptors trafficking through the endosomal system, which all need to escape degradation, it seems likely that additional functions of the CCC‐WASH‐retriever complex and the retromer complex will be discovered. It will be interesting to see how cargo specificity in the retrieving pathway is achieved and how this is physiologically regulated. Recently, an elegant study has suggested that the adaptor protein PID1, which regulates whole body glucose homeostasis,76 is an insulin‐dependent molecular switch that regulates the cellular distribution of another cholesterol transporter, LRP1 (low density lipoprotein particle related protein 1), and functions in the postprandial state by controlling SNX17 binding to LRP1.77 Interestingly, the CCC complex is also required for LRP1 trafficking to the plasma membrane, suggesting that this complex could mediate the insulin‐dependent regulation of LRP1 trafficking via PID1.10, 71 Whether this is regulated through a direct interaction with the CCC‐WASH‐retriever complex with PID1 still needs to be determined.
In addition to LDLR trafficking, endocytosis participates in the regulation of cholesterol flux within cells, which requires the function of Niemann‐Pick type C (NPC) proteins. Indeed, mutations of Vps53 prevent the proper sorting of NPC2 to lysosomes, causing cholesterol accumulation.78 Vps53 is a common component of two complexes, the endosome‐associated recycling protein (EARP) and the Golgi‐associated retrograde protein (GARP), which are implicated in the tethering of cargo‐containing carriers sorted from the endosomes to direct them either to the recycling compartments or the TGN, respectively (for review see Reference 79). Consistent with this finding, mutation of the specific component of the GARP complex, Vps54, in human amyotrophic lateral sclerosis (ALS) patients and in wobbler mice was found to be associated with a dysregulation of energy metabolism.11, 80, 81 In wobbler mice, these energy metabolism defects were linked to a disturbance of cholesterol homeostasis,78 suggesting that the GARP complex may be involved in the regulation of cholesterol flux. However, this needs to be confirmed, because EARP and GARP complexes share one‐quarter of their components, such that affecting one complex may alter the other.
Altogether, these results show that the retrieval of cargoes from endosomes has important implications for lipid and glucose metabolism. This could be a consequence of cargo recycling to the plasma membrane that requires this sorting step. Whether this retrieval is controlled by extracellular stimuli that initiate the recycling of nutrient receptors to the cell surface remains to be seen, but it would provide an interesting mechanism to explain how metabolic cues and conditions could influence cargo retrieval.
3. METABOLIC SIGNAL PROCESSING THROUGH THE ENDOSOMAL SYSTEM
3.1. Regulation of insulin and glucagon receptor trafficking
The regulation of the fasting/feeding transition is mediated by the action of insulin, glucagon and glucocorticoids. While the latter functions mainly through its nuclear receptor,82 insulin and glucagon signaling is mediated through their plasma membrane receptors. Extensive studies have investigated the role of the endosomal system on insulin receptor (IR) signaling and trafficking in cell lines.83 IR is internalized by clathrin‐dependent pathways84, 85 but also through caveolin‐mediated uptake,86, 87 suggesting that both processes exist and the receptor might utilize the different endocytic entry routes depending on the IR isoform, cell types and ligands. Nevertheless, the IR has been shown to localize to caveolae in adipocytes and myotubes,86, 88 and this localization seems to be essential for proper insulin signaling, which was found to require caveolin1 in adipocytes and caveolin3 in muscle cells.88, 89, 90 Indeed, the depletion of CAV1 or CAV3 in mice leads to insulin resistance in adipose tissue and muscle, respectively.91, 92 Importantly, the failure in insulin sensing by adipocytes results in a reduction in GLUT4 translocation (see above) and consequently reduced glucose storage into triglycerides, which could explain the lean phenotype and the resistance to diet‐induced obesity in caveolin1 knockout mice.93
Besides these studies in caveolin knockout mice, there are very few examples that investigate whether components of the endosomal system alter IR signaling and insulin sensitivity in vivo. The reason for this could be that the active internalized IR, which continues to signal on early endosomes,94 is more important for its mitogenic than its metabolic function.95 However, recent data have identified SORLA (sorting‐related receptor with type A repeats, encoded by SORL1) as directing the IR back to the plasma membrane and enhancing IR signaling, thereby contributing to glucose intolerance and obesity.96 This highlights the importance of the balance between IR recycling and degradation for metabolic control.
The glucagon receptor is a prototypical G protein‐coupled receptor, for which prolonged agonist stimulation attenuates signaling (homologous desensitization). However, internalization of the desensitized receptors and their sequestration into intracellular vesicles is a primary mechanism of signal attenuation and is essential for receptor resensitization (for review see Reference 97). Internalization of the glucagon receptor is mediated by clathrin‐dependent endocytosis in hepatocytes,98 as well as through caveolin.99 This dual mode of internalization may differ depending on the cell types and may have important consequences for signaling outcomes. However, it remains unclear whether endocytosis participates in glucagon signaling independently of its role in receptor distribution.
3.2. Signal interpretation through the endosomal system
While signaling receptor trafficking is important for signal transduction and downregulation, specific components of the endosomal system could play important roles in signal interpretation by interacting with downstream signaling receptor targets. Indeed, the surface of endosomes functions as a platform for signaling domains100 that control the spatiotemporal distribution of signaling molecules within the cell.101 Interestingly, the Rab5 effector APPL (adaptor protein containing PH domain, PTB domain and Leucine zipper motif) has been shown to interact with AKT2 on endosomes,100, 102 suggesting a potential metabolic function. In fact, APPL1 was reported to interact with adiponectin, an adipocyte hormone that is important for glucose sensitivity and fatty acid oxidation.103 APPL1 has been shown to be necessary for adiponectin functions, such as lipid oxidation, glucose uptake and GLUT4 translocation in L6‐myotubes.104 There, adiponectin stimulates the interaction between APPL1 and Rab5, leading to increased GLUT4 translocation. It would be interesting to see whether insulin influences this interaction in muscle.
Besides its function in mediating glucose uptake in fat and muscle, insulin also acts on the liver during feeding by reducing the expression of gluconeogenic genes, such as Glucose‐6‐phosphatase (G6Pase), which is rate limiting for glucose output.105 This is achieved by AKT‐mediated phosphorylation of Forkhead box‐O1 (FOXO1), a main transcription factor for gluconeogenesis, resulting in FOXO1 inactivation.106 As APPL1 has been shown to interact with the metabolically active isoform AKT2,100, 102 it is not surprising that liver‐specific overexpression of APPL1 by adenovirus results in increased hepatic insulin sensitivity and enhanced insulin‐induced suppression of the gluconeogenic program,107 while its knockdown has the opposite effect. The improvement of insulin sensitivity is achieved by binding of APPL1 to AKT2, thereby preventing the interaction of AKT2 with its inhibitor Tribble3 (TRB3),108 thus promoting AKT translocation to the plasma membrane and inducing enhanced AKT signaling. Interestingly, conditional deletion of APPL2 in skeletal muscle exhibits the opposite phenotype. Here, insulin‐induced plasma membrane recruitment of GLUT4 and glucose uptake are impaired by APPL2 overexpression but are enhanced upon knockdown, leading to improved glucose tolerance.109 This is achieved by a direct interaction of APPL2 with the Rab‐GTPase activating protein TBC1D1, which is necessary for GLUT4 translocation. Why APPL1 and APPL2 have these opposite effects and interaction partners, and how they are physiologically regulated upon glucose challenge, remains to be elucidated. The pathophysiological significance of APPL1/2 for glucose homeostasis, however, is supported by observations in diabetic patients and patients with NAFLD, where loss‐of‐function mutations in APPL1 and genetic variation in APPL1/APPL2 loci are linked to familial forms of diabetes16 and hepatic steatosis,17 respectively.
As APPL1 is a well‐established Rab5 effector, one would assume that interfering with Rab5 would mimic the APPL1 phenotype in relation to insulin sensitivity and glucose production. Surprisingly, depletion of Rab5 in the liver in vivo had no effect on insulin signaling and insulin‐induced AKT activation.8 Nevertheless, we still observed a strong reduction in gluconeogenic genes causing an almost complete block of glucose output, thus inducing hypoglycemia. Usually, reduced blood glucose levels induce glucagon signaling, causing upregulation of gluconeogenesis through cyclic‐AMP‐mediated activation of protein kinase A (PKA). This results in phosphorylation and activation of the transcription factor cAMP responsive binding protein (CREB) and the expression of FOXO1, which function together to drive the gluconeogenic program.105, 110, 111 Despite activated PKA and enhanced cAMP production under Rab5 knockdown, the expression of FOXO1 and the CREB downstream target PPARγ coactivator protein 1α (PGC1α) is strongly diminished, abolishing G6Pase gene expression.8 However, how Rab5 affects CREB and FOXO1 function still needs to be determined. One possibility is that the endosomal membranes could serve as platforms for factors such as FOXO1 to interact with their signaling kinases, which regulate their activity. Upon loss of the endosomal membranes due to Rab5 knockdown,7 those platforms are not available anymore, which actually leads to the observed redistribution of FOXO1 and CREB in the liver.8 Importantly, G6Pase is highly upregulated in type 2 diabetic livers, which contributes to enhanced glucose production and hyperglycemia. Therefore, it is intriguing to note that depletion of Rab5 in diabetic db/db mice can rescue their elevated blood glucose levels by downregulating G6Pase expression,8 highlighting a potential therapeutic application for metabolic diseases.
Further support for a function of early endosomes in metabolic signaling comes from studies interfering with the class III PI3 kinase, Vps34. Although Vps34 is not directly downstream of insulin signaling, knockouts of Vps34 and Vps15 in mice show enhanced insulin sensitivity and glucose tolerance, with reduced hepatic glucose production.112, 113 The effect on glucose production was found to be due to a strong reduction of gluconeogenic genes in hepatocytes, caused by an alteration in insulin‐induced AKT activation.113 However, none of these approaches seem to reduce the number of endosomes, suggesting an alternative mechanism compared to Rab5 knockdown. Altogether, these data highlight unexpected functions of endocytic components on metabolic gene regulation, where the underlying mechanisms still need to be further explored.
Glucose‐stimulated insulin secretion has been shown to depend on EphrinA (EFNA)/EphrinA‐receptor (EPHA) signaling114 and single‐cell transcriptomics of human pancreatic islets revealed that the EPH‐receptor and EFN‐ligand family was significantly enriched.115 In islets of a β‐cell‐specific, inducible intraflagellar Transport 88 (Ift88)‐knockout mouse model, EphA3 is hyperphosphorylated, blocking glucose‐stimulated insulin secretion, resulting in reduced glucose tolerance.116 In these cells, pEphA is not efficiently internalized and trafficked to the perinuclear region where the main protein tyrosine phosphatase is located that suppresses spontaneous, ligand‐independent activation. This represents one example for metabolic defects related to impaired endosomal signal processing.
There are many more metabolic signaling receptors that are important for glucose and lipid homeostasis, such as the GIP receptor (GIPR), GLP1 receptor, leptin receptor, FGF21 co‐receptors FGFR1 and β‐Klotho, where we have yet to determine how they are trafficked through the endosomal system and whether the transport is important for receptor function. These receptors are expressed in many different tissues, such as adipocytes, which could be used to study their trafficking and signaling. In fact, activation of GIPR by ligand binding in 3T3‐L1 adipocytes has been shown to induce a slow recycling of GIPR without changing internalization, resulting in receptor desensitization.117 Genome‐wide association studies (GWAS) have shown that mutations of this receptor are associated with obesity, cardiovascular disease and an increased risk for bone fractures in humans.118, 119, 120 These mutations cause enhanced receptor desensitization and downregulation,117 underscoring how subtle changes in trafficking of GIPR impact human physiology. It will be interesting to see how these mutations affect the behavior of GIPR in vivo in mice and whether the induced metabolic alterations can explain the human correlations with obesity in these GWAS study.
4. THE ROLE OF ENDOSOMAL DEGRADATION IN METABOLIC CONTROL
Degradation is an essential process in the regulation of energy homeostasis. This includes controlling the cellular amount of nutrient receptors or transporters, which are necessary for cargo entry. The degradation of activated signaling receptors is pivotal for signal downregulation to avoid prolonged signaling. In addition, the release of energy stored as macromolecules may require degradation processes through the lysosome.
4.1. Degradation of nutrient transporters/receptors
All cargo that has not been sorted in early endosomes for recycling through multiple recycling and retrieval complexes6 will be sent to lysosomes for degradation. Therefore, these retrieval complexes play essential roles in regulating the expression of nutrient transporters and receptors at the cell surface. We have already reviewed some of those in the section on nutrient uptake, where retromer was important for GLUT1 recycling,53, 54 WASH complex for GLUT2 and LDLR62, 71 and the CCC complex for LDLR and LRP1.10 With so many nutrient transporters and receptors trafficking through the endosomal pathway, it will be interesting to see how their recycling and degradation are regulated, and whether the fate of these cargoes changes in metabolic diseases. One could imagine that enhanced degradation of particular nutrient receptors, possibly through malfunction of the retrieval proteins, could participate in the metabolic imbalance observed in diabetes and obesity.
4.2. Degradation of signaling receptors
As discussed above, the signaling cascades that emerge following receptor‐ligand binding regulate several metabolic functions. Importantly, these signals need to be transient to induce a proper cellular response and avoid continuous activation of cells. The signaling peaks are mechanistically achieved by desensitization of the signaling receptors mainly via their lysosomal degradation. Therefore, perturbing signaling receptor degradation may have an important metabolic impact. Consistent with this possibility, conditional knockout of EPN1 and EPN2 in lymphatic endothelial cells prevent VEGF‐C‐induced vascular endothelial growth factor receptor 3 (VEGFR3) from endocytosis and degradation.121 VEGFR3 is essential for lymphatic vessel growth and lymphangiogenesis, and was found to drop during diabetes.122 Interestingly, the loss of lymphatic‐specific epsins alleviates insufficient lymphangiogenesis in diabetic mice, indicating a novel approach to treating diabetic complications.122
Signaling receptors (mainly receptor tyrosine kinases) subject to degradation are marked by ubiquitination on lysine‐63 and are recognized by the evolutionarily conserved endosomal sorting complexes required for transport (ESCRT) machinery, which drives membrane deformation and vesicle scission to generate cargo‐enriched intraluminal vesicles.123, 124 In theory, the ESCRT machinery should play an essential role in the degradation of metabolically active signaling receptors. Not many studies have elucidated their function in metabolic homeostasis, however, probably also because some components of this machinery are essential for embryonic development, leading to embryonic lethality in whole‐body knockout mice (eg, TSG101, CHMP3, CHMP6, CHMP7) (IMPC). However, one study has highlighted how activation of the ESCRT machinery seems to protect against NAFLD and non‐alcoholic‐steatohepatitis development in mice and monkeys, by targeting toll‐like receptor 4 (TLR4) for degradation.125 This suggests that interfering with ESCRT complexes in mice in vivo will reveal novel roles for the ESCRT machinery in glucose and lipid metabolism.
4.3. Degradation of macromolecules
To keep whole‐body nutrient supplies constant during periods of fasting and feeding, energy stores in the form of fat and glycogen are built up in postprandial periods and mobilized under starvation. Although many endocytic processes are connected to nutrient uptake and storage (see above), the endocytic machinery is also useful for degrading macromolecular components. This includes processes related to lipid droplet and glycogen degradation. In relation to the endocytic system, Rab7 and Vps34, components that are shared between the endosomal and autophagic machinery, have been shown to be required for lipid droplet degradation via autophagy under nutrient starvation.126, 127 Interestingly, knockdown of Vps34 in mouse liver causes hepatomegaly and liver steatosis, underscoring its physiological relevance.127 However, the role of Rab7 in lipophagy has only been investigated in vitro, and further studies in vivo are still required. Interestingly, caveolin1 has also been implicated in liver steatosis and lipid transport, underscoring the diverse function of caveolae in metabolic control.128
Glycogen is another storage macromolecule that is pivotal to maintain blood glucose concentrations (see above). Strikingly, we previously found that depletion of Rab5 in mouse liver leads to a dramatic accumulation of intrahepatocellular glycogen contents.7, 8 How the depletion of Rab5 could perturb glycogen breakdown is puzzling. One explanation is that the strong reduction in G6Pase expression, which is not only the rate‐limiting gene in gluconeogenesis but also in glycogenolysis, could cause accumulation of glycogen. Alternatively, the endo‐lysosomal system, which is abolished in the Rab5 knockdown, could be involved in glycogenolysis. Supporting this possibility, Pompe disease, a lysosomal storage disorder also known as glycogen storage disease type II, is caused by a mutation in the gene encoding the acid α‐glucosidase, which localizes to the lysosome to hydrolyze glycogen to glucose.129 Cytosolic glycogen seems to be targeted to lysosomes in the process of autophagy to achieve its breakdown.130
Altogether, lysosomal degradation is essential in metabolic homeostasis, which has already been well recognized. The components of the endosomal system that initiate the sorting of cargoes for degradation have not been studied intensively in metabolic control. These components are decision drivers that could alter metabolic outcomes by changing the fate of nutrient transporters and metabolic signaling receptors towards degradation or recycling. It would be interesting to see whether metabolic cues influence their expression. In fact, promotor analysis of over 2000 new knockout mouse strains revealed an unknown link between the ESCRT‐III protein CHMP3 and metabolism. CHMP3 was among those candidates, and its promoter contains common regulatory elements functioning as transcription factor binding sites, which are co‐regulated upon metabolic alterations.131 This suggests a differential expression of at least CHMP3 in metabolic diseases. Whether this has functional consequences needs to be elucidated.
5. CONCLUSIONS AND OUTLOOK
In the present review, we have highlighted how endosomal trafficking integrates into metabolic homeostasis and focused on components of the endocytic system with existing mechanistic implications. Besides those, there are still a large number of endocytic factors that influence metabolism but for which we lack an understanding of how and why. This is especially true in invertebrate model organisms, where several genome‐wide screens have been performed to identify genes implicated in either glucose or lipid metabolism. Strikingly, more than 30 endocytic components located all along the different endocytic pathways were found to impact glucose or lipid metabolism in Caenorhabditis elegans, Drosophila melanogaster or Danio rerio 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144 (see Table 1). Unfortunately, as these screens cannot reveal the mechanisms leading to changes in fat mass, glucose storage or catabolism, the metabolic functions of the identified genes remain unclear. Similarly, numerous other components have been found to play a role in mammalian metabolism, but the mechanisms of action are still not well defined145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157 (see Table 1). Thus, despite the large number of endosomal proteins that are now known to participate in metabolic regulation, there are still many for which a metabolic function is implied but not really mechanistically demonstrated. Why is that and what can we do better?
Table 1.
Involvement of endosomal components in metabolism
| Gene (complex) | Organism | Regulation | Metabolic/endocytic effect | Reference |
|---|---|---|---|---|
| Clathrin | Caenorhabditis elegans | RNAi | Increase glucose metabolism | Lee et al132 |
| Human | Functional study and tissue specific o/e | Clathrin heavy chain isoform CHC22 regulate GLUT4 compartment | Vassilopoulos et al15 | |
| AP‐2 | C. elegans | RNAi | Reduction of whole body fat content | Webster et al134 |
| C. elegans | RNAi | Increase glucose metabolism | Lee et al132 | |
| Dab2 | Drosophila melanogaster | RNAi | Reduction of whole body fat content | Pospisilik et al135 |
| Mice | KO | Defect in adipogenesis. Reduction of fat mass | Tao et al145 | |
| Epsin | C. elegans | RNAi | Reduction of whole body fat content. Abnormal lipid metabolism | Ashrafi et al136 |
| Mice | LEC‐iDKO | Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes | Wu et al122 | |
| Dynamin | Primary rat adipocytes | o/e of wt and GTPase def. dynamin2 | Dynamin 2 is necessary for GLUT4 internalization | Al‐Hasani et al26 |
| Human muscle cells | o/e of mutant dynamin 2 | Dynamin 2 mutations disrupted the formation of new actin filaments as well as the stimulus‐induced translocation of GLUT4 to the plasma membrane | González‐Jamett et al9 | |
| Mice | Small molecule, dynamin 2 activator | Pharmacological targeting of actin‐dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models | Schiffer et al27 | |
| Mice | Dynamin 2 deletion in β cells | Dynamin 2 important for second phase insulin secretion | Fan et al65 | |
| HSC70 | D. melanogaster | RNAi | Reduction of whole body fat content | Pospisilik et al135 |
| STZ‐induced diabetic rats | Expression | Abundance of Hsc70 increased by insulin. Downregulation of Hsc70 in diabetic myocardium was secondary to insulin deficiency | Chen et al146 | |
| Caveolin | C. elegans | Null mutant | Reduction of whole body fat content | Han et al137 |
| Mice | Caveolin 1 KO | Caveolin‐1‐deficient mice are lean, resistant to diet‐induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. Caveolin‐1‐deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue | Razani et al93; Cohen et al91 | |
| Mouse liver | Caveolin‐1 function in glucose and lipid metabolism in the liver | Fernandez‐Rojo and Ramm128 | ||
| Mouse adipocytes | Localization | IR is highly enriched in caveolae structures by EM | Gustavsson et al86 | |
| Mouse adipocytes | RNAi by lentivirus | Caveolin‐1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3‐L1 adipocytes | Gonzalez‐Munoz et al89 | |
| Rho A | D. melanogaster | RNAi | Increase of whole body fat content | Baumbach et al138 |
| C. elegans | RNAi | Increase of whole body fat content | Webster et al134 | |
| Mice | Activation | Lipid‐induced muscle insulin resistance through activation of the RhoA/Rho kinase signaling pathway | Tao et al147 | |
| Mice | Inhibitor | Rho‐kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway | Noda et al149 | |
| Rats | Activation through exercise | Short‐term exercise increased insulin sensitivity and glucose tolerance through increased Rock activity and pIRS1 | Muñoz et al150 | |
| CDC42 | D. melanogaster | RNAi | Hyperglycemia | Ugrankar et al139 |
| Human islets | Activation | Restoration of glucose‐stimulated Cdc42‐Pak1 activation and insulin secretion by a selective Epac activator in type 2 diabetic human islets | Veluthakal et al64 | |
| Human islets; mouse islets; mouse pancreatic cell lines | Review | Cdc42: a novel regulator of insulin secretion and diabetes‐associated diseases | Huang et al151 | |
| Wistar rats, STZ and HFD diet | Expression analysis | Increased cardiac expression of Cdc42 and Pak1 in diabetic hearts and in HG‐treated cardiomyocytes | Raut et al148 | |
| Mouse islets | RNAi | Glucose‐stimulated Cdc42 signaling is essential for the second phase of insulin secretion | Wang et al63 | |
| Rab5 | C. elegans | RNAi | Reduction of whole body fat content | Mukhopadhyay et al140; Webster et al134 |
| C. elegans | RNAi | Abnormal fat localization | Mukhopadhyay et al140 | |
| C. elegans | RNAi | Increase glucose metabolism | Lee et al132 | |
| D. melanogaster | RNAi | Hyperglycemia | Ugrankar et al139 | |
| Mice | RNAi liver | Rab5 KD leads to loss of the endo‐lysosomal system and to the accumulation of glycogen | Zeigerer et al7 | |
| Mice | RNAi liver | Regulation of liver metabolism by the endosomal GTPase Rab5. Rab5 KD mimics Van Gierke's disease | Zeigerer et al8 | |
| 3T3‐L1 adipocytes | Transfected with GFP‐Rab5 | Rab5 is necessary for GLUT4 internalization | Huang et al32 | |
| 3T3‐L1 adipocytes | Overexpression of SN (DN) mutant | Rab5 activity regulates GLUT4 sorting | Tessneer et al33 | |
| Mouse liver | Recruitment | PI3K‐C2g is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signaling | Braccini et al152 | |
| VPS34 (PI3K) | C. elegans | RNAi | Alteration of lipid metabolism | Lapierre et al141 |
| Mice | Vps34 floxed mice | Liver‐specific albumin‐Cre; Vps34f/f mice develop hepatomegaly and hepatic steatosis, and impaired protein turnover | Jaber et al127 | |
| Mice | Mutant | Heterozygous Vps34 kinase‐dead mice show alterations in cellular energy metabolism, activating the AMPK pathway in liver and muscle | Bilanges et al112 | |
| Vps15 (PI3K) | C. elegans | RNAi | Reduction of whole body fat content. Abnormal lipid metabolism | Ashrafi et al136 |
| Hepatocellular carcinoma Hepa1.6 cells; primary hepatocytes; Vps15f/f mice | RNAi | Acute and chronic depletion of hepatic Vps15 increases insulin sensitivity and Akt signaling leading to alleviation of the metabolic syndrome in genetic and diet‐induced models of insulin resistance and diabetes | Nemazanyy et al113 | |
| VPS45 | 3T3‐L1 adipocytes | Expression and RNAi | Sorting of GLUT4 into its insulin‐sensitive store requires the Sec1/Munc18 protein mVps45 | Roccisana et al153 |
| Skeletal muscle | o/e and RNAi | Conditional deletion of APPL2 in skeletal muscles enhances insulin sensitivity, leading to an improvement in glucose tolerance | Cheng et al109 | |
| Diabetic patients | Mutants | Loss‐of‐function mutations in APPL1 in familial diabetes mellitus | Prudente et al16 | |
| L6 myoblasts and myotubes, C2C12 myoblasts | o/e and RNAi | APPL1 binds to adiponectin receptors and mediates adiponectin signaling and function | Mao et al104 | |
| Pancreatic islets | APPL1 KO mice | Deletion of the Appl1 gene leads to impairment of both the first and second phases of insulin secretion | Wang et al158 | |
| Rat hepatocytes and liver | o/e and RNAi | APPL1 increases hepatic insulin sensitivity by potentiating insulin‐mediated suppression of the gluconeogenic program | Cheng et al107 | |
| Human livers | SNP analysis | Association of genetic variation in adaptor protein APPL1/APPL2 loci with non‐alcoholic fatty liver disease | Barbieri et al17 | |
| Rab4b | Helicoverpa armigera | RNAi | Decrease glycogen whole body content. Increase the transcription of FOXO | Hou et al142 |
| AP1G1 | C. elegans | RNAi | Reduction of whole body fat content | Webster et al134 |
| Rab35 | 3T3‐L1 adipocytes | o/e of DN Rab35 | TBC1D13 is a Rab35 specific gap that plays an important role in Glut4 trafficking in adipocytes | Davey et al43 |
| Rab11‐FIP3 | D. melanogaster | RNAi | Reduction of whole body fat content | Pospisilik et al135 |
| VPS51 (GARP/EARP) | Danio rerio | Mutant | Defect in glucose homeostasis | Liu et al143 |
| VPS54 (GARP) | C. elegans | RNAi | Reduction of whole body fat content. Abnormal lipid metabolism | Ashrafi et al136 |
| Mice | Spontaneous recessive point mutation in Vps54 | The wobbler mouse is associated with a dysregulation of energy metabolism | Schmitt‐John et al80; Moser et al81 | |
| VPS53 (GARP) | Mouse and human cell lines and mice | RNAi, Vps54 mutant | The GARP complex is involved in intracellular cholesterol transport | Wei et al78 |
| COMMD1 (CCC) | Mice | Whole body KO | Lethal | Van de Sluis159 |
| Mice | Liver‐specific KO | Hepatic copper accumulation and hypercholesterolemia | Vonk et al75; Bartuzi et al71 | |
| Dogs | Whole body deficiency | Hepatic copper storage disorder and hypercholesterolemia | Bartuzi et al71 | |
| COMMD6 and COMMD9 (CCC) | Mice | Liver‐specific KO | Hypercholesterolemia | Fedoseienko et al10 |
| CCDC22 (CCC) | Patients (X‐linked intellectual disability) | Hypomorphic mutation | Hypercholesterolemia | Bartuzi et al71 |
| WASH | Mouse and human cell lines | RNAi | WASH complex is needed to efficiently recycle the nutrient transporters GLUT1 (also known as SLC2A1) and SLC1A4, and potentially many other surface proteins | Kvainickas et al52 |
| Patients, dogs, liver | Liver‐specific KO | CCC‐ and WASH‐mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL | Bartuzi et al71 | |
| Primary hepatocytes, liver, adipocytes | Whole body; liver‐ and adipocyte‐specific Pid1‐deficient mice | The adaptor protein PID1 regulates receptor dependent endocytosis of postprandial triglyceride‐rich lipoproteins, and whole body glucose homeostasis | Fischer et al77; Chen et al76 | |
| WASHC1 (WASH) | Mouse embryonic fibroblast | KO | Impaired LDLR trafficking, reduced LDLR uptake | Bartuzi et al71 |
| Pancreas | Conditional KO | WASH regulates glucose homeostasis by facilitating Glut2 receptor recycling in pancreatic beta cells | Ding et al62 | |
| WASHC5 (WASH) | Patients (RSS) | Mutation | High plasma LDL‐C | Bartuzi et al71 |
| Retromer complex | Human cells | Interaction and infection | PTEN regulates glucose transporter recycling by impairing SNX27 retromer assembly | Shinde and Maddika53 |
| 3T3‐L1 adipocytes | KD | VPS35 (retromer) regulates GLUT4 trafficking | Pan et al162; Yang et al161 | |
| Human renal proximal tubule cells, rat and mouse kidneys | RNAi | Loss of renal SNX5 results in impaired IDE activity and insulin resistance in mice | Li et al160 | |
| COPI | C. elegans | RNAi | Increase glucose metabolism | Lee et al132 |
| VPS33A/B (CORVET /HOPS) | D. melanogaster | RNAi | Increase of whole body fat content | Baranski et al144 |
| Patients | Expression/mutation analysis and patient characterization | Patients with Vps33b mutations in the ARC syndrome show cholestasis, metabolic acidosis, nephrogenic diabetes insipidus, chronic diarrhea, platelet abnormalities, and central nervous system anomalies | Jang et al154 | |
| Mice | Vps33bfl/fl‐AlfpCre KO | Vps33b is crucial for structural and functional hepatocyte polarity and defects of lipid metabolism | Hanley et al155 | |
| VPS16 (CORVET /HOPS) | C. elegans | RNAi | Increase glucose metabolism | Lee et al132 |
| VPS18 (CORVET) | D. melanogaster | RNAi | Reduction of whole body fat content | Baumbach et al138 |
| Rab7 | C. elegans | RNAi | Abnormal lipid metabolism | Ashrafi et al136 |
| C. elegans | RNAi | Reduction of whole body fat content | Mukhopadhyay et al140; Ashrafi et al136 | |
| D. melanogaster | RNAi | Reduction of whole body fat content | Baumbach et al138 | |
| Hepatic cells | RNAi | The Small GTPase Rab7 as a central regulator of hepatocellular lipophagy | Schroeder et al126 | |
| VPS28 (ESCRT I) | D. melanogaster | RNAi | Increase of whole body fat content | Baumbach et al138 |
| D. melanogaster | RNAi | Reduction of whole body fat content | Pospisilik et al135 | |
| Mouse and monkey | Activation through enhanced ubiquitination | Tmbim1 is a multivesicular body regulator that protects against non‐alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4 | Zhao et al125 | |
| Mouse and human cell lines | siRNA against ESCRT‐0 (STAM1), ESCRT‐I (UBAP1), ESCRT‐II (Vps22), or ESCRT‐III (CHMP4C) | The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4‐promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR | Verma and Marchese156 | |
| VPS36 (ESCRT II) | D. melanogaster | RNAi | Increase of whole body fat content | Baumbach et al138 |
| Chmp3 (ESCRT III) | D. melanogaster | RNAi | Increase of whole body fat content | Baumbach et al138 |
| C. elegans | RNAi | Reduction of whole body fat content | Ashrafi et al136 | |
| Rat adipocytes | DN inhibitor constructs | GLUT4 traffic through an ESCRT‐III‐dependent sorting compartment in adipocytes | Koumanov et al157 | |
| Mice | Promotor analysis | Identification of genetic elements in Chmp3 that are associated with metabolic diseases by high‐throughput mouse phenotyping | Rozman et al131 | |
| Chmp4c (ESCRT III) | C. elegans | RNAi | Reduction of whole body fat content | Webster et al134 |
| Mouse and human cell lines | siRNA against ESCRT‐0 (STAM1), ESCRT‐I (UBAP1), ESCRT‐II (Vps22), or ESCRT‐III (CHMP4C) | The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4‐promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR | Verma and Marchese156 | |
| Chmp1 (ESCRT III) | D. melanogaster | RNAi | Increase of whole body fat content | Baumbach et al138 |
| Vps4 | D. melanogaster | RNAi | Reduction of whole body fat content | Baumbach et al138 |
| Rat adipocytes | DN inhibitor constructs | GLUT4 traffic through an ESCRT‐III‐dependent sorting compartment in adipocytes | Koumanov et al157 | |
| VAMP3 | C. elegans | RNAi | Reduction of whole body fat content. Abnormal lipid metabolism | Ashrafi et al136 |
| Exoc6 (exocyst) | D. melanogaster | RNAi | Reduction of whole body fat content | Baumbach et al138 |
| Rab10 | 3T3‐L1 adipocytes | RNAi | SEC16A is a RAB10 effector required for insulin‐stimulated GLUT4 trafficking in adipocytes | Bruno et al45 |
| Mice | Rab10 KO | The small Rab GTPase, Rab10, is required for insulin‐stimulated GLUT4 translocation in cultured 3T3‐L1 adipocytes | Vazirani et al44 | |
| 3T3‐L1 adipocytes | o/e CA Rab10 and RNAi | Rab10, a target of the AS160 Rab GAP, is required for insulin‐stimulated translocation of GLUT4 to the adipocyte plasma membrane | Sano et al41 | |
| Munc18c | Mice | Heterozygous KO | Altered Glut4 translocation in muscle; defect of beta‐cells insulin secretion; severe glucose intolerance | Oh et al48 |
Despite the recent advances in technology to study both endocytosis and metabolism in great detail, there is still little information on how these two modules are connected. These gaps in knowledge can be largely explained by two main limitations in current research that should be addressed. Firstly, endosomal trafficking and metabolism are mainly studied independently of each other, largely due to the complexity of both processes. Therefore, basic scientists and clinicians of even closely related fields often use different terminology and concepts, leading to an impression of speaking different “languages.” This makes the understanding and appreciation of each other's work challenging. Secondly, endosomal transport processes are mainly studied in cells (in vitro systems). Basic knowledge obtained through these in vitro studies is rarely followed up by studies in mouse models and in humans (clinical samples), which hampers the translation of fundamental knowledge on endosomal transport into (patho‐) physiological information and also potential clinical relevance. This is largely explained by the limited number of model systems available to study the endosomal system at an organismal level. Furthermore, a lot of sophisticated technologies are mainly applied to in vitro systems and not to in vivo systems, because of their restrictions, high costs or lack of knowledge regarding how to use them in more complex model systems (eg, organoids and animal models). While the connection between endocytosis and gluco‐lipid metabolism is becoming more evident, fruitful collaborations with other scientists equipped with animal facilities and established primary cell culture systems will enable a more in vivo oriented characterization of endosomal transport in metabolic control. In addition, shared conferences and research consortia could be initiated to bring together scientists from both fields for discussions, exchange and inspiration. Apart from the collaborations that could arise from such initiatives, this would also help to establish a common language and understanding in the field.
There are many open questions that could be addressed following such a strategy. For instance, what is the function of the endosomal system in different metabolically active tissues? We have seen already that adipose and liver tissue fulfill very different functions. Further work should therefore extend and deepen these initial insights. Likewise, what is the function of the endosomal system in cells with minimal direct contribution to whole body energy metabolism? We recently demonstrated that depleting Rab4b in T cells was sufficient to alter adipose tissue function leading to liver steatosis and insulin resistance.34 Is the recycling of cargo through the retriever pathway, as shown for LDLR in the liver, similar in fat? The interesting data on the role of the WASH complex on GLUT2 trafficking opens up the possibility of a potential function of the retriever‐CCC‐WASH complex on GLUT4 in vivo, as has been suggested from adipocyte cell lines.161, 162 Also, is the control of signaling through the endosomal system comparable between muscle and liver? For instance, AKT2 activation induces very different responses in the muscle, where it induces glucose uptake, than in the liver, where it represses gluconeogenesis. The conflicting data on APPL1 and APPL2 in these tissues could be examples of tissue‐specific regulation.
Interestingly, we also presented evidence that components of the endosomal system are altered upon different metabolic challenges and in metabolic diseases, such as type 2 diabetes and NAFLD. Further studies are necessary to elucidate the mechanisms underlying these observations. This would require culturing cells in more physiological conditions (between 5 and 11 mM glucose, which corresponds to 90 and 200 mg/dL blood glucose concentration, respectively) rather than exposing them to extremely high glucose levels of usually 25 mM, which translates into 450 mg/dL blood glucose values, usually only observed in severely diabetic and obese mice. Metabolically active cells will respond, when primed. For instance, studies in yeast have already shown that high glucose stimulations induce the assembly of adapters and clathrin on the TGN and endosomes.163 In addition, expression analysis and functional assays can be performed in human tissues and mouse models of metabolic disease, respectively, to elucidate whether endosomal components and endocytosis functions properly in a diabetic or obese condition. There are many more endosomal components waiting to be studied, where exciting new functions of these components and endocytosis per se in metabolic control can be explored and unraveled. There are exciting times ahead for this emerging field.
ACKNOWLEDGMENTS
We thank B. van de Sluis, P.J. Cullen and J. Klumperman for helpful discussions and critical comments to the review. This work was supported by the DFG Grant ZE1037/1‐1, ZE1037/3‐1, the DZD Grant 920.561‐82DZD0032G, the EFSD Grant 01KU1501C and the BMBF Grant 031L0114A to A.Z., and by INSERM, the Université Cote d'azur and the Young Investigator Program of the ANR to J.G. (ANR18‐CE14‐0035‐01‐GILLERON).
Gilleron J, Gerdes JM, Zeigerer A. Metabolic regulation through the endosomal system. Traffic. 2019;20:552–570. 10.1111/tra.12670
Funding information Agence Nationale de Recherches, Grant/Award Number: ANR18‐CE14‐0035‐01‐GILLERON; Bundesministerium für Bildung und Forschung, Grant/Award Number: 031L0114A; Deutsche Forschungsgemeinschaft, Grant/Award Numbers: ZE1037/1‐1, ZE1037/3‐1; European Foundation for the Study of Diabetes, Grant/Award Number: 01KU1501C; DZD, Grant/Award Number: 920.561‐82DZD0032G
The copyright line for this article was changed on 14 August 2019 after original online publication.
REFERENCES
- 1. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9(2):125‐138. [DOI] [PubMed] [Google Scholar]
- 2. Antonescu CN, McGraw TE, Klip A. Reciprocal regulation of endocytosis and metabolism. Cold Spring Harb Perspect Biol. 2014;6(7):a016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maxfield FR. Role of endosomes and lysosomes in human disease. Cold Spring Harb Perspect Biol. 2014;6(5):a016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mellman I, Yarden Y. Endocytosis and cancer. Cold Spring Harb Perspect Biol. 2013;5(12):a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19(11):679‐696. [DOI] [PubMed] [Google Scholar]
- 6. McNally KE, Cullen PJ. Endosomal retrieval of cargo: Retromer is not alone. Trends Cell Biol. 2018;28(10):807‐822. [DOI] [PubMed] [Google Scholar]
- 7. Zeigerer A, Gilleron J, Bogorad RL, et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature. 2012;485(7399):465‐470. [DOI] [PubMed] [Google Scholar]
- 8. Zeigerer A, Bogorad RL, Sharma K, et al. Regulation of liver metabolism by the endosomal GTPase Rab5. Cell Rep. 2015;11(6):884‐892. [DOI] [PubMed] [Google Scholar]
- 9. Gonzalez‐Jamett AM, Baez‐Matus X, Olivares MJ, et al. Dynamin‐2 mutations linked to centronuclear myopathy impair actin‐dependent trafficking in muscle cells. Sci Rep. 2017;7(1):4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fedoseienko A, Wijers M, Wolters JC, et al. The COMMD family regulates plasma LDL levels and attenuates atherosclerosis through stabilizing the CCC complex in endosomal LDLR trafficking. Circ Res. 2018;122(12):1648‐1660. [DOI] [PubMed] [Google Scholar]
- 11. Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80(4):616‐625. [DOI] [PubMed] [Google Scholar]
- 12. Lizunov VA, Lee JP, Skarulis MC, Zimmerberg J, Cushman SW, Stenkula KG. Impaired tethering and fusion of GLUT4 vesicles in insulin‐resistant human adipose cells. Diabetes. 2013;62(9):3114‐3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High‐fat feeding impairs insulin‐stimulated GLUT4 recruitment via an early insulin‐signaling defect. Diabetes. 1997;46(2):215‐223. [DOI] [PubMed] [Google Scholar]
- 14. Maianu L, Keller SR, Garvey WT. Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin‐regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J Clin Endocrinol Metab. 2001;86(11):5450‐5456. [DOI] [PubMed] [Google Scholar]
- 15. Vassilopoulos S, Esk C, Hoshino S, et al. A role for the CHC22 clathrin heavy‐chain isoform in human glucose metabolism. Science. 2009;324(5931):1192‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prudente S, Jungtrakoon P, Marucci A, et al. Loss‐of‐function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. 2015;97(1):177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barbieri M, Esposito A, Angellotti E, Rizzo MR, Marfella R, Paolisso G. Association of genetic variation in adaptor protein APPL1/APPL2 loci with non‐alcoholic fatty liver disease. PLoS One. 2013;8(8):e71391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med. 2012;18(9):524‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475‐2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yao Y, Jones E, Inoki K. Lysosomal regulation of mTORC1 by amino acids in mammalian cells. Biomolecules. 2017;7(3):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaldin‐Fincati JR, Pavarotti M, Frendo‐Cumbo S, Bilan PJ, Klip A. Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol Metab. 2017;28(8):597‐611. [DOI] [PubMed] [Google Scholar]
- 22. Stockli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124(pt 24):4147‐4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13(6):383‐396. [DOI] [PubMed] [Google Scholar]
- 24. Foley K, Boguslavsky S, Klip A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry. 2011;50(15):3048‐3061. [DOI] [PubMed] [Google Scholar]
- 25. Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem. 2012;81:507‐532. [DOI] [PubMed] [Google Scholar]
- 26. Al‐Hasani H, Hinck CS, Cushman SW. Endocytosis of the glucose transporter GLUT4 is mediated by the GTPase dynamin. J Biol Chem. 1998;273(28):17504‐17510. [DOI] [PubMed] [Google Scholar]
- 27. Schiffer M, Teng B, Gu C, et al. Pharmacological targeting of actin‐dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat Med. 2015;21(6):601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cormont M, Gautier N, Ilc K, le Marchand‐Brustel Y. Expression of a prenylation‐deficient Rab4 inhibits the GLUT4 translocation induced by active phosphatidylinositol 3‐kinase and protein kinase B. Biochem J. 2001;356(pt 1):143‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vollenweider P, Martin SS, Haruta T, et al. The small guanosine triphosphate‐binding protein Rab4 is involved in insulin‐induced GLUT4 translocation and actin filament rearrangement in 3T3‐L1 cells. Endocrinology. 1997;138(11):4941‐4949. [DOI] [PubMed] [Google Scholar]
- 30. Cormont M, Bortoluzzi MN, Gautier N, Mari M, van Obberghen E, Le Marchand‐Brustel Y. Potential role of Rab4 in the regulation of subcellular localization of Glut4 in adipocytes. Mol Cell Biol. 1996;16(12):6879‐6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaddai V, Gonzalez T, Keslair F, et al. Rab4b is a small GTPase involved in the control of the glucose transporter GLUT4 localization in adipocyte. PLoS One. 2009;4(4):e5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang J, Imamura T, Olefsky JM. Insulin can regulate GLUT4 internalization by signaling to Rab5 and the motor protein dynein. Proc Natl Acad Sci U S A. 2001;98(23):13084‐13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tessneer KL, Jackson RM, Griesel BA, Olson AL. Rab5 activity regulates GLUT4 sorting into insulin‐responsive and non‐insulin‐responsive endosomal compartments: a potential mechanism for development of insulin resistance. Endocrinology. 2014;155(9):3315‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilleron J, Bouget G, Ivanov S, et al. Rab4b deficiency in T cells promotes adipose Treg/Th17 imbalance, adipose tissue dysfunction, and insulin resistance. Cell Rep. 2018;25(12):3329‐3341.e3325. [DOI] [PubMed] [Google Scholar]
- 35. Zeigerer A, Lampson MA, Karylowski O, et al. GLUT4 retention in adipocytes requires two intracellular insulin‐regulated transport steps. Mol Biol Cell. 2002;13(7):2421‐2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takata K, Matsuzaki T, Tajika Y, Ablimit A, Hasegawa T. Localization and trafficking of aquaporin 2 in the kidney. Histochem Cell Biol. 2008;130(2):197‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L, Boyer JL. The maintenance and generation of membrane polarity in hepatocytes. Hepatology. 2004;39(4):892‐899. [DOI] [PubMed] [Google Scholar]
- 38. Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci U S A. 2010;107(46):19909‐19914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klip A, Sun Y, Chiu TT, Foley KP. Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am J Physiol Cell Physiol. 2014;306(10):C879‐C886. [DOI] [PubMed] [Google Scholar]
- 40. Li H, Ou L, Fan J, et al. Rab8A regulates insulin‐stimulated GLUT4 translocation in C2C12 myoblasts. FEBS Lett. 2017;591(3):491‐499. [DOI] [PubMed] [Google Scholar]
- 41. Sano H, Eguez L, Teruel MN, et al. Rab10, a target of the AS160 Rab GAP, is required for insulin‐stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5(4):293‐303. [DOI] [PubMed] [Google Scholar]
- 42. Chen Y, Lippincott‐Schwartz J. Insulin triggers surface‐directed trafficking of sequestered GLUT4 storage vesicles marked by Rab10. Small GTPases. 2013;4(3):193‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davey JR, Humphrey SJ, Junutula JR, et al. TBC1D13 is a RAB35 specific GAP that plays an important role in GLUT4 trafficking in adipocytes. Traffic. 2012;13(10):1429‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vazirani RP, Verma A, Sadacca LA, et al. Disruption of adipose Rab10‐dependent insulin signaling causes hepatic insulin resistance. Diabetes. 2016;65(6):1577‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bruno J, Brumfield A, Chaudhary N, Iaea D, McGraw TE. SEC16A is a RAB10 effector required for insulin‐stimulated GLUT4 trafficking in adipocytes. J Cell Biol. 2016;214(1):61‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramalingam L, Yoder SM, Oh E, Thurmond DC. Munc18c: a controversial regulator of peripheral insulin action. Trends Endocrinol Metab. 2014;25(11):601‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tamori Y, Kawanishi M, Niki T, et al. Inhibition of insulin‐induced GLUT4 translocation by Munc18c through interaction with syntaxin4 in 3T3‐L1 adipocytes. J Biol Chem. 1998;273(31):19740‐19746. [DOI] [PubMed] [Google Scholar]
- 48. Oh E, Spurlin BA, Pessin JE, Thurmond DC. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes. 2005;54(3):638‐647. [DOI] [PubMed] [Google Scholar]
- 49. Klumperman J, Raposo G. The complex ultrastructure of the endolysosomal system. Cold Spring Harb Perspect Biol. 2014;6(10):a016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van de Sluis B, Wijers M, Herz J. News on the molecular regulation and function of hepatic low‐density lipoprotein receptor and LDLR‐related protein 1. Curr Opin Lipidol. 2017;28(3):241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steinberg F, Gallon M, Winfield M, et al. A global analysis of SNX27‐retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15(5):461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kvainickas A, Orgaz AJ, Nagele H, et al. Retromer‐ and WASH‐dependent sorting of nutrient transporters requires a multivalent interaction network with ANKRD50. J Cell Sci. 2017;130(2):382‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shinde SR, Maddika S. PTEN regulates glucose transporter recycling by impairing SNX27 retromer assembly. Cell Rep. 2017;21(6):1655‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. Autophagy‐dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell. 2017;67(1):84‐95.e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang J, Fedoseienko A, Chen B, Burstein E, Jia D, Billadeau DD. Endosomal receptor trafficking: retromer and beyond. Traffic. 2018;19(8):578‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wen L, Tang FL, Hong Y, et al. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol. 2011;195(5):765‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58(2):221‐232. [DOI] [PubMed] [Google Scholar]
- 58. McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab. 2011;104(4):648‐653. [DOI] [PubMed] [Google Scholar]
- 59. De Vos A, Heimberg H, Quartier E, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest. 1995;96(5):2489‐2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matschinsky FM, Ellerman JE. Metabolism of glucose in the Islets of Langerhans. J Biol Chem. 1968;243(10):2730‐2736. [PubMed] [Google Scholar]
- 61. Yang SN, Berggren PO. The role of voltage‐gated calcium channels in pancreatic beta‐cell physiology and pathophysiology. Endocr Rev. 2006;27(6):621‐676. [DOI] [PubMed] [Google Scholar]
- 62. Ding L, Han L, Dube J, Billadeau DD. WASH regulates glucose homeostasis by facilitating Glut2 receptor recycling in pancreatic beta‐cells. Diabetes. 2019;68(2):377‐386. [DOI] [PubMed] [Google Scholar]
- 63. Wang Z, Oh E, Thurmond DC. Glucose‐stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem. 2007;282(13):9536‐9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Veluthakal R, Chepurny OG, Leech CA, Schwede F, Holz GG, Thurmond DC. Restoration of glucose‐stimulated Cdc42‐Pak1 activation and insulin secretion by a selective Epac activator in type 2 diabetic human islets. Diabetes. 2018;67(10):1999‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fan F, Ji C, Wu Y, et al. Dynamin 2 regulates biphasic insulin secretion and plasma glucose homeostasis. J Clin Invest. 2015;125(11):4026‐4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129‐157. [DOI] [PubMed] [Google Scholar]
- 67. Boyer JL. Bile formation and secretion. Compr Physiol. 2013;3(3):1035‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5(2):121‐132. [DOI] [PubMed] [Google Scholar]
- 69. Scotti E, Calamai M, Goulbourne CN, et al. IDOL stimulates clathrin‐independent endocytosis and multivesicular body‐mediated lysosomal degradation of the low‐density lipoprotein receptor. Mol Cell Biol. 2013;33(8):1503‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gilleron J, Querbes W, Zeigerer A, et al. Image‐based analysis of lipid nanoparticle‐mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638‐646. [DOI] [PubMed] [Google Scholar]
- 71. Bartuzi P, Billadeau DD, Favier R, et al. CCC‐ and WASH‐mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL. Nat Commun. 2016;7:10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11(2):165‐173. [DOI] [PubMed] [Google Scholar]
- 73. Maine GN, Burstein E. COMMD proteins: COMMing to the scene. Cell Mol Life Sci. 2007;64(15):1997‐2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Phillips‐Krawczak CA, Singla A, Starokadomskyy P, et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol Biol Cell. 2015;26(1):91‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vonk WI, Bartuzi P, de Bie P, et al. Liver‐specific Commd1 knockout mice are susceptible to hepatic copper accumulation. PLoS One. 2011;6(12):e29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen L, Wang XY, Zhu JG, et al. PID1 in adipocytes modulates whole‐body glucose homeostasis. Biochim Biophys Acta Gene Regul Mech. 2018;1861(2):125‐132. [DOI] [PubMed] [Google Scholar]
- 77. Fischer AW, Albers K, Krott LM, et al. The adaptor protein PID1 regulates receptor‐dependent endocytosis of postprandial triglyceride‐rich lipoproteins. Mol Metab. 2018;16:88‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei J, Zhang YY, Luo J, et al. The GARP complex is involved in intracellular cholesterol transport via targeting NPC2 to lysosomes. Cell Rep. 2017;19(13):2823‐2835. [DOI] [PubMed] [Google Scholar]
- 79. Spang A. Membrane tethering complexes in the endosomal system. Front Cell Dev Biol. 2016;4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schmitt‐John T, Drepper C, Mussmann A, et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37(11):1213‐1215. [DOI] [PubMed] [Google Scholar]
- 81. Moser JM, Bigini P, Schmitt‐John T. The wobbler mouse, an ALS animal model. Mol Genet Genomics. 2013;288(5–6):207‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling‐more than just a ligand‐binding receptor. Front Endocrinol (Lausanne). 2017;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bergeron JJ, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI. Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu Rev Biochem. 2016;85:573‐597. [DOI] [PubMed] [Google Scholar]
- 84. Paccaud JP, Siddle K, Carpentier JL. Internalization of the human insulin receptor. The insulin‐independent pathway. J Biol Chem. 1992;267(18):13101‐13106. [PubMed] [Google Scholar]
- 85. Fan JY, Carpentier JL, Gorden P, et al. Receptor‐mediated endocytosis of insulin: role of microvilli, coated pits, and coated vesicles. Proc Natl Acad Sci U S A. 1982;79(24):7788‐7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gustavsson J, Parpal S, Karlsson M, et al. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999;13(14):1961‐1971. [PubMed] [Google Scholar]
- 87. Fagerholm S, Ortegren U, Karlsson M, Ruishalme I, Stralfors P. Rapid insulin‐dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS One. 2009;4(6):e5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG Jr, Ishikawa Y. Caveolin is an activator of insulin receptor signaling. J Biol Chem. 1998;273(41):26962‐26968. [DOI] [PubMed] [Google Scholar]
- 89. Gonzalez‐Munoz E, Lopez‐Iglesias C, Calvo M, Palacin M, Zorzano A, Camps M. Caveolin‐1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3‐L1 adipocytes. Endocrinology. 2009;150(8):3493‐3502. [DOI] [PubMed] [Google Scholar]
- 90. Regazzetti C, Peraldi P, Gremeaux T, et al. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes. 2009;58(1):95‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cohen AW, Razani B, Wang XB, et al. Caveolin‐1‐deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003;285(1):C222‐C235. [DOI] [PubMed] [Google Scholar]
- 92. Oshikawa J, Otsu K, Toya Y, et al. Insulin resistance in skeletal muscles of caveolin‐3‐null mice. Proc Natl Acad Sci U S A. 2004;101(34):12670‐12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Razani B, Combs TP, Wang XB, et al. Caveolin‐1‐deficient mice are lean, resistant to diet‐induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277(10):8635‐8647. [DOI] [PubMed] [Google Scholar]
- 94. Jensen M, De Meyts P. Molecular mechanisms of differential intracellular signaling from the insulin receptor. Vitam Horm. 2009;80:51‐75. [DOI] [PubMed] [Google Scholar]
- 95. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;19(1):31‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schmidt V, Schulz N, Yan X, et al. SORLA facilitates insulin receptor signaling in adipocytes and exacerbates obesity. J Clin Invest. 2016;126(7):2706‐2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Authier F, Desbuquois B. Glucagon receptors. Cell Mol Life Sci. 2008;65(12):1880‐1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Watanabe J, Kanamura S, Asada‐Kubota M, Kanai K, Oka M. Receptor‐mediated endocytosis of glucagon in isolated mouse hepatocytes. Anat Rec. 1984;210(4):557‐567. [DOI] [PubMed] [Google Scholar]
- 99. Krilov L, Nguyen A, Miyazaki T, et al. Dual mode of glucagon receptor internalization: role of PKCalpha, GRKs and beta‐arrestins. Exp Cell Res. 2011;317(20):2981‐2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116(3):445‐456. [DOI] [PubMed] [Google Scholar]
- 101. Villasenor R, Nonaka H, Del Conte‐Zerial P, Kalaidzidis Y, Zerial M. Regulation of EGFR signal transduction by analogue‐to‐digital conversion in endosomes. Elife. 2015;4:1‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schenck A, Goto‐Silva L, Collinet C, et al. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133(3):486‐497. [DOI] [PubMed] [Google Scholar]
- 103. Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol. 2016;8(2):101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8(5):516‐523. [DOI] [PubMed] [Google Scholar]
- 105. Yabaluri N, Bashyam MD. Hormonal regulation of gluconeogenic gene transcription in the liver. J Biosci. 2010;35(3):473‐484. [DOI] [PubMed] [Google Scholar]
- 106. Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14(1):9‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cheng KK, Iglesias MA, Lam KS, et al. APPL1 potentiates insulin‐mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab. 2009;9(5):417‐427. [DOI] [PubMed] [Google Scholar]
- 108. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574‐1577. [DOI] [PubMed] [Google Scholar]
- 109. Cheng KK, Zhu W, Chen B, et al. The adaptor protein APPL2 inhibits insulin‐stimulated glucose uptake by interacting with TBC1D1 in skeletal muscle. Diabetes. 2014;63(11):3748‐3758. [DOI] [PubMed] [Google Scholar]
- 110. Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC‐1. Nature. 2001;413(6852):179‐183. [DOI] [PubMed] [Google Scholar]
- 111. Montminy MR, Gonzalez GA, Yamamoto KK. Characteristics of the cAMP response unit. Metabolism. 1990;39(9, suppl 2):6‐12. [DOI] [PubMed] [Google Scholar]
- 112. Bilanges B, Alliouachene S, Pearce W, et al. Vps34 PI 3‐kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat Commun. 2017;8(1):1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nemazanyy I, Montagnac G, Russell RC, et al. Class III PI3K regulates organismal glucose homeostasis by providing negative feedback on hepatic insulin signalling. Nat Commun. 2015;6:8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Konstantinova I, Nikolova G, Ohara‐Imaizumi M, et al. EphA‐Ephrin‐A‐mediated β cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129(2):359‐370. [DOI] [PubMed] [Google Scholar]
- 115. Dorrell C, Schug J, Lin CF, et al. Transcriptomes of the major human pancreatic cell types. Diabetologia. 2011;54(11):2832‐2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Volta F, Scerbo MJ, Seelig A, et al. Glucose homeostasis is regulated by pancreatic beta‐cell cilia via endosomal EphA‐processing. Nat Commun. 2019. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Abdullah N, Beg M, Soares D, Dittman JS, McGraw TE. Downregulation of a GPCR by beta‐Arrestin2‐mediated switch from an endosomal to a TGN recycling pathway. Cell Rep. 2016;17(11):2966‐2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nitz I, Fisher E, Weikert C, et al. Association analyses of GIP and GIPR polymorphisms with traits of the metabolic syndrome. Mol Nutr Food Res. 2007;51(8):1046‐1052. [DOI] [PubMed] [Google Scholar]
- 119. Sauber J, Grothe J, Behm M, et al. Association of variants in gastric inhibitory polypeptide receptor gene with impaired glucose homeostasis in obese children and adolescents from Berlin. Eur J Endocrinol. 2010;163(2):259‐264. [DOI] [PubMed] [Google Scholar]
- 120. Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42(2):142‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pasula S, Cai X, Dong Y, et al. Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest. 2012;122(12):4424‐4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wu H, Rahman HNA, Dong Y, et al. Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J Clin Invest. 2018;128(9):4025‐4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. 2013;5(9):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Szymanska E, Budick‐Harmelin N, Miaczynska M. Endosomal "sort" of signaling control: the role of ESCRT machinery in regulation of receptor‐mediated signaling pathways. Semin Cell Dev Biol. 2018;74:11‐20. [DOI] [PubMed] [Google Scholar]
- 125. Zhao GN, Zhang P, Gong J, et al. Tmbim1 is a multivesicular body regulator that protects against non‐alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4. Nat Med. 2017;23(6):742‐752. [DOI] [PubMed] [Google Scholar]
- 126. Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61(6):1896‐1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109(6):2003‐2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fernandez‐Rojo MA, Ramm GA. Caveolin‐1 function in liver physiology and disease. Trends Mol Med. 2016;22(10):889‐904. [DOI] [PubMed] [Google Scholar]
- 129. Raben N, Plotz P, Byrne BJ. Acid alpha‐glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med. 2002;2(2):145‐166. [DOI] [PubMed] [Google Scholar]
- 130. Zirin J, Nieuwenhuis J, Perrimon N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol. 2013;11(11):e1001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Rozman J, Rathkolb B, Oestereicher MA, et al. Identification of genetic elements in metabolism by high‐throughput mouse phenotyping. Nat Commun. 2018;9(1):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lee D, Jeong DE, Son HG, et al. SREBP and MDT‐15 protect C. elegans from glucose‐induced accelerated aging by preventing accumulation of saturated fat. Genes Dev. 2015;29(23):2490‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lee HC, Kubo T, Kono N, et al. Depletion of mboa‐7, an enzyme that incorporates polyunsaturated fatty acids into phosphatidylinositol (PI), impairs PI 3‐phosphate signaling in Caenorhabditis elegans . Genes Cells. 2012;17(9):748‐757. [DOI] [PubMed] [Google Scholar]
- 134. Webster CM, Pino EC, Carr CE, et al. Genome‐wide RNAi screen for fat regulatory genes in C. elegans identifies a Proteostasis‐AMPK axis critical for starvation survival. Cell Rep. 2017;20(3):627‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pospisilik JA, Schramek D, Schnidar H, et al. Drosophila genome‐wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140(1):148‐160. [DOI] [PubMed] [Google Scholar]
- 136. Ashrafi K, Chang FY, Watts JL, et al. Genome‐wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421(6920):268‐272. [DOI] [PubMed] [Google Scholar]
- 137. Han M, Chang H, Zhang P, et al. C13C4.5/spinster, an evolutionarily conserved protein that regulates fertility in C. elegans through a lysosome‐mediated lipid metabolism process. Protein Cell. 2013;4(5):364‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Baumbach J, Hummel P, Bickmeyer I, et al. A Drosophila in vivo screen identifies store‐operated calcium entry as a key regulator of adiposity. Cell Metab. 2014;19(2):331‐343. [DOI] [PubMed] [Google Scholar]
- 139. Ugrankar R, Berglund E, Akdemir F, et al. Drosophila glucome screening identifies Ck1alpha as a regulator of mammalian glucose metabolism. Nat Commun. 2015;6:7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mukhopadhyay A, Pan X, Lambright DG, Tissenbaum HA. An endocytic pathway as a target of tubby for regulation of fat storage. EMBO Rep. 2007;8(10):931‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lapierre LR, Gelino S, Melendez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline‐less C. elegans . Curr Biol. 2011;21(18):1507‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hou L, Cai MJ, Liu W, Song Q, Zhao XF. Small GTPase Rab4b participates in the gene transcription of 20‐hydroxyecdysone and insulin pathways to regulate glycogen level and metamorphosis. Dev Biol. 2012;371(1):13‐22. [DOI] [PubMed] [Google Scholar]
- 143. Liu HY, Lee N, Tsai TY, Ho SY. Zebrafish fat‐free, a novel Arf effector, regulates phospholipase D to mediate lipid and glucose metabolism. Biochim Biophys Acta. 2010;1801(12):1330‐1340. [DOI] [PubMed] [Google Scholar]
- 144. Baranski TJ, Kraja AT, Fink JL, et al. A high throughput, functional screen of human body mass index GWAS loci using tissue‐specific RNAi Drosophila melanogaster crosses. PLoS Genet. 2018;14(4):e1007222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tao W, Moore R, Meng Y, Yeasky TM, Smith ER, Xu XX. Disabled‐2 determines commitment of a pre‐adipocyte population in juvenile mice. Sci Rep. 2016;6:35947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chen HS, Jia J, Su HF, et al. Downregulation of the constitutively expressed Hsc70 in diabetic myocardium is mediated by insulin deficiency. J Endocrinol. 2006;190(2):433‐440. [DOI] [PubMed] [Google Scholar]
- 147. Tao W, Wu J, Xie BX, et al. Lipid‐induced muscle insulin resistance is mediated by GGPPS via modulation of the RhoA/rho kinase signaling pathway. J Biol Chem. 2015;290(33):20086‐20097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Raut SK, Kumar A, Singh GB, et al. miR‐30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther. 2015;33(3):89‐97. [DOI] [PubMed] [Google Scholar]
- 149. Noda K, Nakajima S, Godo S, et al. Rho‐kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS One. 2014;9(11):e110446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Munoz VR, Gaspar RC, Kuga GK, et al. Exercise increases rho‐kinase activity and insulin signaling in skeletal muscle. J Cell Physiol. 2018;233(6):4791‐4800. [DOI] [PubMed] [Google Scholar]
- 151. Huang QY, Lai XN, Qian XL, et al. Cdc42: a novel regulator of insulin secretion and diabetes‐associated diseases. Int J Mol Sci. 2019;20(1):1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Braccini L, Ciraolo E, Campa CC, et al. PI3K‐C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat Commun. 2015;6:7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Roccisana J, Sadler JB, Bryant NJ, Gould GW. Sorting of GLUT4 into its insulin‐sensitive store requires the Sec1/Munc18 protein mVps45. Mol Biol Cell. 2013;24(15):2389‐2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Jang JY, Kim KM, Kim GH, et al. Clinical characteristics and VPS33B mutations in patients with ARC syndrome. J Pediatr Gastroenterol Nutr. 2009;48(3):348‐354. [DOI] [PubMed] [Google Scholar]
- 155. Hanley J, Dhar DK, Mazzacuva F, et al. Vps33b is crucial for structural and functional hepatocyte polarity. J Hepatol. 2017;66(5):1001‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Verma R, Marchese A. The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4‐promoted lysosomal degradation of the mammalian target of rapamycin antagonist DEPTOR. J Biol Chem. 2015;290(11):6810‐6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Koumanov F, Pereira VJ, Whitley PR, Holman GD. GLUT4 traffic through an ESCRT‐III‐dependent sorting compartment in adipocytes. PLoS One. 2012;7(9):e44141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wang C, Li X, Mu K, Li L, et al. Deficiency of APPL1 in mice impairs glucose‐stimulated insulin secretion through inhibition of pancreatic beta cell mitochondrial function. Diabetologia 2013;56(9):1999‐2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. van de Sluis B, Muller P, Duran K, et al. Increased activity of hypoxia‐inducible factor 1 is associated with early embryonic lethality in Commd1 null mice. Mol Cell Biol. 2007;27(11):4142‐4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Li F, Yang J, Villar VAM, et al. Loss of renal SNX5 results in impaired IDE activity and insulin resistance in mice. Diabetologia 2018;61(3):727‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Yang Z, Hong LK, Follett J, et al. Functional characterization of retromer in GLUT4 storage vesicle formation and adipocyte differentiation. FASEB J. 2016;30(3):1037‐1050. [DOI] [PubMed] [Google Scholar]
- 162. Pan X, Zaarur N, Singh M, Morin P, Kandror KV. Sortilin and retromer mediate retrograde transport of Glut4 in 3T3‐L1 adipocytes. Mol Biol Cell. 2017;28(12):1667‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Aoh QL, Hung CW, Duncan MC. Energy metabolism regulates clathrin adaptors at the trans‐Golgi network and endosomes. Mol Biol Cell. 2013;24(6):832‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]