

Abstract
Aims
Enteropeptidase is a serine protease localized on the duodenal brush border that catalyzes the conversion of inactive trypsinogen into active trypsin, thereby regulating protein breakdown in the gut. We evaluated the effects of SCO‐792, a novel enteropeptidase inhibitor, in mice.
Materials and methods
In vivo inhibition of enteropeptidase was evaluated via an oral protein challenge. Pharmacological effects were evaluated in normal mice, in diet‐induced obese (DIO) mice and in obese and diabetic ob/ob mice.
Results
A single oral administration of SCO‐792 inhibited plasma branched‐chain amino acids (BCAAs) in an oral protein challenge test in mice, indicating in vivo inhibition of enteropeptidase. Repeated treatment with SCO‐792 induced reduction in food intake and decrease in body weight in DIO and ob/ob mice. Plasma FGF21 levels were increased in SCO‐792‐treated DIO mice, an observation that was probably independent of reduction in food intake. Hyperglycaemia was markedly improved in SCO‐792‐treated ob/ob mice. A hyperinsulinaemic‐euglycaemic clamp study revealed improved muscle insulin sensitivity in SCO‐792‐treated ob/ob mice. SCO‐792 also improved plasma and liver lipid profiles and decreased plasma alanine transaminase, suggesting a potential treatment for liver diseases. Dietary supplementation with essential amino acids attenuated the effect of SCO‐792 on reduction in food intake and decrease in body weight in normal mice, suggesting a pivotal role for enteropeptidase in these biological phenomena.
Conclusions
SCO‐792 inhibited enteropeptidase in vivo, reduced food intake, decreased body weight, increased insulin sensitivity, improved glucose and lipid control, and ameliorated liver parameters in mouse models with obesity and/or diabetes. SCO‐792 may exhibit similar effects in patients.
Keywords: animal pharmacology, diabetes, drug development, mouse model, obesity therapy
1. INTRODUCTION
Obesity is an abnormal condition with excessive body fat accumulation, mostly the result of a chronic imbalance between energy intake and energy expenditure; it is a primary factor in all obesity‐related complications such as type 2 diabetes, dyslipidaemia and cardiovascular diseases.1, 2, 3, 4 Although individual and genetic factors influence its onset and severity, the major causative factor for obesity is excessive calorie intake from the three macronutrients, fat, carbohydrate and protein, which provide nine, four and four calories per gram, respectively.5, 6
From the therapeutic perspective, researchers have developed inhibitors to block gut nutrient absorption, thereby inducing treatment effects for various diseases. For instance, pancreatic and intestinal lipase inhibitors, which inhibit fat breakdown in the gut, were developed and used clinically as a potential treatment for dyslipidaemia and obesity.7 Similarly, alpha‐glucosidase inhibitors are well‐known anti‐diabetic drugs that inhibit the digestion of carbohydrates, such as starch and sugar, thereby blocking glucose uptake in the gut.8 These approaches are reasonable for patients who over ingest foods with high fat and/or high carbohydrate content, but the mechanism‐driven gastrointestinal side‐effects, including oily spotting, fecal incontinence and diarrhea, limit patient compliance.9, 10 Although proteins contain the same number of calories as carbohydrates, drugs that inhibit dietary protein absorption, unlike carbohydrate and fat absorption, have not been developed for treating metabolic diseases.
Enteropeptidase (enterokinase, EC3.4.21.9) is a transmembrane serine protease, localized at the brush border of the duodenal and jejunal mucosa, which is involved in food digestion in mammals.11, 12, 13 Enteropeptidase converts inactive trypsinogen into its active form, trypsin, resulting in the subsequent activation of digestive enzyme precursors produced in the pancreas (eg, chymotrypsinogen, proelastase and procarboxypeptidases A and B).11, 12, 13 These activated enzymes facilitate protein breakdown, resulting in amino acid absorption in the gut.11, 12, 13 Notably, a congenital enteropeptidase deficiency is known to be associated with a lean phenotype in humans.14, 15 In addition, it has been reported that OBE‐2008, an enteropeptidase inhibitor, inhibited body weight gain during a 2‐month study involving the growth phase in DIO mice.15 This suggests that enteropeptidase inhibition may constitute a novel strategy to improve body weight control in obesity. However, the pharmacological effects of enteropeptidase inhibition on food intake, body weight and metabolic parameters are not well characterized. SCO‐792 is a novel, orally available enteropeptidase inhibitor that is currently undergoing evaluation in clinical trials.16 It is reported to show potent inhibitory activity against human enteropeptidase when tested in vitro (complete enteropeptidase inhibition at 3.3 μM SCO‐792 in vitro).16
Here, we evaluated the therapeutic effects of SCO‐792 on obesity and diabetes in mouse models. First, we evaluated the plasma biomarker changes induced by SCO‐792. Next, the body weight lowering effects of SCO‐792 were evaluated in diet‐induced obese (DIO) mice. The metabolic effects of SCO‐792 were evaluated in genetically obese and diabetic mice. Additional experiments were performed to reveal effects of SCO‐792 on FGF21 levels and whole body insulin sensitivity, and to elucidate a role of essential amino acid intake on SCO‐792‐induced food intake and body weight reduction in mouse models.
2. MATERIALS AND METHODS
2.1. Chemicals
All reagents were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) or Sigma‐Aldrich (Tokyo, Japan) unless otherwise indicated. SCO‐792 and pioglitazone were synthesized by Takeda Pharmaceutical Company Limited (Tokyo, Japan). Sibutramine was purchased from Enzo Life Sciences, Inc (Farmingdale, New York). Doses of compounds are expressed in free base form.
2.2. Animals
All animals were housed in a room with controlled temperature (23°C), humidity (55%) and lighting (illumination between 7:00 am and 7:00 pm). The care of the animals and use of the experimental protocols in the current studies were approved by the Takeda Institutional Animal Care and Use Committee (IACUC) in a facility accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC). All animals were obtained from Charles River Laboratories (Yokohama, Japan). The DIO model was established by using a high‐fat diet (HFD) (41 kcal% from fat, 17 kcal% from protein and 43 kcal% from carbohydrate; reference: D12079B; Research Diets, Inc., New Brunswick, New Jeersey) for male C57BL/6J mice. Male ob/ob mice (B6.Cg‐Lep ob/J, genotype: Lep ob/Lep ob) and their untyped non‐diabetic ?/+ mice (B6.Cg‐Lep ob/J, genotype: Lep ob /+ or +/+) were maintained using a laboratory chow diet (CE‐2 powder; 25% kcals from protein) (CLEA, Tokyo, Japan). Male C57BL/6J mice were maintained using a laboratory chow diet (CE‐2). Throughout all animal studies, animal randomization was performed to keep conditions identical in each group.
2.3. Oral protein challenge test in DIO mice
DIO mice that received the D12079B HFD diet from 6 weeks of age, with a baseline body weight of 53.5 ± 3.7 g, were used throughout the study until approximately 64 weeks of age. Mice were randomized into three groups (n = 5) based on body weight and body weight changes, assessed 1 day prior to initiation of the experiment. Non‐fasted mice received orally either SCO‐792 (10 and 30 mg/kg) or vehicle (0.5% [w/v] methylcellulose) 6 hours before an oral protein challenge (2.5 g/kg; SAVAS whey protein 100) (Meiji, Tokyo, Japan). Blood samples were collected at the time points indicated in the Results section and in the associated Figures. Plasma branched‐chain amino acid (BCAA) levels were determined.
2.4. Repeated dosing in DIO mice
The DIO model was established by using an HFD (D12079B) from 5 weeks of age until initiation of the study. Male DIO mice, 46 weeks of age, with a baseline body weight of 49.3 ± 2.9 g, were divided into four groups (n = 6 for vehicle and SCO‐792; n = 4 for sibutramine) based on plasma glucose, triglyceride, cholesterol, alanine transaminase, body fat and lean mass, food intake and body weight. Mice received orally either SCO‐792 (20 and 59 mg/kg), sibutramine (10 mg/kg) or vehicle (0.5% [w/v] methylcellulose) once daily at approximately 5:00 to 6:00 pm and received an HFD (D12079B) for 4 weeks (first treatment day designated as Day 0). Sibutramine was selected as a control drug, as this agent was demonstrated to be effective in both humans17 and mice (data not shown) within similar dosage levels. Food intake and body weight were measured regularly. Plasma glucose, triglyceride, cholesterol, alanine transaminase and insulin levels, as well as body composition, were determined at Day 28. Plasma glucagon‐like peptide‐1 (GLP‐1) and peptide YY (PYY) levels, as well as liver lipid content, were measured using plasma and tissue samples obtained at Day 31.
2.5. ob/ob mouse evaluation
Six‐week‐old male ob/ob mice, with a baseline body weight of 33.2 ± 1.4 g, were randomized into five groups (n = 5) based on plasma glucose, triglyceride, cholesterol, insulin and blood glycosylated haemoglobin levels, as well as food intake and body weight. Untyped ?/+ mice were used as normal controls (n = 5). Mice had free access to the CE‐2 powder diet containing each compound ([w/w]: 0.003, 0.01 and 0.03% SCO‐792; 0.009% pioglitazone) or diet alone (ob/ob vehicle and ?/+ vehicle) for 2 weeks (first treatment day designated as Day 0). Pioglitazone was selected as a control drug as this agent is known to improve metabolic control by increasing insulin sensitivity.18 Food intake and body weight were monitored regularly at the time points indicated in the Results section and in the associated Figures. Blood samples were collected at Day 14 to evaluate metabolic parameters, after which mice were sacrificed for measurement of tissue weight and liver lipid content.
2.6. Short‐term study for measuring fibroblast growth factor 21 (FGF21) in DIO mice
The DIO model was established by using an HFD (D12079B) from 5 weeks of age until initiation of the study. Male DIO mice, 27 weeks of age, with a baseline body weight of 48.0 ± 2.1 g, were randomized into three groups (n = 5) based on body weight and food intake. Mice received orally either SCO‐792 (60 mg/kg) or vehicle (0.5% [w/v] methylcellulose) once daily at approximately 5:00 to 6:00 pm and received an HFD (D12079B) for 3 days (first treatment day designated as Day 0). The pair‐fed group received the same amount of food consumed by the SCO‐792 group. At the end of the study, mice were sacrificed for tissue samples.
2.7. Hyperinsulinaemic‐euglycaemic clamp in ob/ob mice
Male ob/ob mice, 8 weeks of age, with a baseline body weight of 44.6 ± 1.5 g, were randomized into two groups (n = 14 and n = 16 for vehicle and SCO‐792, respectively) based on glycosylated haemoglobin, plasma glucose, body weight, plasma insulin and plasma alanine transaminase levels. Mice had free access to the CE‐2 powder diet containing the compound ([w/w]: 0.01% SCO‐792) or diet alone for 6 days (first treatment day designated as Day 0). Body weight and food intake were measured regularly. Blood samples were collected at Day 6 for determination of blood parameters. The left jugular veins of mice were catheterized after blood sampling. Hyperinsulinaemic‐euglycaemic clamps were performed after overnight fasting in conscious and unrestrained mice at Day 7. The protocol comprised a 15‐minute priming period, followed by a 125‐minute hyperinsulinaemic‐euglycaemic clamp period. Throughout the study, plasma glucose levels were measured every 5 minutes. Insulin (250 mU/kg/min) plus [3H]glucose (25 μCi/kg/min) was infused for glucose level equilibration during the priming period. After this period, hyperinsulinaemic‐euglycaemic clamp was initiated at 0 minutes, with a constant infusion of insulin (25 mU/kg/min) plus [3H]glucose (2.5 μCi/kg/min). During hyperinsulinaemic‐euglycaemic clamp, glucose (50%) was infused at variable rates to maintain euglycaemia (target glucose level, 150 mg/dL). To estimate insulin‐stimulated glucose uptake in muscle, liver and fat, [14C]2‐deoxy‐D‐glucose was administered as a bolus (10 μCi) at 80 minutes. Blood samples were taken at 85, 100 and 120 minutes for measurement of 3H and 14C radioactivity. After final blood sampling, mice were anesthetized with sodium pentobarbital (65 mg/kg, intraperitoneal injection) and gastrocnemius muscle, liver and epididymal fat were immediately extracted for 14C radioactivity measurement. The glucose disposal rate was calculated as the ratio of the labelled glucose infusion rate (dpm/kg/min) to plasma glucose‐specific activity (dpm/mg). Hepatic glucose production was calculated by subtracting the glucose infusion rate from the glucose disposal rate.
2.8. Evaluation of amino acid supplementation in C57BL/6J mice
Male C57/BL 6J mice, 9 weeks of age, with a baseline body weight of 23.8 ± 0.9 g, were randomized into four groups (n = 5) based on food intake and body weight. Mice received a powder diet (CE‐2) only or a diet containing 0.01% (w/w) SCO‐792 in the presence of essential amino acids for 8 days. Nine essential amino acids (histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine) were mixed at 0.92% (w/w) each into a normal chow diet. Food intake and body weight were measured at the time points indicated in the Results section and in the associated Figures.
2.9. Statistical analysis
Statistical significance was first analysed using Bartlett's test for homogeneity of variances, followed by the Williams' test (P > .05) and the Shirley‐Williams test (P ≤ .05) for dose‐dependent studies and the Dunnett's test (P > .05) and the Steel test (P ≤ .05) for multiple comparisons. Alternatively, statistical significance was analysed using the F test for homogeneity of variances, followed by the Student's t‐test (P > .2) or the Aspin‐Welch test (P ≤ .2). The Williams' and Shirley‐Williams tests were conducted using a one‐tailed significance level of 2.5% (0.025). Other tests were conducted using a two‐tailed significance level of 5% (0.05). All data are presented as mean ± standard deviation (SD).
Methods for biochemical measurement, measurement of hepatic triglyceride and total cholesterol contents, BCAA measurement and measurement of fecal protein content are included in the Appendix S1.
3. RESULTS
3.1. SCO‐792 was an in vivo efficacious enteropeptidase inhibitor
Given that SCO‐792 is a potent inhibitor against enteropeptidase16 and that enteropeptidase activity, expressed exclusively in the duodenum, is essential for dietary protein digestion and absorption, inhibiting this duodenal enzyme should result in the inhibition of amino acid absorption in the gut. In vivo effects of the oral administration of SCO‐792 on plasma BCAA levels, a reflection of the gut's absorption of digested protein manifested in blood circulation, in C57BL/6J mice revealed a dose‐dependent inhibition of plasma BCAA levels induced by oral protein dosing in mice (Figure 1).
Figure 1.
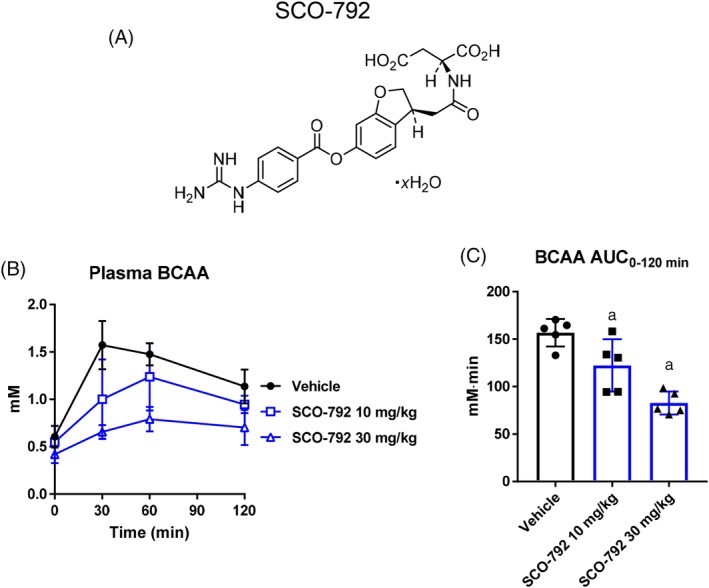
Effects of SCO‐792 on plasma BCAA change in an oral protein challenge test in DIO mice. A, Chemical structure of SCO‐792. B and C, Plasma BCAA levels during a protein challenge test in mice. SCO‐792 was administered to DIO mice 6 hours before a 2.5 g/kg protein meal challenge. SCO‐792 inhibited elevation in plasma BCAA in an oral protein challenge test. Values are expressed as mean ± SD (n = 6). a P ≤ .025 vs vehicle by one‐tailed Williams' test
3.2. SCO‐792 improved obesity‐related parameters in DIO mice
To evaluate the anti‐obesity potential of enteropeptidase inhibition, SCO‐792 was orally administered to DIO mice once‐daily for 4 weeks. As shown in Figure 2A, SCO‐792 dose‐dependently increased fecal protein levels. The average protein‐derived calorie loss to the feces against total caloric intake was 0.6% (vehicle), 1.7% (SCO‐792 20 mg/kg) and 3.0% (SCO‐792 59 mg/kg). The decrease in food intake was much more substantial in the early phase of the study compared to that in later phases (Figure 2B). Total food intake decreased significantly in SCO‐792‐treated mice (Figure 2C). Despite a partial attenuation of food intake‐reduction in later stages of the experiment, SCO‐792 continued to decrease body weight in DIO mice, with body weight change reaching over 15% in the high‐dose SCO‐792 group (Figure 2D,E). Plasma glucose remained unchanged, while insulin decreased dose‐dependently, GLP‐1 decreased slightly and PYY remained unaltered in SCO‐792‐treated DIO mice (Figure 2F‐I). Although plasma triglyceride levels did not change, plasma cholesterol and alanine transaminase levels decreased as the result of SCO‐792 treatment (Figure 2J‐L). SCO‐792 decreased fat mass while exhibiting a neutral effect on lean mass (Figure 2M,N). Upon evaluation of liver lipid content, SCO‐792 was found to decrease liver triglyceride levels and cholesterol content at the end of the study (Figure 2O,P). Although sibutramine acutely suppressed food intake in early stages of the study, this effect was reversed within a week, and total food intake thereafter remained unchanged (Figure 2B,C). The other parameters tested in this study were unchanged by sibutramine. Throughout the study, no abnormal findings were noted in SCO‐792‐treated mice.
Figure 2.
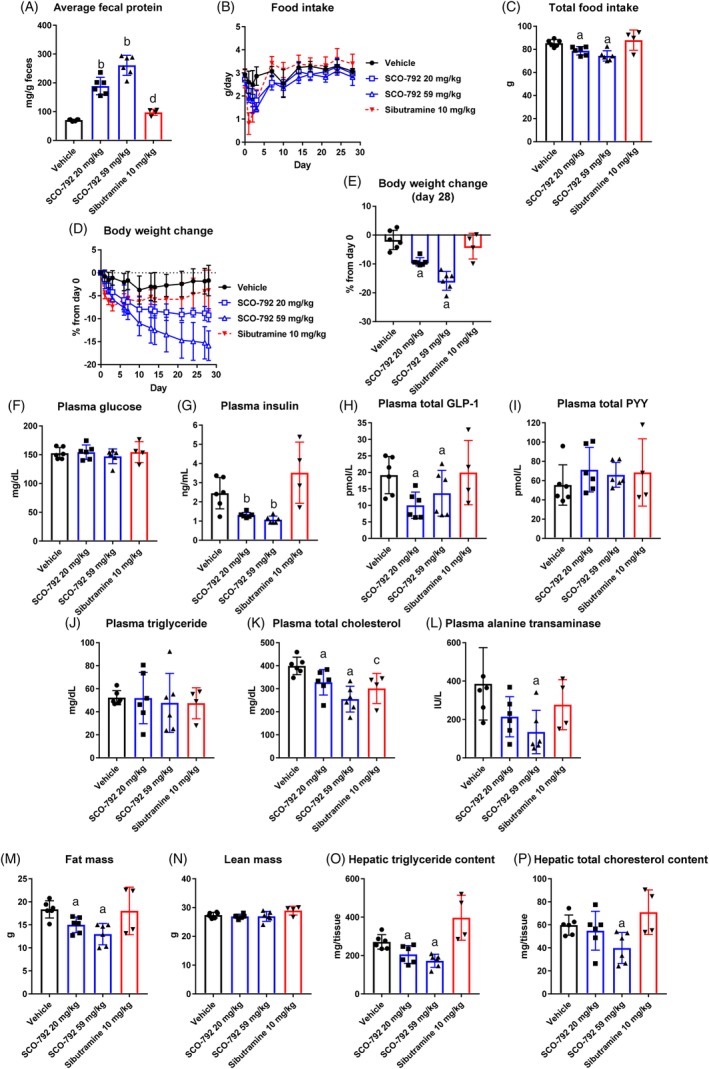
Effects of repeated administration of SCO‐792 on body weight and metabolic parameters in DIO mice. Average fecal protein; A, feces collected at four independent periods: Days 2‐3, Days 6‐7, Days 13‐14 and Days 27‐28. B, food intake during the study. C, total food intake. D, body weight change during the study. E, body weight change at Day 28. At end of study, levels of F, plasma glucose. G, insulin. H, total GLP‐1. I, total PYY. J, triglyceride. K, total cholesterol. L, alanine transaminase. M, fat mass. N, lean mass. O, hepatic triglyceride content and P, hepatic total cholesterol content. SCO‐792 increased fecal protein, a biomarker, inhibited food intake and continued to reduce body weight. Plasma insulin, cholesterol and alanine transaminase levels decreased, along with reductions in fat mass and hepatic lipid content. Data are expressed as mean ± SD (n = 4‐5). a P ≤ .025 vs vehicle by one‐tailed Williams' test. b P ≤ .025 vs vehicle by one‐tailed Shirley‐Williams test. c P ≤ .05 vs vehicle by Student's t‐test. d P ≤ .05 vs vehicle by Aspin‐Welch test
3.3. SCO‐792 increased FGF21 levels in DIO mice
To evaluate the mechanism by which SCO‐792 continuously decreases body weight, FGF21 levels, which reportedly increase energy expenditure in mice,19, 20 were compared in SCO‐792‐treated DIO mice and pair‐fed DIO mice. As shown in Figure 3A,B, the SCO‐792‐treated and pair‐fed groups show similar levels of food intake and body weight reduction in a 3‐day study. At the end of the study, Fgf21 mRNA in liver and plasma FGF21 levels were increased in SCO‐792‐treated DIO mice, whereas these increases were not observed in pair‐fed DIO mice (Figure 3C,D).
Figure 3.
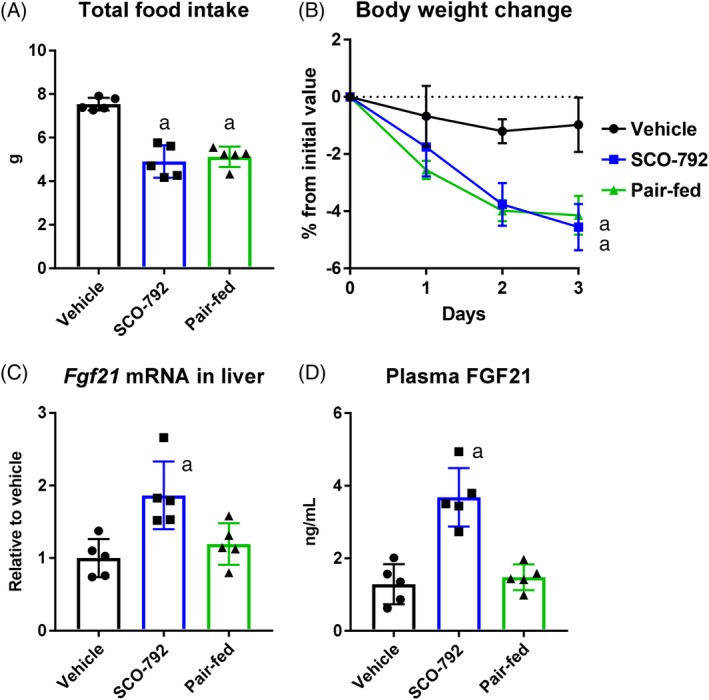
Effects of short‐term administration of SCO‐792 on FGF21 levels in DIO mice. A, Total food intake. B, body weight change. C, liver Fgf21 mRNA levels; and D, plasma FGF21 levels. DIO mice were treated with SCO‐792 (60 mg/kg) for 3 days. Pair‐fed group was fed the same amount of food consumed by the SCO‐792 group. SCO‐792 increased liver and circulating FGF21 levels. Data are expressed as mean ± SD (n = 5). a P ≤ .01 vs vehicle by Dunnett's test
3.4. SCO‐792 improved disease status of diabetes and obesity in ob/ob mice
To evaluate the effect of sustained administration of SCO‐792 on diabetic and obesity parameters, ob/ob mice were fed with a diet containing SCO‐792 (0.003%, 0.01% and 0.03% [w/w]) or with diet alone. The pioglitazone (0.009% [w/w]) group was set as a reference. The average doses of SCO‐792 (0.003%, 0.01% and 0.03%) were calculated as 3.9 ± 0.2, 9.9 ± 0.5 and 23.8 ± 2.1 mg/kg/d, respectively, and the average dose of pioglitazone (0.009%) was 13.1 ± 0.3 mg/kg/d. Consistent with results for DIO mice, SCO‐792 dose‐dependently increased fecal protein levels at Day 7 and Day 14 in ob/ob mice (Figure 4A,B). The average protein‐derived calorie loss to feces against total caloric intake was 6.5% (vehicle), 9.6% (SCO‐792 0.003%), 14.9% (SCO‐792 0.01%) and 15.2% (SCO‐792 0.03%), respectively. Food intake decreased dose‐dependently in SCO‐792‐treated ob/ob mice (Figure 4C,D), along with dose‐dependent decreases in body weight that were in‐line with decreased food intake (Figure 4E,F). Metabolic analyses revealed that SCO‐792 improved hyperglycaemia in ob/ob mice; 0.01% and 0.03% of SCO‐792 in the diet normalized glucose levels and dose‐dependently decreased glycosylated haemoglobin (Figure 4G,H). As shown in Figure 4I, SCO‐792 also improved hyperinsulinaemia in ob/ob mice, with treatment levels of 0.01% and 0.03% of SCO‐792 in the diet yielding nearly normal levels of circulating insulin. Plasma triglyceride and total cholesterol levels also decreased in SCO‐792‐treated ob/ob mice (Figure 4J,K), as did the plasma alanine transaminase level (Figure 4L). Fat weight also decreased in SCO‐792‐treated ob/ob mice (Figure 4M‐O). Abnormal liver weight, as well as triglyceride and total cholesterol content, decreased in SCO‐792‐treated ob/ob mice (Figure 4P‐R). Pioglitazone improved both glucose and lipid profiles, as well as insulin resistance in ob/ob mice. However, liver weight and other hepatic parameters were unchanged by pioglitazone treatment. Throughout the study, no abnormal findings were noted in SCO‐792‐treated mice.
Figure 4.
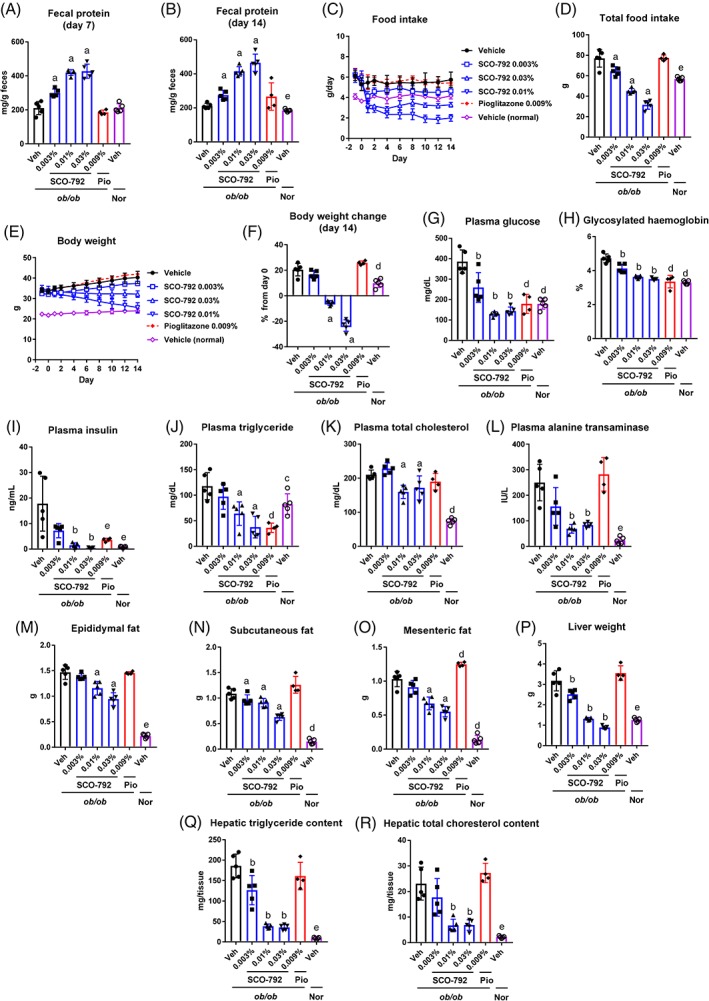
Effects of repeated administration of SCO‐792 on metabolic parameters in ob/ob mice. ob/ob mice were treated with either SCO‐792 (0.003%‐0.01% added to the diet), pioglitazone (0.009% added to the diet) or vehicle alone for 2 weeks. A, Fecal protein at Day 7 (Days 6‐7) and B, Day 14 (Days 13‐14); C, food intake during the study period; D, total food intake; E, body weight during the study period; and F, body weight change at Day 14. Metabolic parameters were determined at the end of the study: G, plasma levels of glucose; H, glycosylated haemoglobin; I, insulin; J, triglycerides; K, total cholesterol; L, alanine transaminase; M, epididymal fat; N, subcutaneous fat; O, mesenteric fat; P, liver weight; Q, hepatic triglyceride content; and R hepatic total cholesterol content. Data are expressed as mean ± SD (n = 4‐5). a P ≤ .025 vs ob/ob vehicle by one‐tailed Williams' test. b P ≤ .025 vs ob/ob vehicle by one‐tailed Shirley‐Williams test. c P ≤ .05 vs ob/ob vehicle by Dunnett's test. d P ≤ .01 vs ob/ob vehicle by Dunnett's test. e P ≤ .05 vs ob/ob vehicle by Steel test. Abbreviations: Nor, ?/+ mice; Pio, pioglitazone
3.5. SCO‐792 improved insulin sensitivity in muscles
To test whether SCO‐792 interferes with insulin action in ob/ob mice, hyperinsulinaemic‐euglycaemic clamps were performed after 6 days of treatment, followed by overnight fasting. Metabolic analyses confirmed reductions in food intake, body weight, plasma glucose and plasma insulin in SCO‐792‐treated ob/ob mice immediately prior to a hyperinsulinaemic‐euglycaemic clamp study (Figure 5A‐D). A clamp study (Figure 5E) revealed that ob/ob mice treated with SCO‐792 required a higher glucose infusion rate to maintain euglycaemia as compared to mice that received vehicle (Figure 5F‐H). Hepatic glucose production during the clamp appeared to be similar between SCO‐792‐treated mice and those that received vehicle (Figure 5I). The glucose disposal rate was increased in SCO‐792‐treated ob/ob mice (Figure 5J). Glucose uptake, measured by [14C]2‐deoxy‐D‐glucose, was increased in muscles, unchanged in liver and decreased in fat following treatment with SCO‐792 (Figure 5K‐M).
Figure 5.
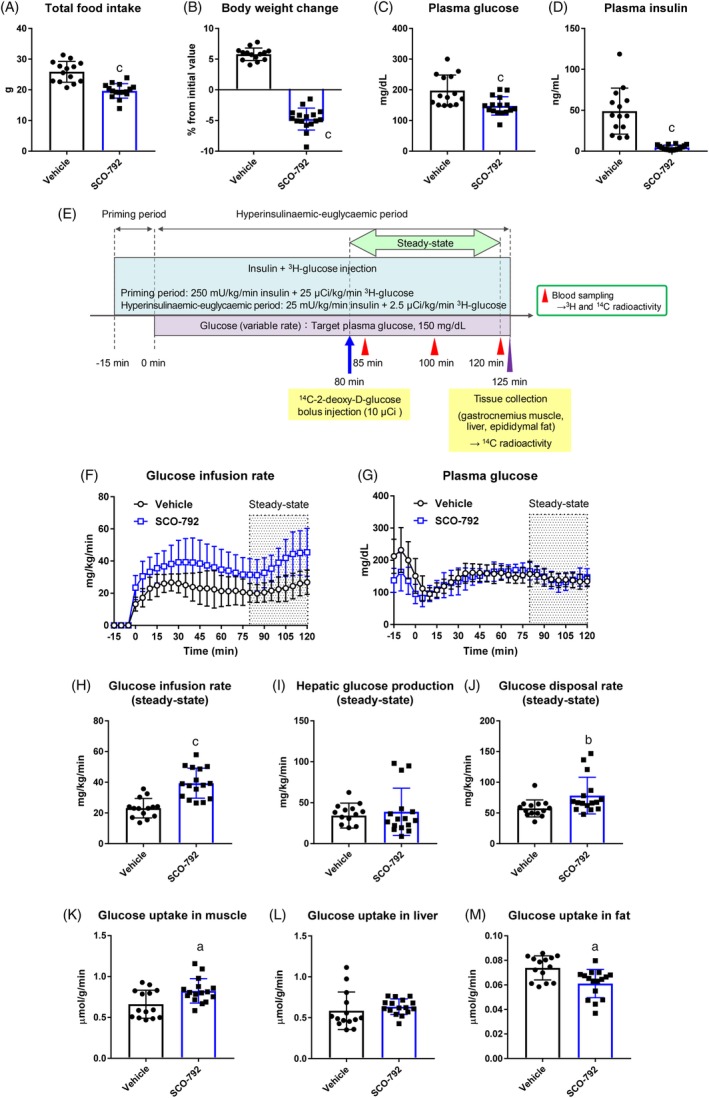
Effects of repeated administration of SCO‐792 on insulin sensitivity in ob/ob mice. ob/ob mice were treated with either SCO‐792 (0.01% added to the diet) or vehicle alone for 6 days. A, Total food intake and B, body weight change; C, plasma glucose and D, insulin at end of study. E, After 6 days of dosing, mice were fasted overnight and hyperinsulinaemic‐euglycaemic clamp was performed. F, Glucose infusion rate during hyperinsulinaemic‐euglycaemic clamp. G, Plasma glucose levels maintained at c. 150 mg/dL during clamp procedure. H, Averaged glucose infusion rate; I, hepatic glucose production; and J, glucose disposal rate during steady state. Glucose uptake in K, muscle, L, liver and M, fat at end of clamp procedure. Data are expressed as mean ± SD (n = 14 and n = 16 for vehicle and SCO‐792, respectively). a P ≤ .01 vs vehicle by Student's t‐test. b P ≤ .05 vs vehicle by Aspin‐Welch test. c P ≤ .01 vs vehicle by Aspin‐Welch test
3.6. Amino acid supplementation of diet partially reversed efficacy of SCO‐792 in mice
Inhibiting enteropeptidase decreases the generation of essential amino acids from protein, which may have a role in controlling food intake and body weight. To explore the role of amino acid intake on overall food intake and body weight control, SCO‐792‐treated mice and those that received vehicle were housed with free‐access to diets with or without supplementation of essential amino acids. SCO‐792 decreased food intake and body weight in normal diet‐fed mice, as expected (Figure 6A,C,D and F). With essential amino acid supplementation of the diet, reduction in food intake was evident only at Day 1 and total food intake remained unchanged in SCO‐792‐treated mice (Figure 6B,C). Consistent with this, body weight remained unaltered in SCO‐792‐treated mice that received the diet supplemented with essential amino acids (Figure 6E,F).
Figure 6.
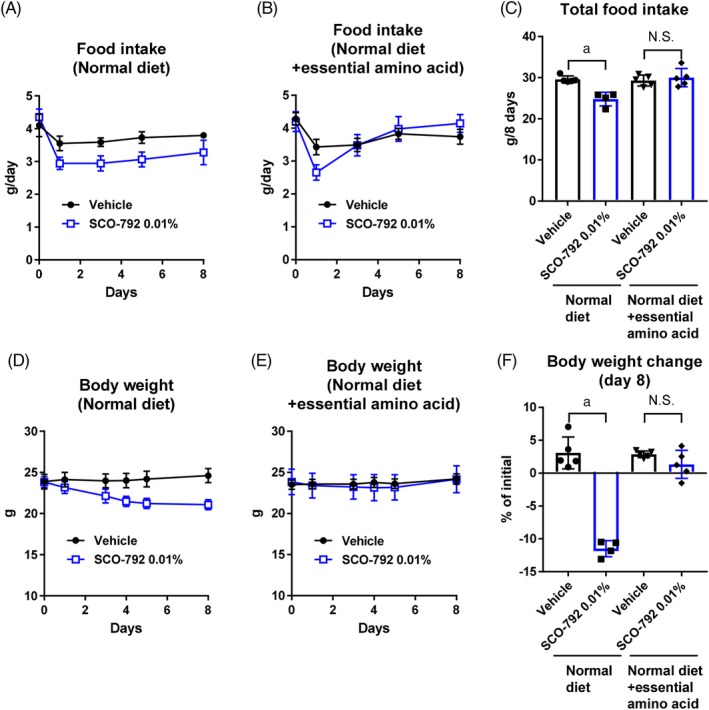
Effects of essential amino acid supplementation on SCO‐792‐induced food intake and body weight reduction in C57BL/6J mice. Food intake of A, normal diet group and B, essential amino acid‐supplemented group were monitored throughout the study and C, total food intake was calculated. Body weight of D, normal diet group and E, essential amino acid‐supplemented group were monitored throughout the study and F, percent change in body weight from initial values was determined. Data are expressed as mean ± SD (n = 4‐5). a P ≤ .01 by Student's t‐test. Abbreviations: EAA, essential amino acid; N.S., not significant
4. DISCUSSION
Enteropeptidase is a key enzyme that regulates protein breakdown in the gut. In this study, we evaluated the effects of SCO‐792, a novel and highly effective enteropeptidase inhibitor, in mouse models. As expected, SCO‐792 was demonstrated to inhibit protein breakdown in the gut. Of note, administration of SCO‐792 resulted in inhibition of food intake and reduction in body weight in obese mouse models. It also markedly improved abnormal metabolic parameters, including glucose, lipid and liver parameters, in obese/diabetic mice. Studies have also revealed that SCO‐792 increased FGF21 levels in obese mice and increased muscular insulin sensitivity in obese/diabetic mice. An amino acid supplementation of the diet partially reversed the effect of SCO‐792, suggesting the role of enteropeptidase in control of metabolism and body weight. Thus, we propose the SCO‐792‐mediated inhibition of enteropeptidase as a novel treatment option for diabetes and obesity.
In a 4‐week study of DIO mice, once‐daily oral dosing of SCO‐792 inhibited food intake, which was more evident in the early phase of the study, up to approximately 1 week, as compared to the rest of the study. Although the inhibition of food intake was attenuated at later stages, after approximately 1 week, body weight continued to decrease as a result of treatment with SCO‐792. This suggests that a food intake inhibition‐independent mechanism is likely to contribute to the SCO‐792‐induced decrease in body weight. Continued fecal exclusion of protein‐derived calories may contribute, in part, to the sustained loss of body weight observed in mice. In a short‐term study, treatment with SCO‐792, but not pair‐fed treatment, increased FGF21 levels in DIO mice. FGF21 reportedly increases energy expenditure in mice, thereby correcting obesity.19, 20 Thus, SCO‐792‐mediated FGF21 elevation may have a role in body weight reduction. Gut GLP‐1 and PYY are physiological hormones known to regulate satiety and body weight.21 In the current study, plasma levels of total GLP‐1 were slightly lower and plasma PYY remained unchanged in SCO‐792‐treated DIO mice. Thus, plasma GLP‐1 and PYY are unlikely to play a significant role in body weight reduction in SCO‐792‐treated mice. Considering that SCO‐792 alters protein conditions inside the gut, the microbiome may play a role in this parameter. In fact, gut microbiota has been associated with human obesity,22 as has antibiotic therapy.23 In addition to DIO mice, in which it was demonstrated that once‐daily oral dosing of SCO‐792 was therapeutically effective, body weight in ob/ob mice decreased drastically upon sustained administration of SCO‐792, suggesting that continued inhibition of enteropeptidase may be even more effective in improving disease parameters. Enteropeptidase stimulates protein breakdown, thereby generating amino acids. Adding essential amino acids to the diet partially reversed inhibition of food intake and decrease in body weight in SCO‐792‐treated mice. This suggests that enteropeptidase inhibition may regulate these changes by modulating amino acid generation, at least in part.
To the best of our knowledge, this report is the first to demonstrate that enteropeptidase inhibition acts to correct abnormally high glucose levels. In addition to its body weight‐lowering effect, SCO‐792 significantly improved hyperglycaemia in diabetic ob/ob mice, that also showed very low levels of circulating insulin. A hyperinsulinaemic‐euglycaemic clamp study demonstrated that SCO‐792 increases insulin sensitivity, mainly in muscles. Amino acids are known to play a role in metabolism, which is dysregulated in diabetes and obesity disease states.24, 25, 26 For example, protein ingestion is known to induce muscle insulin resistance in humans.27 Thus, the modulation of amino acid absorption by enteropeptidase inhibition may be an early event that eventually results in improved insulin sensitivity in the diabetic condition. Considering the pivotal role of insulin signaling in the maintenance of muscle mass,28 increased insulin sensitivity in muscle may explain why SCO‐792‐treated mice show unaltered lean mass despite body weight loss. Interestingly, glucose uptake in fat was decreased in the hyperinsulinaemic‐euglycaemic condition. Although the reason for this observation remains unclear, it may prove beneficial in obesity, as reports indicate that glucose uptake is a key step in lipogenesis in fat.29 Unlike in diabetic ob/ob mice, SCO‐792 did not alter glucose levels in DIO mice that exhibited normal glucose levels, suggesting that SCO‐792 may not induce unfavourable hypoglycaemia in non‐diabetic conditions.
SCO‐792‐treatment improved abnormal lipid profiles in ob/ob and DIO mice. SCO‐792‐treatment decreased plasma triglyceride levels in ob/ob mice and total cholesterol levels in ob/ob and DIO mice. In addition to plasma levels, hepatic lipid contents and plasma alanine transaminase levels decreased in ob/ob and DIO mice. Furthermore, liver weight, which increased abnormally in ob/ob mice, decreased upon treatment with SCO‐792. As it is widely accepted that liver lipotoxicity induces nonalcoholic steatohepatitis,30 SCO‐792‐treatment may also offer benefits for this condition.
SCO‐792‐treatment decreased fat mass in DIO and ob/ob mice. Lean mass was unaltered in SCO‐792‐treated DIO mice, indicating favourable changes in body composition as a result of this treatment regimen for obesity. The recommended dietary allowance (RDA) for protein is 0.8 g/kg/d31; however, obese patients usually consume excess amounts of nutrients including protein.32 Therefore, inhibiting protein uptake presents an appropriate strategy in the treatment of metabolic diseases and obesity.
In a previous study, Harosh et al reported that the boropeptide enteropeptidase inhibitor OBE‐2008 did not alter food intake but decreased body weight, probably by inhibiting protein and fat absorption in DIO mice.15 In contrast to this study, inhibition of food intake was repeatedly observed in mice treated with SCO‐792 in our study. This discrepancy may be explained by model differences in DIO mice. Harosh et al used DIO mice that exhibited diet‐induced additional body weight gain, whereas we used DIO mice with established obesity that did not exhibit body weight gain. Furthermore, considering that SCO‐792 demonstrated favourable safety profiles in preclinical studies (unpublished observations), reduction in food intake is unlikely to be the result of unfavourable side effects.
Plasma BCAA levels and fecal protein content are easily measured and significantly atered by treatment with SCO‐792. This suggests that both parameters may be suitable as favourable biomarkers of enteropeptidase inhibition in future clinical studies.
Although we demonstrated that treatment with SCO‐792 was highly effective in improving diabetes and obesity disease states in mice, the key biological mechanisms/events by which SCO‐792 induces these benefits remain unclear. Thus, further studies are essential to gain an understanding of how enteropeptidase inhibition results in the beneficial results observed in the current study.
In conclusion, the enteropeptidase inhibitor SCO‐792 effectively improved insulin resistance and glucose control in a diabetic mouse model, while also showing substantial efficacy in lowering body weight in obese models. In addition, the favourable effect of treatment with SCO‐792 in improving plasma and liver lipid profiles was demonstrated. These findings suggest that the inhibition of gut enteropeptidase activity with the concomitant protein breakdown modulation in the gut may offer a novel treatment option for obesity, diabetes and liver diseases.
CONFLICT OF INTEREST
H. Y., K. H., H. H., K. T. and M. S. are or were employees of Takeda Pharmaceutical Company Ltd. J. S., F. Y., Y. M., T. M., Y. Y. and M. W. are employees of SCOHIA PHARMA, Inc.
AUTHOR CONTRIBUTIONS
The research was designed by all authors. Experiments were conducted by H. Y., K. H., H. H., J. S., K. T. and F. Y. Data were analysed and interpreted by all authors. The manuscript was written by Y. M., H. Y. and M. W. and important intellectual content of the manuscript was reviewed and revised by all authors. All authors have agreed to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Supporting information
Appendix S1. Biochemical measurements, Measurement of hepatic triglyceride and total cholesterol contents, BCAA measurement, Measurement of fecal protein content, Measurement of Fgf21 mRNA and FGF21 protein levels.
ACKNOWLEDGMENTS
The authors would like to acknowledge all project members for the study of SCO‐792.
Yashiro H, Hamagami K, Hiyoshi H, et al. SCO‐792, an enteropeptidase inhibitor, improves disease status of diabetes and obesity in mice. Diabetes Obes Metab. 2019;21:2228–2239. 10.1111/dom.13799
Funding information The study was conducted with financial support from SCOHIA PHARMA, Inc. and Takeda Pharmaceutical Company Ltd.
REFERENCES
- 1. Mendis S, Lindholm LH, Mancia G, et al. World Health Organization (WHO) and International Society of Hypertension (ISH) risk prediction charts: assessment of cardiovascular risk for prevention and control of cardiovascular disease in low and middle‐income countries. J Hypertens. 2007;25:1578‐1582. [DOI] [PubMed] [Google Scholar]
- 2. Chopra M, Galbraith S, Darnton‐Hill I. A global response to a global problem: the epidemic of overnutrition. Bull World Health Organ. 2002;80:952‐958. [PMC free article] [PubMed] [Google Scholar]
- 3. Monteiro CA, Moura EC, Conde WL, Popkin BM. Socioeconomic status and obesity in adult populations of developing countries: a review. Bull World Health Organ. 2004;82:940‐946. [PMC free article] [PubMed] [Google Scholar]
- 4. Haidar YM, Cosman BC. Obesity epidemiology. Clin Colon Rectal Surg. 2011;24:205‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mozaffarian D. Foods, obesity, and diabetes‐are all calories created equal? Nutr Rev. 2017;75(suppl 1):19‐31. [DOI] [PubMed] [Google Scholar]
- 6. Ludwig DS. Lowering the bar on the low‐fat diet. JAMA. 2016;316(20):2087‐2088. [DOI] [PubMed] [Google Scholar]
- 7. Padwal R. Cetilistat, a new lipase inhibitor for the treatment of obesity. Curr Opin Investig Drugs. 2008;9:414‐421. [PubMed] [Google Scholar]
- 8. Joshi SR, Standl E, Tong N, Shah P, Kalra S, Rathod R. Therapeutic potential of alpha‐glucosidase inhibitors in type 2 diabetes mellitus: an evidence‐based review. Expert Opin Pharmacother. 2015;16:1959‐1981. [DOI] [PubMed] [Google Scholar]
- 9. Godbout A, Chiasson JL. Who should benefit from the use of alpha‐glucosidase inhibitors? Curr Diab Rep. 2007;7:333‐339. [DOI] [PubMed] [Google Scholar]
- 10. Heck AM, Yanovski JA, Calis KA. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy. 2000;20:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mann NS, Mann SK. Enterokinase. Proc Soc Exp Biol Med. 1994;206(2):114‐118. [DOI] [PubMed] [Google Scholar]
- 12. Zheng XL, Kitamoto Y, Sadler JE. Enteropeptidase, a type II transmembrane serine protease. Front Biosci (Elite Ed). 2009;1:242‐249. [DOI] [PubMed] [Google Scholar]
- 13. Light A, Janska H. Enterokinase (enteropeptidase): comparative aspects. Trends Biochem Sci. 1989;14:110‐112. [DOI] [PubMed] [Google Scholar]
- 14. Hadorn B, Haworth JC, Gourley B, Prasad A, Troesch V. Intestinal enterokinase deficiency. Occurrence in two sibs and age dependency of clinical expression. Arch Dis Child. 1975;50:277‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braud S, Ciufolini MA, Harosh I. Enteropeptidase: a gene associated with a starvation human phenotype and a novel target for obesity treatment. PLoS One. 2012;7:e49612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sasaki M, Kakegawa K, Kikuchi F, Ikeda Z, Nishikawa Y. Fused heterocyclic compound. 2015; WO2015/122187.
- 17. McNulty SJ, Ur E, Williams G, Multicenter Sibutramine Study Group . A randomized trial of sibutramine in the management of obese type 2 diabetic patients treated with metformin. Diabetes Care. 2003;26:125‐131. [DOI] [PubMed] [Google Scholar]
- 18. Devchand PR, Liu T, Altman RB, FitzGerald GA, Schadt EE. The Pioglitazone Trek via Human PPAR Gamma: from discovery to a medicine at the FDA and beyond. Front Pharmacol. 2018;9:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu J, Lloyd DJ, Hale C, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet‐induced obese mice. Diabetes. 2009;58:250‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coskun T, Bina HA, Schneider MA, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018‐6027. [DOI] [PubMed] [Google Scholar]
- 21. Moran TH. Gut peptide signaling in the controls of food intake. Obesity (Silver Spring). 2006;14(suppl 5):250S‐253S. [DOI] [PubMed] [Google Scholar]
- 22. Bjorneklett A, Viddal KO, Midtvedt T, Nygaard K. Intestinal and gastric bypass. Changes in intestinal microecology after surgical treatment of morbid obesity in man. Scand J Gastroenterol. 1981;16:681‐687. [DOI] [PubMed] [Google Scholar]
- 23. Thuny F, Richet H, Casalta JP, Angelakis E, Habib G, Raoult D. Vancomycin treatment of infective endocarditis is linked with recently acquired obesity. PLoS One. 2010;5:e9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Badoud F, Lam KP, DiBattista A, et al. Serum and adipose tissue amino acid homeostasis in the metabolically healthy obese. J Proteome Res. 2014;13:3455‐3466. [DOI] [PubMed] [Google Scholar]
- 25. Jang C, Oh SF, Wada S, et al. A branched‐chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22:421‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newgard CB, An J, Bain JR, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith GI, Yoshino J, Stromsdorfer KL, et al. Protein ingestion induces muscle insulin resistance independent of leucine‐mediated mTOR activation. Diabetes. 2015;64:1555‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rhoads RP, Baumgard LH, El‐Kadi SW, Zhao LD. Physiology and Endocrinology Symposium: roles for insulin‐supported skeletal muscle growth. J Anim Sci. 2016;94:1791‐1802. [DOI] [PubMed] [Google Scholar]
- 29. Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2:282‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity and pro‐inflammatory fatty liver disease. Adv Exp Med Biol. 2018;1061:19‐44. [DOI] [PubMed] [Google Scholar]
- 31. Wolfe RR, Cifelli AM, Kostas G, Kim IY. Optimizing protein intake in adults: interpretation and application of the recommended dietary allowance compared with the acceptable macronutrient distribution range. Adv Nutr. 2017;8:266‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smethers AD, Rolls BJ. Dietary management of obesity: cornerstones of healthy eating patterns. Med Clin North Am. 2018;102:107‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Biochemical measurements, Measurement of hepatic triglyceride and total cholesterol contents, BCAA measurement, Measurement of fecal protein content, Measurement of Fgf21 mRNA and FGF21 protein levels.