

Abstract
Background
Patients with peritoneal metastases from colorectal cancer have a poor prognosis. If the intraperitoneal tumour load is limited, patients may be eligible for cytoreductive surgery followed by hyperthermic intraperitoneal chemotherapy (HIPEC). This treatment has improved overall survival, but recurrence rates are high. The aim of this study was to create a preclinical platform for the development of more effective intraperitoneal chemotherapy strategies.
Methods
Using organoid technology, five tumour cultures were generated from malignant ascites and resected peritoneal metastases. These were used in an in vitro HIPEC model to assess sensitivity to mitomycin C (MMC) and oxaliplatin, the drugs used most commonly in HIPEC. The model was also used to test a rational combination treatment involving MMC and inhibitors of the checkpoint kinase ATR.
Results
MMC was more effective in eliminating peritoneal metastasis‐derived organoids than oxaliplatin at clinically relevant concentrations. However, the drug concentrations required to eliminate 50 per cent of the tumour cells (IC50) were higher than the median clinical dose in two of five organoid lines for MMC, and all five lines for oxaliplatin, indicating a general resistance to monotherapy. ATR inhibition increased the sensitivity of all peritoneal metastasis‐derived organoids to MMC, as the IC50 decreased 2·6–12·4‐fold to well below concentrations commonly attained in clinical practice. Live‐cell imaging and flow cytometric analysis showed that ATR inhibition did not release cells from MMC‐induced cell cycle arrest, but caused increased replication stress and accelerated cell death.
Conclusion
Peritoneal metastasis‐derived organoids can be used to evaluate existing HIPEC regimens on an individual‐patient level and for development of more effective treatment strategies.
Surgical relevance.
Cytoreductive surgery followed by hyperthermic intraperitoneal chemotherapy (HIPEC) has improved prognosis of patients with peritoneal metastases from colorectal cancer, but disease recurrence is common. More effective and personalized HIPEC is urgently needed. Organoid technology is frequently used for drug screens, as patient‐derived organoids can accurately predict clinical therapeutic response in vitro.
A panel of organoids was established from peritoneal metastases from colorectal cancer and used to develop a model for testing HIPEC regimens in vitro. Patient‐derived organoids differed in sensitivity to commonly used chemotherapeutics, in line with variable clinical outcomes following cytoreductive surgery–HIPEC. Combining MMC with an ATR inhibitor improved the efficacy of MMC.
Peritoneal metastasis‐derived organoids can be used as a platform to test novel (combination) strategies that increase HIPEC efficacy. In the future, organoids could be used to select patent‐tailored HIPEC regimens.
Abstract
Antecedentes
Los pacientes con metástasis peritoneales (peritoneal metastasis, PM) de cáncer colorrectal tienen un mal pronóstico. Si la carga tumoral intraperitoneal es reducida, los pacientes pueden ser candidatos a cirugía citorreductora seguida de quimioterapia intraperitoneal hipertérmica (hyperthermic intraperitoneal chemotherapy, HIPEC). Este tratamiento ha mejorado la supervivencia global, pero las tasas de recidiva son altas. El objetivo de este estudio fue crear una plataforma preclínica para el desarrollo de las estrategias de quimioterapia intraperitoneal más efectivas.
Métodos
Mediante la utilización de la tecnología de organoides, se generaron cinco cultivos tumorales a partir de ascitis maligna y PM resecadas. Se utilizó un modelo de HIPEC in vitro para evaluar la sensibilidad a la mitomicina C (mitomycin C, MMC) y al oxaliplatino, los fármacos más utilizados en la HIPEC. El modelo también se usó para probar un tratamiento combinado de MMC e inhibidores de control inmunitario de la quinasa ATR.
Resultados
A concentraciones clínicamente relevantes, la MMC fue más efectiva que el oxaliplatino para eliminar los organoides derivados de PM. Sin embargo, las concentraciones de fármaco necesarias para eliminar el 50% de las células tumorales (IC50) fueron más elevadas que la mediana de la dosis clínica en 2/5 (MMC) o 5/5 (oxaliplatino) de las líneas de organoides, lo que indica una resistencia general a la monoterapia. La inhibición de ATR aumentó la sensibilidad a MMC de todos los organoides derivados de PM, ya que la IC50 disminuyó (2,6‐12,4 veces) a concentraciones muy por debajo de las que se alcanzan comúnmente en la práctica clínica. Los análisis de viabilidad celular y de citometría de flujo (FACS) mostraron que la inhibición de ATR no liberaba células tras la detención del ciclo celular inducida por la MMC, sino que causaba un aumento en el estrés replicativo y muerte celular acelerada.
Conclusión
Se pueden usar los organoides derivados de PM para evaluar los regímenes HIPEC existentes a nivel del paciente individual y para desarrollar estrategias terapéuticas más efectivas.
Introduction
Peritoneal metastases (PMs) develop in approximately 10 per cent of patients with colorectal cancer1. These patients generally have a very poor prognosis, with a median survival of only 6–9 months without treatment2. For selected patients with limited PMs and treatable extraperitoneal disease, cytoreductive surgery (CRS) combined with hyperthermic intraperitoneal chemotherapy (HIPEC) is a potentially curative localized treatment option3. The goal is to remove all macroscopic PMs surgically, while eliminating any residual microscopic disease by HIPEC. CRS–HIPEC has significantly improved median overall survival of patients with PMs from colorectal cancer to 36 months4, 5, 6, 7.
Retrospective comparisons between oxaliplatin and mitomycin C (MMC), the two drugs most commonly used in HIPEC for PMs from colorectal cancer, generated contradictory results7, 8, 9, 10. As a consequence, there is currently no consensus on the choice of chemotherapy, the dose administered or the duration of perfusion3. Regardless of the chemotherapy drug used in HIPEC, recurrence rates after CRS–HIPEC are high and more than half of patients experience disease recurrence within 2 years7, 11, 12. The added value of HIPEC after CRS has recently been questioned in light of results from the Prodige 7 RCT12, which showed that adding oxaliplatin‐based HIPEC to CRS provided no survival benefit12. However, clinical proof that HIPEC after CRS can effectively target minimal residual disease in the peritoneum has been provided by a large phase III study in ovarian cancer13. Rather than omitting HIPEC from the treatment of PMs from colorectal cancer, efforts should be made to improve the efficacy of intraperitoneal chemotherapy.
Simply increasing the HIPEC dose is not feasible, as a higher perfusate concentration leads to a higher plasma concentration, which has been associated with increased toxicity14. A rational combination treatment of MMC with inhibition of ataxia telangiectasia and Rad3‐related protein kinase (ATR) could enhance HIPEC efficacy. ATR inhibitors are currently being tested in a number of clinical trials, either alone or in combination with chemotherapy15. MMC causes cytotoxicity mainly through formation of interstrand crosslinks16. This type of DNA damage activates ATR, which induces cell cycle arrest to allow DNA repair. Inhibition of ATR after exposure to MMC could lead to fatal collapse of replication forks and increased cell death17.
To evaluate current HIPEC agents and novel combination therapies, a representative in vitro model of colorectal PM is required. Tumour‐derived organoids are ideally suited for this purpose, as they retain the genetic and phenotypic characteristics of the original cancers, and can accurately predict clinical therapeutic response in vitro 18, 19, 20. The aim of this project was to create an organoid‐based preclinical model of PM from colorectal cancer to study current HIPEC regimens and to test novel combination strategies that augment the efficacy of HIPEC.
Methods
A detailed description of the methodology is provided in Appendix S1 (supporting information).
Tissue collection and organoid culture
Tissue samples from PMs were collected during CRS or diagnostic laparoscopy within biobanking protocol HUB‐Cancer TcBio#12‐09, which was approved by the medical ethics committee of the University Medical Centre Utrecht. Ascites was collected within the context of the CRC‐PIPAC trial (NCT03246321) at the Catherina Hospital, Eindhoven. Before treatment with electrostatically precipitated pressurized intraperitoneal aerosol chemotherapy, 40 ml ascites was aspirated using a gastronasal tube and a syringe through one of the laparoscopic trocar openings.
Chemotherapy regimens and drug concentrations
Before undertaking drug screens on the PM‐derived organoids, the concentrations of MMC and oxaliplatin that are commonly attained in the peritoneal cavity during HIPEC procedures were investigated. Data from 40 HIPEC procedures for PMs from colorectal cancer or pseudomyxoma peritonei carried out at the University Medical Centre Utrecht between January 2016 and July 2018 were collected. Perfusion volume, body surface area (BSA) and chemotherapy dose were extracted from intraoperative recordings of the perfusion pump system Performer HT® (RanD, Medolla, Italy). At University Medical Centre Utrecht, chemotherapy dose is determined by BSA, at 35 mg/m2 for MMC and 460 mg/m2 for oxaliplatin. The concentration of intraperitoneal chemotherapy further depends on the volume used for dialysis. During HIPEC, MMC is added to the total perfusion fluid in three separate doses. First, 50 per cent of the total dose is added, followed by 25 per cent after 30 and 60 min. This compensates for systemic uptake of MMC and results in a relatively stable perfusate concentration21. As a result, the maximum concentration reached in the perfusate is roughly equal to the starting concentration (50 per cent of the total dose divided by the total perfusate volume).
Subsequent drug screens were performed across a wide range of concentrations including the calculated clinical concentrations. MMC (S8146; Selleckchem, Munich, Germany) was dissolved in dimethylsulphoxide (DMSO) to a stock concentration of 60 mmol/l, and was further diluted in basal culture medium without niche factors to a concentration range of 0·4–300 μmol/l. Oxaliplatin (5 mg/ml solution; Fresenius Kabi, Bad Homburg, Germany) was diluted in 0·45 per cent sodium chloride/2·5 per cent glucose solution to preserve solubility in a physiological carrier solution22, to a concentration range of 8·6 μmol/l to 6·29 mmol/l.
In an effort to improve MMC‐based HIPEC, combination treatment of MMC with VE‐821, an inhibitor of ATR, was assessed. The clinical derivative of VE‐821 (VX‐970) was also assessed for enhancement of response to MMC, to evaluate whether this rational combination strategy could be translated to the clinical setting. VE‐821 (Sigma Aldrich, Saint Louis, Missouri, USA) and VX‐970 (VE‐822; Selleckchem) were dissolved in DMSO at a stock concentration of 10 mmol/l, and added to basal growth medium at the concentrations indicated.
Hyperthermic intraperitoneal chemotherapy in vitro
The clinical presentation of microscopic tumour cell deposits attached to the peritoneum was simulated by allowing multicellular organoids to adhere to a bottom layer of basement membrane extract (BME). As such, organoids were in direct contact with the chemotherapy solution, similar to micrometastases during HIPEC. Bottom layers of 40 μl BME were dispensed in a 96‐well plate (Costar® 3904; Corning, New York, USA) by use of a Multidrop™ Combi Reagent Dispenser with a small tubing cassette (ThermoFisher Scientific, Waltham, Massachusetts, USA). Approximately 3000 2‐day‐old organoids suspended in 100 μl basal culture medium were dispensed on top of the BME. Two days after plating, medium was aspirated, and the organoids were incubated with 100 μl preheated chemotherapy at a range of concentrations for 30 min (oxaliplatin) or 90 min (MMC) at 42°C, in line with current standard clinical treatment protocols10. ATR inhibitors VE‐821 and VX‐970 were added during the 72 h after HIPEC. As a measure of cell viability, ATP levels were assessed using a CellTiterGlo® 2.0 kit (Promega, Fitchburg, Wisconsin, USA) on a Spectramax M5e reader (Molecular Devices, San Jose, California, USA). Viability was normalized to the mean of three control wells per batch, which were treated with 0·5 per cent DMSO in MMC and glucose/sodium chloride solution in oxaliplatin experiments, and included the respective inhibitors in the drug combination experiments. Viability assays were performed in triplicate and repeated multiple times on different days, yielding a total of 1422 data points from 61 separate experiments.
Live‐cell imaging
TOR14 organoids expressing mNeon‐tagged histone 2B were incubated with either basal culture medium, 1 μmol/l VE821, 0·5 μmol/l MMC, or 0·5 μmol MMC + 1 μmol VE821 for 90 min at 42°C. The chemotherapy agent was washed away, medium was replaced and 1 μmol/l VE‐821 was added to the appropriate wells. The plate was mounted on an inverted confocal microscope (Nikon TiE‐based CSU‐W1 Spinning Disk; Nikon Minato, Tokyo, Japan) equipped with a culture chamber at 37°C and 5 per cent carbon dioxide. Organoids were imaged every 15 min over 72 h to follow cell fate after treatment.
Results
Organoids derived from colorectal peritoneal metastases
A panel of five patient‐derived PM organoid lines was established for this study; four were generated from solid lesions and the fifth from malignant ascites (Fig. 1; Table S1, supporting information). The five organoid lines showed divergent morphologies in vitro, with either predominant tubular formation (TOR10MII, TOR22 and p02‐1), a papillary growth pattern with some tubular formation (TOR14) or a solid growth pattern (TOR17) (Fig. 2 a). These growth patterns are common in colorectal adenocarcinoma histology. Organoid morphology closely resembled the histology of the original metastases. The orthotopic xenograft of TOR10 showed the same histological features as the original carcinoma, with evident mucin lakes comprising more than 50 per cent of the tumour mass, classifying them as mucinous carcinomas. The organoid TOR10MII also produced extracellular mucin, as confirmed by a positive periodic acid Schiff stain (data not shown). Comparison of hotspot mutations between the original tumours and the organoids showed that 13 of 14 mutations found in the tumours were also identified in the matched organoid (Fig. 2 b). Thus, the PM‐derived organoid lines faithfully recapitulated the histological and genetic characteristics of the cancers from which they were derived.
Figure 1.
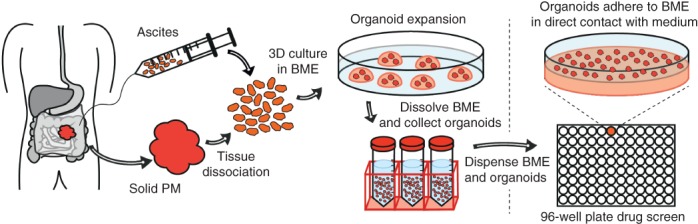
Organoid generation and testing Organoids were generated directly from malignant ascites or from solid lesions by first digesting the tissue enzymatically. Three‐dimensional (3D) tumour organoids were grown out by culturing single cells in 3D basement membrane extract (BME) immersed in basal culture medium. For the in vitro HIPEC experiments, organoids were plated on top of a layer of BME, rather than inside the matrix, to maximize drug exposure and simulate the clinical features of microscopic peritoneal metastases (PMs).
Figure 2.
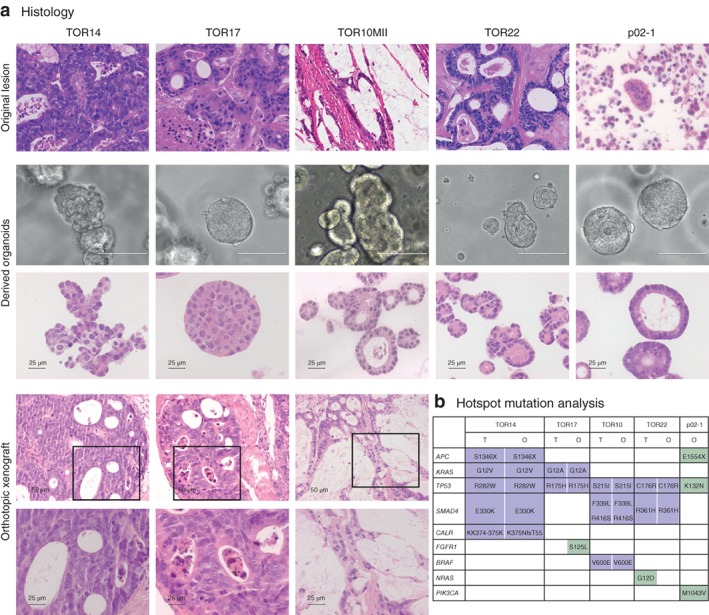
Morphological and molecular comparison of organoids of peritoneal metastases with parent tumours from which they were derived a Histological or cytological (p02‐1) comparison of parent tumours (haematoxylin and eosin stain; TOR14, TOR17, TOR22 and p02‐1: original magnification × 40; TOR10: original magnification × 20) with matched organoids (bright field, scale bar 100 μm; haematoxylin and eosin stain, scale bar 25 μm) and orthotopic xenografts in mouse caecum (haematoxylin and eosin stain, original magnification × 10 and × 40). b Comparison of hotspot mutations in original tumours (T) and matched organoids (O). The tumour cell content of solid peritoneal lesions of p02‐1 was too low (less than 5 per cent) to call mutations reliably.
In vivo models system for hyperthermic intraperitoneal chemotherapy in vitro
Before performing drug screens on the organoid panel, the concentrations of intraperitoneal chemotherapy drugs in 40 consecutive HIPEC procedures were evaluated. Both BSA and perfusate volumes varied widely between patients (Table 1). Importantly, BSA was a poor predictor of abdominal volume in both men and women (Fig. 3 a), which led to variable perfusate concentrations. The calculated median starting concentration was 13·6 (range 9·7–19·8) μmol/l for MMC (Fig. 3 b) and 306 (215–439) μmol/l for oxaliplatin.
Table 1.
Perfusion data for patients undergoing hyperthermic chemotherapy
| No. of patients* | |
|---|---|
| Sex ratio (M : F) | 18 : 22 |
| Body surface area (m2)† | 1·84 (1·55–2·36) |
| Patient volume (ml)† | 6055 (3000–9300) |
| Total perfusate volume (ml)† | 7060 (4800–11 018) |
Unless indicated otherwise;
values are median (range).
Figure 3.
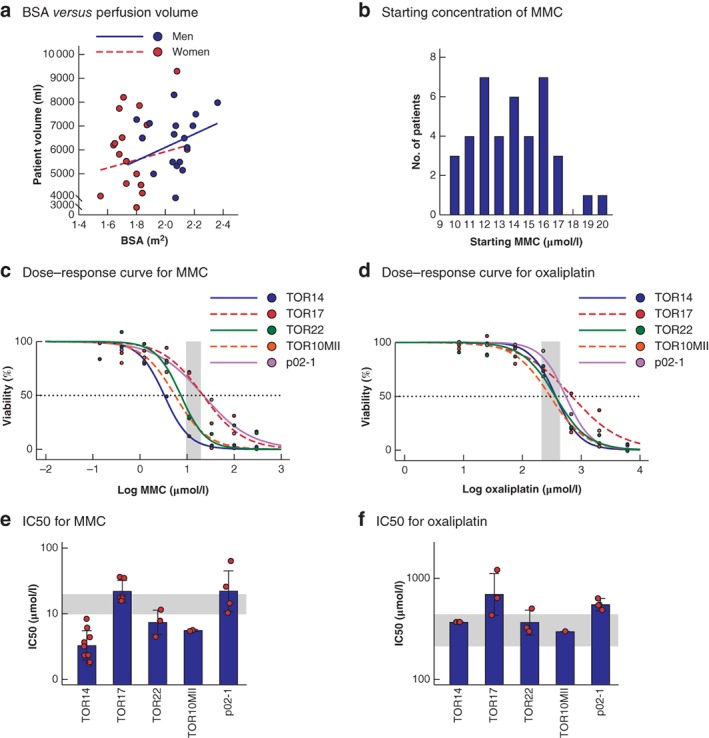
Organoids as an in vitro model to study the efficacy of hyperthermic intraperitoneal chemotherapy regimens a Scatter plot of body surface area (BSA) versus intra‐abdominal perfusion volume, which together determine the concentration of hyperthermic intraperitoneal chemotherapy (HIPEC) drug in the clinic (men: R 2 = 0·019, P = 0·540; women: R 2 = 0·119, P = 0·161). b Frequency distribution histogram of calculated starting concentrations of mitomycin C (MMC) in 40 HIPEC procedures. c,d Dose–response curves illustrating the variation in sensitivity to MMC (c) and oxaliplatin (d) on the individual patient‐derived organoids. The shaded area represents the clinically relevant concentration ranges determined in the clinical cohort. The dashed line crosses the individual curves at the concentration required to eliminate 50 per cent of the tumour cells (IC50). e,f Estimated between‐batch variation (variation between separate in vitro HIPEC experiments) for MMC (e) and oxaliplatin (f). Bars represent the estimated IC50 values pooled over all batches using a non‐linear mixed‐effect model as determined in c and d; error bars show estimated standard deviation in IC50 values between batches, and symbols indicate estimated per‐batch IC50 values (both based on random batch effects included in the model). The shaded area represents the clinically relevant concentration ranges determined in the clinical cohort.
The viability of organoids in response to a range of drug concentrations, including the calculated clinical concentrations, was then assessed. The experimental set‐up of the in vitro drug screen is shown in Fig. 1. Dose–response curves to MMC and oxaliplatin for each organoid showed that drug sensitivity varied considerably between individual patient‐derived organoids (Fig. 3 c,d). The concentration required to eliminate 50 per cent of the tumour cells (IC50) for MMC ranged from 3·3 to 22·2 μmol/l; TOR14 was most sensitive to MMC and p02‐1 least sensitive. The IC50 for oxaliplatin ranged from 297 to 692 μmol/l, with TOR10MII as the most sensitive organoid line (Table 2). The assay proved robust and reproducible, with limited variation in IC50 values in repeated experiments over several months (Fig. 3 e,f). The dose–response curves also showed that the doses of MMC and oxaliplatin achieved clinically were insufficient to eliminate all cancer cells completely in these organoid lines. This highlights the need for improving the efficacy of HIPEC treatment.
Table 2.
Half‐maximal inhibitory concentration for mitomycin C and oxaliplatin by organoid line
| Organoid line | IC50 (μmol/l) | |
|---|---|---|
| Mitomycin C | Oxaliplatin | |
| TOR 14 | 3·3 (2·3, 4·6) | 368 (326, 414) |
| TOR17 | 12·9 (15·5, 30·9) | 692 (375, 1277) |
| TOR22 | 7·4 (4·4, 12·5) | 364 (243, 544) |
| TOR10MII | 5·6 (4·7, 6·6) | 297 (253, 348) |
| P02‐1 | 22·2 (10·8, 45·5) | 549 (449, 672) |
Estimated half‐maximal inhibitory concentrations (IC50) were pooled over all batches using a non‐linear mixed‐effect model. Values in parentheses are 95 per cent confidence intervals.
ATR inhibition increases the efficacy of mitomycin C
Given the recent finding that oxaliplatin‐based HIPEC is not effective in a clinical setting12 and that MMC appeared more effective at clinical concentrations than oxaliplatin in the PM‐organoid model, further improvement of MMC HIPEC was considered more promising, and combination treatment with ATR inhibition was assessed. The ATR inhibitor VE‐821 showed limited toxicity as monotherapy (Fig. S1, supporting information), but greatly sensitized all PM organoids to MMC, as shown by statistically significant reductions of 2·6–12·4‐fold in IC50 values (Fig. 4 a; Table S2, supporting information). Importantly, for each organoid line, the IC50 values for MMC in the combination treatment were below the concentrations of MMC attained clinically.
Figure 4.
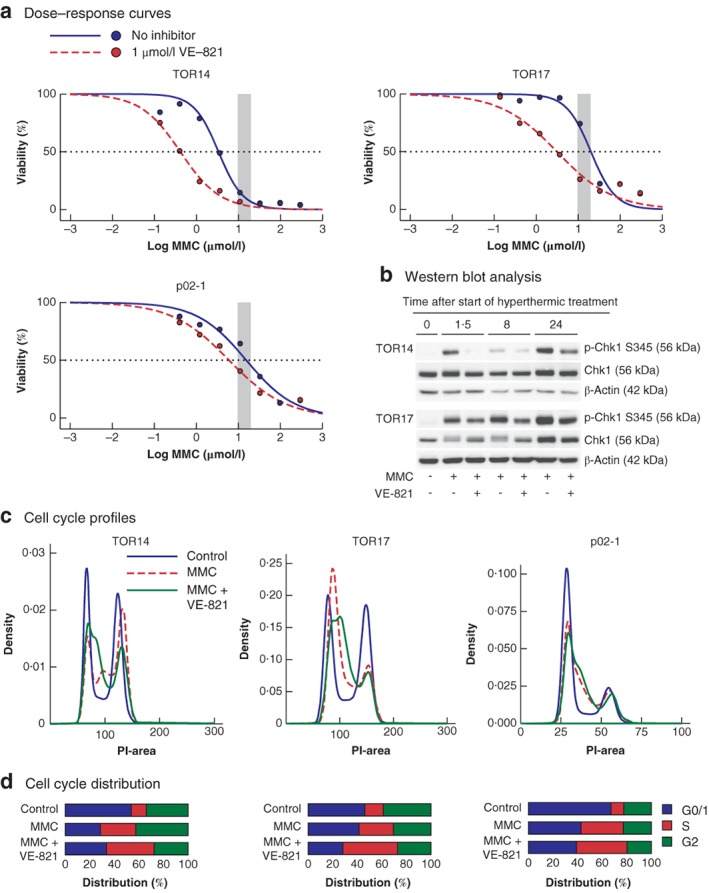
Effects of mitomycin C and ATR inhibition on organoid viability and cell cycle distribution a Dose–response curves comparing mitomycin C (MMC) monotherapy and the combination of MMC and ATR inhibitor VE‐821 in the individual organoids. The shaded area represents the clinically relevant concentration ranges determined in the clinical cohort. The dashed line crosses the individual curves at the concentration required to eliminate 50 per cent of the tumour cells. b Western blot analysis of checkpoint kinase 1 (Chk1) phosphorylation over time, following 90 min of treatment with hyperthermic MMC or a combination of MMC and VE‐821; p‐Chk1 S345, Chk1 phosphorylated on serine 345. c Cell cycle profiles of the different organoids 24 h after treatment (90 min at 42°C) with either no drugs (control), MMC (TOR14, 3 μmol/l; TOR17, 20 μmol/l; p02‐1, 24 μmol/l), or MMC followed by 24 h of treatment with 1 μmol/l VE‐821. The DNA content of individual cells was measured by propidium iodide (PI) intensity. d Quantification of cell cycle distribution. Data for TOR22 and TOR10MII are shown in Fig. S2 (supporting information).
To understand the mechanism of action of the combination therapy, the effects of the drugs on cell cycle distribution and cell fate were characterized. MMC treatment at IC50 concentrations induced rapid phosphorylation of the ATR substrate Checkpoint kinase 1 (Chk1) (Fig. 4 b) and led to cell cycle arrest in the S phase (Fig. 4 c,d). Chk1 phosphorylation was reduced by combining MMC with VE‐821 (Fig. 4 b). However, rather than inducing a shift from S phase towards G2/M phase, co‐treatment with VE‐821 and MMC induced a more pronounced S‐phase arrest than MMC alone (Fig. 4 c,d). Live‐cell imaging of TOR14 over 72 h after MMC treatment confirmed that combination treatment did not increase the fraction of death in mitosis compared with monotherapy with MMC (Videos S1–S4, supporting information). In both conditions, most cell death occurred during a period of interphase (Fig. 5 a,b). Cell death appeared to occur earlier in cells co‐treated with MMC and ATR inhibitors than in cultures treated with MMC alone (Figs S3 and S4, supporting information).
Figure 5.
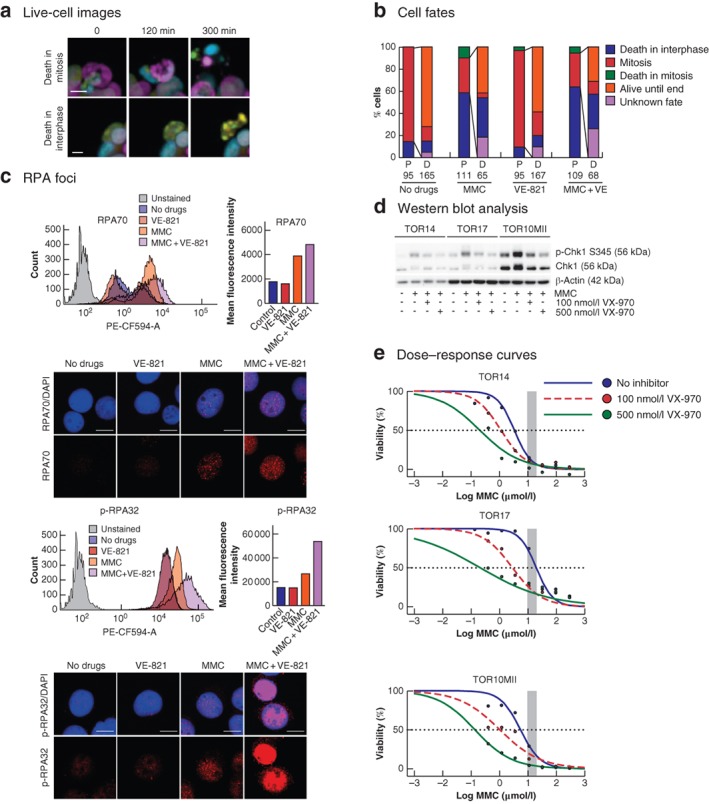
Effects of mitomycin C and ATR inhibition on cell fate and replication stress a Examples of cell death events observed during live‐cell imaging. Upper panel shows cell death in mitosis (cell death is preceded by mitotic chromosome condensation) and lower panel death in interphase (no chromosomal condensation observed before the formation of apoptotic bodies) (scale bar 10 μm). b Quantification of TOR14 cell fates observed during 72 h of live‐cell imaging after treatment with either no drugs, 0·5 μmol/l mitomycin C (MMC) for 90 min, 1 μmol/l VE‐821 for 72 h, or 0·5 μmol/l MMC for 90 min + 1 μmol/l VE‐821 for 72 h. If a parent cell (P) underwent mitosis, the fates of the resulting daughter cells (D) were also followed. c Analysis of replication protein A (RPA) foci in response to MMC or the combination treatment of MMC + VE‐821 in TOR14. Flow cytometry profiles for the different conditions are shown in the frequency distribution plots, and the mean intensities of the RPA or phosphorylated RPA (p‐RPA) signal per cell are plotted separately. PE‐CF594‐A, fluorescence area. Maximum projection confocal images of cells from the same experiment illustrate the increased number of RPA or p‐RPA foci in the combination treatment. The red signal represents the RPA or p‐RPA, and the blue signal shows 4′,6‐diamidino‐2‐phenylindole staining (scale bar 10 μm). d Western blot analysis of checkpoint kinase 1 (Chk1) phosphorylation after 90 min of treatment with hyperthermic MMC or the combination of MMC and VX‐970 (100 and 500 nmol/l); p‐Chk1 S345, Chk1 phosphorylated on serine 345. e Dose–response curves comparing MMC monotherapy and the combination of MMC and VX‐970 (100 and 500 nmol/l) in the individual organoids. The shaded area represents the clinically relevant concentration ranges determined in the clinical cohort. The dashed line crosses the individual curves at the concentration required to eliminate 50 per cent of the tumour cells.
Flow cytometric analysis showed that MMC induced a profound increase in replication protein A (RPA) foci. RPA binds to single‐strand DNA at sites of DNA damage, and the increased presence of RPA foci is indicative of replication stress23, 24. Combined treatment with MMC and VE‐821 slightly increased the number of RPA foci per cell as reflected by the mean fluorescence of RPA70, and induced a pronounced increase in RPA phosphorylation (RPA32, phosphorylated on either serine 4 or 8) (Fig. 5 c). Replication stress can lead to massive chromosome breakage and subsequent cell death23.
To evaluate whether this combination strategy could be translated to the clinical setting, VX‐970, the clinical derivative of VE‐821, was tested in combination with MMC. VX‐970 also inhibited Chk1 phosphorylation induced by MMC in a dose‐dependent fashion (Fig. 5 d), and dramatically lowered the IC50 value of MMC (Fig. 5 e; Table S2, supporting information), similar to VE‐821. This suggests that MMC plus ATR inhibition is a feasible combination strategy in HIPEC for PM from colorectal cancer.
Discussion
Retrospective comparisons between MMC and oxaliplatin as intraperitoneal chemotherapy agents in HIPEC for PM from colorectal cancer are contradictory7, 8, 9, 10. This may be due to heterogeneous patient populations, variation in selection criteria for CRS–HIPEC and differences in the application of perioperative systemic chemotherapy. Furthermore, the intraperitoneal drug concentration during HIPEC varies widely as a consequence of BSA‐based chemotherapeutic dose. This study has shown that BSA correlates poorly with intraperitoneal volume, and perfusate volume influences the concentration to which peritoneal micrometastases are exposed. Finally, even though there may not be a clear superiority of either chemotherapy agent at group level, one drug might perform better than the other in individual patients, depending on the tumour biology of the metastases.
In the present study, organoid technology was used to develop a model system for evaluating the efficacy of HIPEC. Considerable differences in sensitivity to MMC and oxaliplatin were observed between patient‐derived organoid lines. Moreover, hyperthermic chemotherapy at commonly attained clinical doses had only a minor effect on the viability of several organoid lines. This indicates that current HIPEC regimens are insufficient to eradicate residual microscopic disease at least in a subgroup of patients with colorectal PMs, and this is consistent with the high recurrence rates observed after CRS–HIPEC and the negative result from the Prodige 7 trial7, 11, 12.
In an effort to improve MMC efficacy, combined treatment with MMC and inhibitors of the replication checkpoint kinase ATR was assessed in this study. The addition of ATR inhibitors to MMC was effective in eliminating microscopic metastases at clinical doses of MMC in all PM‐derived organoids tested. These findings are in line with a model in which ATR inhibition causes unrestrained replication origin firing despite the presence of DNA damage caused by MMC. Excess origin firing increases the number of RPA foci and exhausts the nuclear RPA pool. Ultimately, this leads to massive chromosome breakage and a type of cell death known as replication catastrophe23.
This study provides insights into the mechanism of action of ATR inhibition in combination with a standard chemotherapy used in HIPEC. Understanding how a drug combination works before it enters the clinical setting is important as it can increase the chance of success. It can yield biomarkers for monitoring therapeutic response as well as predictive biomarkers for trial stratification. Of note, all organoids in this cohort have a mutation in TP53. ATR inhibition is particularly potent in tumours with deficiencies in the ATM pathway, such as TP53‐mutant cancers25. An important future goal is to further optimize organoid establishment and long‐term expansion, in order to create an organoid panel of PMs from colorectal cancer that completely reflects the diversity of mutational and histological subtypes. Further research is also required to find the optimal timing, dose and route of administration (intraperitoneal or systemic) of ATR inhibitors in the clinical HIPEC setting. By modelling the effects of sequential or concomitant treatment, the in vitro model could aid in rational trial design for ATR inhibition in HIPEC.
Several factors could potentially limit the predictive value of the present model. First, the culture matrix used in the model is an abstraction of the microenvironment of the peritoneum, which includes fibroblasts and other stromal and immune cells. These components potentially influence drug response. Second, HIPEC is nearly always preceded by CRS which induces cytokine production26 that could also influence drug response. Third, intrapatient interlesion heterogeneity may provide an additional level of complexity that is not modelled in the present organoid collection27, 28. Despite these limitations, this model of PM from colorectal cancer is a robust in vitro system that allows rapid evaluation of existing and novel HIPEC strategies in a biologically relevant setting.

Supporting information
Appendix S1 Additional methods
Table S1 Clinical and tumour characteristics of the patient‐derived PM organoids
Table S2 Decrease in IC50 by combining MMC with ATR inhibitors
Fig. S1 Toxicity of 72 hours monotherapy with VE‐821 or VX‐970 in TOR14 & TOR17
Fig. S3 Live‐cell imaging fate profiles of individual cells in TOR14 organoids treated with no drugs, MMC, ATR inhibitor VE‐821 or both
Fig. S4 Percentage of all cell deaths that occurred in each 10‐hour interval after HIPEC, to compare timing of death in interphase, in mitosis and post‐mitotic death in the four treatment conditions. The peak of cell death in interphase occurs earlier in co‐treatment of MMC + VE‐821, compared with MMC alone. N = total number of cell death events
Video S1 Live‐cell imaging of TOR14 following 90‐minute treatment with no drugs <MP4 file>
Video S2 Live‐cell imaging of TOR14 following 90‐minute treatment with VE‐821 < MP4 file>
Video S3 Live‐cell imaging of TOR14 following 90‐minute treatment with MMC < MP4 file>
Video S4 Live‐cell imaging of TOR14 following 90‐minute treatment with MMC combined with VE‐821 < MP4 file>
Acknowledgements
The authors thank O. Imhof for the HIPEC perfusion reports, J. Laoukili and E. Küçükköse for help with organoid transfection, L. Wijler and J. Laoukili for setting up high‐throughput drug screening in the authors' laboratory, and C. Huysentruyt for the microscopic image of p02‐1. This work was supported by the Dutch Cancer Society (UU2014–6617 to I.U.), and the Marie Curie Network Ploidynet which is funded by the European Union Seventh Framework Programme (FP7/2007‐2013) under Grant Agreement number 607722 (to A.C.F.B.). The sponsors had no influence on the study design, or collection, analysis and interpretation of data.
Disclosure: The authors declare no conflict of interest.
Presented in part to the Peritoneal Surface Oncology Group International meeting, Paris, France, September, 2018
References
- 1. Segelman J, Granath F, Holm T, Machado M, Mahteme H, Martling A. Incidence, prevalence and risk factors for peritoneal carcinomatosis from colorectal cancer. Br J Surg 2012; 99: 699–705. [DOI] [PubMed] [Google Scholar]
- 2. Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP. Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Ann Surg 2006; 243: 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bushati M, Rovers KP, Sommariva A, Sugarbaker PH, Morris DL, Yonemura Y et al The current practice of cytoreductive surgery and HIPEC for colorectal peritoneal metastases: results of a worldwide web‐based survey of the Peritoneal Surface Oncology Group International (PSOGI). Eur J Surg Oncol 2018; 44: 1942–1948. [DOI] [PubMed] [Google Scholar]
- 4. Franko J, Ibrahim Z, Gusani NJ, Holtzman MP, Bartlett DL, Zeh HJ III. Cytoreductive surgery and hyperthermic intraperitoneal chemoperfusion versus systemic chemotherapy alone for colorectal peritoneal carcinomatosis. Cancer 2010; 116: 3756–3762. [DOI] [PubMed] [Google Scholar]
- 5. Elias D, Gilly F, Boutitie F, Quenet F, Bereder JM, Mansvelt B et al Peritoneal colorectal carcinomatosis treated with surgery and perioperative intraperitoneal chemotherapy: retrospective analysis of 523 patients from a multicentric French study. J Clin Oncol 2010; 28: 63–68. [DOI] [PubMed] [Google Scholar]
- 6. Verwaal VJ, Bruin S, Boot H, van Slooten G, van Tinteren H. 8‐year follow‐up of randomized trial: cytoreduction and hyperthermic intraperitoneal chemotherapy versus systemic chemotherapy in patients with peritoneal carcinomatosis of colorectal cancer. Ann Surg Oncol 2008; 15: 2426–2432. [DOI] [PubMed] [Google Scholar]
- 7. van Eden WJ, Kok NFM, Woensdregt K, Huitema ADR, Boot H, Aalbers AGJ. Safety of intraperitoneal Mitomycin C versus intraperitoneal oxaliplatin in patients with peritoneal carcinomatosis of colorectal cancer undergoing cytoreductive surgery and HIPEC. Eur J Surg Oncol 2018; 44: 220–227. [DOI] [PubMed] [Google Scholar]
- 8. Prada‐Villaverde A, Esquivel J, Lowy AM, Markman M, Chua T, Pelz J et al The American Society of Peritoneal Surface Malignancies evaluation of HIPEC with Mitomycin C versus Oxaliplatin in 539 patients with colon cancer undergoing a complete cytoreductive surgery. J Surg Oncol 2014; 110: 779–785. [DOI] [PubMed] [Google Scholar]
- 9. Leung V, Huo YR, Liauw W, Morris DL. Oxaliplatin versus Mitomycin C for HIPEC in colorectal cancer peritoneal carcinomatosis. Eur J Surg Oncol 2017; 43: 144–149. [DOI] [PubMed] [Google Scholar]
- 10. Hompes D, D'Hoore A, Wolthuis A, Fieuws S, Mirck B, Bruin S et al The use of Oxaliplatin or Mitomycin C in HIPEC treatment for peritoneal carcinomatosis from colorectal cancer: a comparative study. J Surg Oncol 2014; 109: 527–532. [DOI] [PubMed] [Google Scholar]
- 11. Kuijpers AM, Mirck B, Aalbers AG, Nienhuijs SW, de Hingh IH, Wiezer MJ et al Cytoreduction and HIPEC in The Netherlands: nationwide long‐term outcome following the Dutch protocol. Ann Surg Oncol 2013; 20: 4224–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quenet F, Elias D, Roca L, Goere D, Ghouti L, Pocard M et al A UNICANCER phase III trial of hyperthermic intra‐peritoneal chemotherapy (HIPEC) for colorectal peritoneal carcinomatosis (PC): PRODIGE 7. J Clin Oncol 2018; 36: LBA3503. [Google Scholar]
- 13. van Driel WJ, Koole SN, Sikorska K, Schagen van Leeuwen JH, Schreuder HWR, Hermans RHM et al Hyperthermic intraperitoneal chemotherapy in ovarian cancer. N Engl J Med 2018; 378: 230–240. [DOI] [PubMed] [Google Scholar]
- 14. Kemmel V, Mercoli HA, Meyer N, Brumaru D, Romain B, Lessinger JM et al Mitomycin C pharmacokinetics as predictor of severe neutropenia in hyperthermic intraperitoneal therapy. Ann Surg Oncol 2015; 22(Suppl 3): S873–S879. [DOI] [PubMed] [Google Scholar]
- 15. Lecona E, Fernandez‐Capetillo O. Targeting ATR in cancer. Nat Rev Cancer 2018; 18: 586–595. [DOI] [PubMed] [Google Scholar]
- 16. Gargiulo D, Kumar GS, Musser SS, Tomasz M. Structural and function modification of DNA by mitomycin C. Mechanism of the DNA sequence specificity of mitomycins. Nucleic Acids Symp Ser 1995: 169–170. [PubMed] [Google Scholar]
- 17. Toledo L, Neelsen KJ, Lukas J. Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell 2017; 66: 735–749. [DOI] [PubMed] [Google Scholar]
- 18. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A et al Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015; 161: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández‐Mateos J, Khan K et al Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018; 359: 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verissimo CS, Overmeer RM, Ponsioen B, Drost J, Mertens S, Verlaan‐Klink I et al Targeting mutant RAS in patient‐derived colorectal cancer organoids by combinatorial drug screening. Elife 2016; 5: e18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Ruth S, Mathôt RA, Sparidans RW, Beijnen JH, Verwaal VJ, Zoetmulder FA. Population pharmacokinetics and pharmacodynamics of mitomycin during intraoperative hyperthermic intraperitoneal chemotherapy. Clin Pharmacokinet 2004; 43: 131–143. [DOI] [PubMed] [Google Scholar]
- 22. Mehta AM, Van den Hoven JM, Rosing H, Hillebrand MJ, Nuijen B, Huitema AD et al Stability of oxaliplatin in chloride‐containing carrier solutions used in hyperthermic intraperitoneal chemotherapy. Int J Pharm 2015; 479: 23–27. [DOI] [PubMed] [Google Scholar]
- 23. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK et al ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013; 155: 1088–1103. [DOI] [PubMed] [Google Scholar]
- 24. Byrne BM, Oakley GG. Replication protein A, the laxative that keeps DNA regular: the importance of RPA phosphorylation in maintaining genome stability. Semin Cell Dev Biol 2019; 86: 112–120. [DOI] [PubMed] [Google Scholar]
- 25. Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA et al Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7: 428–430. [DOI] [PubMed] [Google Scholar]
- 26. Leijte GP, Custers H, Gerretsen J, Heijne A, Roth J, Vogl T et al Increased plasma levels of danger‐associated molecular patterns are associated with immune suppression and postoperative infections in patients undergoing cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. Front Immunol 2018; 9: 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ubink I, van Eden WJ, Snaebjornsson P, Kok NFM, van Kuik J, van Grevenstein WMU et al Histopathological and molecular classification of colorectal cancer and corresponding peritoneal metastases. Br J Surg 2018; 105: e204–e211. [DOI] [PubMed] [Google Scholar]
- 28. Roerink SF, Sasaki N, Lee‐Six H, Young MD, Alexandrov LB, Behjati S et al Intra‐tumour diversification in colorectal cancer at the single‐cell level. Nature 2018; 556: 457–462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Additional methods
Table S1 Clinical and tumour characteristics of the patient‐derived PM organoids
Table S2 Decrease in IC50 by combining MMC with ATR inhibitors
Fig. S1 Toxicity of 72 hours monotherapy with VE‐821 or VX‐970 in TOR14 & TOR17
Fig. S3 Live‐cell imaging fate profiles of individual cells in TOR14 organoids treated with no drugs, MMC, ATR inhibitor VE‐821 or both
Fig. S4 Percentage of all cell deaths that occurred in each 10‐hour interval after HIPEC, to compare timing of death in interphase, in mitosis and post‐mitotic death in the four treatment conditions. The peak of cell death in interphase occurs earlier in co‐treatment of MMC + VE‐821, compared with MMC alone. N = total number of cell death events
Video S1 Live‐cell imaging of TOR14 following 90‐minute treatment with no drugs <MP4 file>
Video S2 Live‐cell imaging of TOR14 following 90‐minute treatment with VE‐821 < MP4 file>
Video S3 Live‐cell imaging of TOR14 following 90‐minute treatment with MMC < MP4 file>
Video S4 Live‐cell imaging of TOR14 following 90‐minute treatment with MMC combined with VE‐821 < MP4 file>