

Abstract
Regorafenib 160 mg orally once daily (QD) 3 weeks on/1 week off is approved in colorectal cancer, gastrointestinal stromal tumors and hepatocellular carcinoma. We established the safety and pharmacokinetics (PK) of regorafenib combined with cetuximab in advanced refractory solid tumors. This was a phase 1, open‐label, dose‐escalation study (NCT01973868) in patients with advanced/metastatic solid tumors who progressed after standard therapy. Regorafenib was administered at various dose levels QD continuously or intermittently (3 weeks on/1 week off) combined with intravenous cetuximab 250 mg/m2 weekly. The primary objectives were safety, PK and maximum tolerated dose (MTD). The secondary objective was tumor response. Dose‐limiting toxicities (DLTs) were evaluated in Cycle 1. Of 42 treated patients, 31 received regorafenib intermittently (120 mg, n = 8; 160 mg, n = 23) and 11 continuously (60 mg, n = 5; 100 mg, n = 6) plus cetuximab. The continuous arm was terminated due to low tolerable dose. In the intermittent arm, one DLT (grade 3 hand–foot skin reaction) was observed at 120 mg but none at 160 mg, therefore 160 mg/day was declared as the MTD in combination with cetuximab. The most common all‐grade treatment‐emergent adverse events were fatigue (52%), hypophosphatemia (48%) and diarrhea (40%). One grade 3 cetuximab‐related dermatitis acneiform was observed. No clinically relevant drug–drug interactions were observed. Five patients (21%) had a partial response. Regorafenib 160 mg QD (3 weeks on/1 week off) plus standard dose of cetuximab was well tolerated with no unexpected toxicities and promising signs of efficacy.
Keywords: regorafenib, cetuximab, metastatic colorectal cancer, phase 1
Short abstract
What's new?
Cancer treatment approaches frequently target protein kinases active in cancer cells, but resistance development limits long‐term success. In this phase 1b study, the authors combined two kinase inhibitors, regorafenib (oral) and cetuximab (intravenous), to overcome intrinsic and acquired resistance in epidermal growth factor receptor‐sensitive and ‐resistant tumors. The combination was well tolerated with no unexpected toxicities and promising initial signals of efficacy at the approved doses of both drugs in patients with advanced solid tumors.
Abbreviations
- AE
adverse event
- AUC
area under the plasma concentration–time curve
- Cmax
maximum observed drug concentration
- CV
geometric coefficient of variation
- DLT
dose‐limiting toxicity
- EGFR
epidermal growth factor receptor
- HFSR
hand–foot skin reaction
- IV
intravenous
- mCRC
metastatic colorectal cancer
- MTD
maximum tolerated dose
- PK
pharmacokinetics
- PR
partial response
- QD
once daily
- TEAE
treatment‐emergent adverse event
- VEGFR
vascular endothelial growth factor receptor.
Introduction
Constitutive or increased activity of receptor tyrosine kinases, including vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR), as well as their downstream signaling pathways, play an important role in cancer pathogenesis.1 Although targeting these kinase‐signaling pathways have demonstrated therapeutic benefit in solid tumors, drug resistance leading to disease relapse can occur, highlighting the need for multitargeted treatment options that can overcome or delay resistance.
Regorafenib is an oral multikinase inhibitor shown in preclinical studies to target VEGFR1–3, platelet‐derived growth factor receptors, RAF, TIE2 and other kinases involved in angiogenesis, proliferation, the tumor microenvironment, metastasis and tumor immunity.2, 3 The efficacy and safety of regorafenib has been demonstrated in randomized, placebo‐controlled, phase 3 trials in patients with metastatic colorectal cancer (mCRC) refractory to standard therapies, advanced gastrointestinal stromal tumors refractory to standard therapies and hepatocellular carcinoma (HCC) previously treated with sorafenib, which formed the basis for its regulatory approval as a single agent in these indications.4, 5, 6, 7, 8, 9 A consistent safety profile was observed across these studies, with hand–foot skin reaction (HFSR), hypertension, fatigue and diarrhea among the most common treatment‐emergent adverse events (TEAEs). The approved dose for single‐agent regorafenib across indications is 160 mg once daily (QD) on an intermittent schedule of 3 weeks on followed by 1 week off therapy to comprise a cycle of 4 weeks.4, 5 The dose was derived from a phase 1 dose‐escalation study in patients with advanced solid tumors,10 while a continuous dosing schedule evaluated in a parallel, phase 1 study, which established 100 mg regorafenib QD as the maximum tolerated dose (MTD), was not taken forward in clinical development.11
Cetuximab is an anti‐EGFR monoclonal antibody indicated (400 mg/m2 loading dose followed by 250 mg/m2 weekly infusion) as a single agent or in combination for the treatment of patients with EGFR‐expressing, KRAS wild‐type mCRC and those with squamous cell carcinoma of the head and neck.12, 13 The most common adverse reactions associated with cetuximab include cutaneous adverse reactions (including rash, pruritus and nail changes), headache, diarrhea and infection. Preclinical models have demonstrated that the combination of regorafenib and cetuximab may overcome intrinsic and acquired resistance in EGFR‐sensitive and EGFR‐resistant tumors, and may provide an improved clinical benefit over either drug alone in certain tumor types.14 Furthermore, in vivo preclinical data have demonstrated decreased angiogenesis and increased tumor and endothelial cell apoptosis with combined inhibition of VEGF and EGFR.15 In a recently published phase 1 study of regorafenib plus cetuximab in patients with metastatic cancer refractory to standard therapies, regorafenib 80 mg QD plus cetuximab 200 mg/m2 loading dose, followed by cetuximab 150 mg/m2 every week, was determined as the MTD and demonstrated preliminary activity in mCRC.16
In this phase 1, dose‐finding study, we aimed to establish the safety and pharmacokinetics (PK) of regorafenib (continuous and intermittent dosing) in combination with the standard dose of cetuximab in patients with advanced solid tumors.
Materials and Methods
Patient population
Patients ≥18 years of age with a histologically or cytologically confirmed locally advanced or metastatic solid tumors who were unsuitable for, or no longer responding to standard therapy, or for whom regorafenib or cetuximab was considered as a standard treatment, were eligible for inclusion. Other key inclusion criteria included an Eastern Cooperative Oncology Group performance status of 0 or 1, no KRAS mutation in patients with mCRC, a life expectancy of ≥3 months and adequate bone marrow (platelet ≥100,000/mm3, absolute neutrophil count ≥1,000/mm3), liver (aspartate aminotransferase ≤2.5 × upper limit of normal) and renal function (creatinine clearance ≥30 ml/min). Patients were excluded if they had received prior treatment with regorafenib; previously discontinued cetuximab due to toxicity or intolerance; had known metastatic brain or meningeal tumors; a history of organ allograft or cardiac disease; or were diagnosed with human immunodeficiency virus or active hepatitis B/C. Further exclusion criteria included major surgery within 4 weeks of start of study treatment; a nonhealing wound, ulcer, or bone fracture; uncontrolled hypertension or significant acute gastrointestinal disorders with diarrhea as a major symptom; arterial or venous thrombotic or embolic events; and pregnancy or breastfeeding. In addition, other anticancer treatments were not permitted during the study.
Study design and treatment
This was an open‐label, dose‐escalation, phase 1b study of regorafenib in combination with cetuximab conducted at four sites in the USA (NCT01973868; Supporting Information Fig. S1). The primary objectives were to determine the safety, tolerability and MTD of regorafenib in combination with cetuximab, and to characterize the PK of this combination. The secondary objective was the preliminary evaluation of tumor response for this combination. All patients provided written informed consent before any study procedure. The trial was approved by each center's ethics committee or institutional review board and complied with Good Clinical Practice guidelines, the Declaration of Helsinki and applicable local laws.
Before the start of combination treatment (run‐in period), patients received a single dose of regorafenib at the assigned dose level on Day −14 (±2 days) for PK evaluation only, followed by a loading dose of intravenous (IV) cetuximab 400 mg/m2 on Day −7. The single regorafenib dose on Day −14 was later removed from the study. In the combination period, regorafenib was administered at various dose levels QD intermittently (3 weeks on/1 week off) or continuously in combination with a standard dose of IV cetuximab 250 mg/m2 weekly in a cycle of 4 weeks. After one cycle with a starting dose of regorafenib 120 mg intermittently, the decision to escalate or reduce the regorafenib dose was determined by the investigator based on toxicity/tolerability. In the intermittent arm (3 weeks on/1 week off), if tolerable, regorafenib was escalated to 160 mg QD plus cetuximab; if not tolerable, regorafenib was reduced to 80 mg QD plus cetuximab. In the continuous arm, if tolerable at the starting dose level of regorafenib 120 mg intermittently plus cetuximab, regorafenib was started at 100 mg QD continuously in a 4‐week cycle plus cetuximab; if not tolerable at regorafenib 120 mg intermittently or 100 mg continuously, regorafenib was reduced to 60 mg QD continuously plus cetuximab. The continuous treatment arm was prematurely terminated in the study. Patients could continue therapy until tumor progression, unacceptable toxicity, consent withdrawal or withdrawal from the study.
Determination of the MTD was based on the conventional 3 + 3 study design, with three patients per dose level, and up to three additional patients enrolled at the same dose level if one dose‐limiting toxicity (DLT) occurred; if two DLTs occurred in up to six patients, the dose would be declared not tolerable. If no DLT occurred, the dose was escalated in the next cohort of three patients. The MTD was determined as the highest dose at which no more than one out of the six treated patients experienced a DLT.
Assessments
Patients valid for DLT assessment were defined as those who completed Cycle 1 and received at least 80% of the planned dose. DLTs, evaluated in Cycle 1, were regarded by the investigator and/or sponsor to be causally related to the study drug combination, according to hematologic and nonhematologic criteria. Hematologic criteria were absolute neutrophil count <500/mm3 for >7 days, grade 4 febrile neutropenia, platelet count <25,000/mm3 and grade 3 hemorrhage associated with thrombocytopenia of grade ≥3. Nonhematologic criteria were nonhematologic grade 3 or 4 toxicity, except for grade 3 electrolyte imbalances without clinical symptoms, grade 3 liver function test recovered within 7 days, grade 3 hypertension recovered within 7 days with antihypertensive therapy, grade 3 infusion reaction controlled within 24 hr, gastrointestinal toxicity responsive to antiemetics and grade 3 vasovagal reaction. Adverse events (AEs) were recorded from the time written informed consent was provided for study participation. AEs were classified in accordance with the Medical Dictionary for Regulatory Activities (MedDRA) version 20.0 and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‐CTCAE) version 4.03. Drug safety was assessed continuously throughout the study, including a 30‐day follow‐up period after the last study drug administration.
Blood samples for PK assessment of regorafenib and its two active metabolites M‐2 and M‐5 were collected predose and at 2, 4, 8 and 24 hr postdose on Day −14 and Days 1 and 15 of Cycle 1. Blood samples for PK assessment of cetuximab were collected preinfusion, at the end of infusion (approximately 1 hr after start of infusion) and at 4, 6, 10 and 26 hr postinfusion on Days 1 and 15 of Cycle 1. Primary PK parameters included area under the plasma concentration–time curve (AUC) and maximum observed drug concentration (C max).
Tumor response was evaluated based on investigator‐assessed Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 at baseline and in the last week of Cycle 2.
Statistical analysis
All analyses were descriptive in nature. All patients who received at least one dose of study drug were included in the safety analysis; those who also had postbaseline efficacy data or adequate samples were included in the efficacy and PK analyses, respectively.
Results
Of the 66 patients assessed for eligibility (study start November 21, 2013 to last patient last visit February 2, 2018), 42 patients received study treatment. A total of 31 patients received regorafenib intermittently plus cetuximab (regorafenib 120 mg, n = 8; 160 mg, n = 23) and 11 patients received regorafenib continuously plus cetuximab (regorafenib 60 mg, n = 5; 100 mg, n = 6).
Demographics and baseline patient characteristics are shown for each cohort in Table 1. Overall, the median age was 60 years (range: 23–79), and the most common cancers were colorectal cancer (CRC; n = 10; 24%) and pancreatic adenocarcinoma (n = 7; 17%). All 42 patients had received prior systemic anticancer therapy and 22 (52%) had received prior radiotherapy.
Table 1.
Demographics and baseline characteristics (safety analysis set)
| Continuous regorafenib 60 mg plus cetuximab (n = 5) | Continuous regorafenib 100 mg plus cetuximab (n = 6) | Intermittent regorafenib 120 mg plus cetuximab (n = 8) | Intermittent regorafenib 160 mg plus cetuximab (n = 23) | Total (n = 42) | |
|---|---|---|---|---|---|
| Median age, years (range) | 63 (53–72) | 57 (40–65) | 57 (40–69) | 60 (23–79) | 60 (23–79) |
| Male, n (%) | 0 | 1 (17) | 7 (88) | 13 (57) | 21 (50) |
| Ethnicity, n (%) | |||||
| American Indian or Alaska Native | 0 | 0 | 1 (13) | 0 | 1 (2) |
| Black or African American | 0 | 0 | 0 | 2 (9) | 2 (5) |
| White | 4 (80) | 5 (83) | 7 (88) | 19 (83) | 35 (83) |
| Not reported | 1 (20) | 1 (17) | 0 | 2 (9) | 4 (10) |
| Median BMI, kg/m2 (range) | 21.6 (17.6–28.8) | 25.1 (19.3–30.4) | 26.2 (18.4–33.2) | 23.4 (18.4–35.1) | 23.6 (17.6–35.1) |
| Alcohol use, n (%) | |||||
| Abstinent | 3 (60) | 4 (67) | 2 (25) | 13 (57) | 22 (52) |
| Light | 2 (40) | 2 (33) | 5 (63) | 10 (43) | 19 (45) |
| Moderate | 0 | 0 | 1 (13) | 0 | 1 (2) |
| Smoking status, n (%) | |||||
| Never | 1 (20) | 5 (83) | 1 (13) | 13 (57) | 20 (48) |
| Former | 4 (80) | 1 (17) | 5 (63) | 9 (39) | 19 (45) |
| Current | 0 | 0 | 2 (25) | 1 (4) | 3 (7) |
| Cancer type, n (%) | |||||
| Colorectal cancer | 3 (60) | 4 (67) | 0 | 3 (13) | 10 (24) |
| Pancreatic adenocarcinoma | 1 (20) | 0 | 0 | 6 (26) | 7 (17) |
| Hepatocellular carcinoma | 0 | 0 | 1 (13) | 2 (9) | 3 (7) |
| Non‐small cell lung cancer | 0 | 0 | 2 (25) | 1 (4) | 3 (7) |
| Breast cancer | 0 | 0 | 0 | 2 (9) | 2 (5) |
| Bladder cancer | 0 | 0 | 2 (25) | 0 | 2 (5) |
| Other1 | 1 (20) | 2 (33) | 3 (38) | 9 (39) | 15 (36) |
| Histology, n (%) | |||||
| Adenocarcinoma 2 | 3 (60) | 5 (83) | 1 (13) | 14 (61) | 23 (55) |
| Hepatocellular carcinoma | 0 | 0 | 1 (13) | 2 (9) | 3 (7) |
| Squamous cell carcinoma | 1 (20) | 0 | 2 (25) | 0 | 3 (7) |
| Adenoid cystic carcinoma | 0 | 0 | 1 (13) | 1 (4) | 2 (5) |
| Neuroendocrine carcinoma | 0 | 0 | 1 (13) | 1 (4) | 2 (5) |
| Urothelial (transitional cell) carcinoma in situ | 0 | 0 | 2 (25) | 0 | 2 (5) |
| Small cell carcinoma, NOS | 0 | 1 (17) | 0 | 0 | 1 (2) |
| Carcinoma NOS | 0 | 0 | 0 | 1 (4) | 1 (2) |
| Other | 1 (20) | 0 | 0 | 4 (17) | 5 (12) |
| Median time since initial diagnosis, months (range) | 25 (17–49) | 26 (9–42) | 22 (10–54) | 37 (4–125) | 25 (4–125) |
| Any prior systemic anticancer therapy | |||||
| Yes | 5 (100) | 6 (100) | 8 (100) | 23 (100) | 42 (100) |
| No | 0 | 0 | 0 | 0 | 0 |
| Any prior radiotherapy | |||||
| Yes | 2 (40) | 3 (50) | 5 (63) | 12 (52) | 22 (52) |
| No | 3 (60) | 3 (50) | 3 (38) | 11 (48) | 20 (48) |
Other tumor types include adenoid cystic carcinoma, high‐grade neuroendocrine tumor, submandibular gland adenoid cystic carcinoma, cholangiocarcinoma, primary malignant neuroendocrine tumor of the pancreas, anal adenocarcinoma, metastatic basal cell carcinoma, prostate cancer, adenocarcinoma of the rectum, adenocarcinoma of the gastroesophageal junction and adenocarcinoma of the distal esophagus.
Adenocarcinoma includes adenocarcinoma, adenocarcinoma NOS, adenocarcinoma in situ NOS, adenocarcinoma of the distal esophagus (poorly differentiated), ductal adenocarcinoma and invasive, ductal adenocarcinoma.
Abbreviations: BMI, body mass index; NOS, not otherwise specified.
Regorafenib was administered for a median of two cycles (range: 0–19) over a median treatment duration of 49 days (range: 0–539); cetuximab was administered for a median of two cycles (range: 0–19) over a treatment duration of 8 weeks (range: 0–76; Table 3). Reasons for discontinuation were radiologic disease progression (n = 22; 52%), withdrawal of consent (n = 7; 17%), clinical disease progression (n = 5; 12%), AE associated with clinical disease progression (n = 3, 7%), AE not associated with clinical disease progression (n = 3; 7%), death (n = 1; 2%) and other (lost to follow up; n = 1; 2%).
Table 3.
Overview of treatment duration and TEAEs during the combination treatment phase
| Continuous regorafenib 60 mg plus cetuximab (n = 5) | Continuous regorafenib 100 mg plus cetuximab (n = 6) | Intermittent regorafenib 120 mg plus cetuximab (n = 8) | Intermittent regorafenib 160 mg plus cetuximab (n = 23) | Total (n = 42) | |
|---|---|---|---|---|---|
| Treatment duration, median (range) | |||||
| Regorafenib, days | 70 (1–139) | 28 (1–70) | 63 (32–539) | 35 (0–277) | 49 (0–539) |
| Cetuximab, weeks | 9.0 (0–19) | 4.5 (0–9) | 9.0 (4–76) | 6.0 (1–39) | 8.0 (0–76) |
| Any TEAE, n (%) | 5 (100) | 6 (100) | 8 (100) | 22 (96) | 41 (98) |
| Grade 3 or 4 | 3 (60) | 4 (67) | 8 (100) | 17 (74) | 32 (76) |
| Grade ≥3 | 5 (100) | 6 (100) | 8 (100) | 19 (83) | 38 (90) |
| Grade 5 | 2 (40) | 2 (33)1 | 0 | 2 (9) | 6 (14) |
| Serious | 5 (100) | 3 (50) | 4 (50) | 15 (65) | 27 (64) |
| Leading to dose modification | 3 (60) | 2 (33) | 7 (88) | 10 (44) | 22 (52) |
| Leading to treatment discontinuation | 0 | 2 (33) | 0 | 3 (13) | 5 (12) |
TEAEs reported in the run‐in period are reported in the manuscript text.
One death was assessed as related to regorafenib (acute hepatic failure).
Abbreviation: TEAE, treatment‐emergent adverse event.
Maximum tolerated dose
In the continuous arm, four out of six treated patients were evaluable for DLT at regorafenib 100 mg plus standard cetuximab dose, and two DLTs were reported in two patients (grade 3 hoarseness and fatal liver failure). The regorafenib dose was reduced to 60 mg continuously, and three out of five treated patients were evaluable without DLTs. This arm was terminated because a higher regorafenib dose was tolerable in the intermittent arm for the combination.
In the intermittent regorafenib 120 mg arm, one DLT of grade 3 HFSR was reported in seven evaluable patients and no DLT was reported at the regorafenib 160 mg dose (Table 2). Therefore, the MTD was declared at 160 mg regorafenib taken orally QD for 3 weeks on followed by 1 week off therapy to comprise a cycle of 4 weeks (standard approved monotherapy dose for regorafenib) plus the standard approved monotherapy dose of cetuximab (initial 400 mg/m2 followed by 250 mg/m2 weekly).
Table 2.
Summary of DLTs
| Continuous regorafenib 60 mg plus cetuximab (n = 5) | Continuous regorafenib 100 mg plus cetuximab (n = 6) | Intermittent regorafenib 120 mg plus cetuximab (n = 8) | Intermittent regorafenib 160 mg plus cetuximab (n = 23) | Total (n = 42) | |
|---|---|---|---|---|---|
| Patients evaluable for DLT1, n | 3 | 4 | 7 | 13 | 27 |
| DLT observed, n | 0 | 2 | 1 | 0 | 3 |
| Event details | – | Grade 3 hoarseness; Fatal liver failure | Grade 3 HFSR | – | – |
Patients valid for DLT assessment were defined as those who completed Cycle 1 and received at least 80% of the planned doses; includes escalation and expansion cohorts.
Abbreviations: DLT, dose‐limiting toxicity; HFSR, hand–foot skin reaction.
Safety
All 42 patients were evaluable for safety analyses and 98% of patients experienced at least one TEAE (Table 3). The most common all‐grade TEAEs were fatigue (52%), hypophosphatemia (48%) and diarrhea (40%; Supporting Information Table S1). The most common regorafenib‐related TEAEs were hypophosphatemia (38%), fatigue (29%) and diarrhea (24%), while the most common cetuximab‐related TEAEs were dermatitis acneiform (31%), diarrhea (29%) and fatigue (29%). The most common grade ≥3 drug‐related TEAEs were hypophosphatemia (24%), fatigue, and lipase increase (both 10%) for regorafenib and hypophosphatemia (7%), diarrhea, fatigue, hypertension and gamma‐glutamyltransferase increase (5% each) for cetuximab (Table 4). Skin and subcutaneous tissue disorders related to regorafenib were reported in 18 patients (43%), of which HFSR was the most common, occurring in seven patients (17%), one of which was grade 3. A case of grade 3 drug eruption was also reported. Skin and subcutaneous tissue disorders related to cetuximab were reported in 24 patients (57%). Of those, dermatitis acneiform was reported in 13 patients (31%), of which five (12%) were grade 2 and one was grade 3 (2%). One grade ≥3 cetuximab‐related pruritus was reported. The other skin‐related toxicities attributed to cetuximab were grade 1 and 2, with the most frequent being rash (n = 8, 19%), dry skin (n = 6, 14%), drug eruption (n = 3, 7%), pruritus (n = 3, 7%) and skin fissures (n = 2, 5%).
Table 4.
Most frequent drug‐related grade ≥3 TEAEs during the combination treatment phase (≥2 patients; safety analysis set, n = 42)
| Drug‐related TEAE (by MedDRA), n (%) | Regorafenib (n = 42) | Cetuximab (n = 42) |
|---|---|---|
| Hypophosphatemia | 10 (24) | 3 (7) |
| Fatigue | 4 (10) | 2 (5) |
| Lipase increased | 4 (10) | 1 (2) |
| AST increase | 2 (5) | 1 (2) |
| Gamma‐glutamyltransferase increased | 2 (5) | 2 (5) |
| Hypertension | 2 (5) | 2 (5) |
| Hypotension | 2 (5) | 1 (2) |
| Decreased appetite | 2 (5) | 1 (2) |
| Nausea | 2 (5) | 1 (2) |
| Diarrhea | 1 (2) | 2 (5) |
The following AEs are counted in both arms: skin reactions (rash, hand–foot skin reaction, Stevens–Johnson syndrome), hepatobiliary disorders (increase in liver enzyme), metabolism and nutrition disorders (hypomagnesemia, hypocalcemia, anorexia/weight decrease), nervous system disorders (headache), gastrointestinal disorders (diarrhea, nausea, vomiting) and general disorders (mucositis, fatigue).
Abbreviations: AE, adverse event; AST, aspartate aminotransferase; MedDRA, Medical Dictionary for Regulatory Activities; TEAE, treatment‐emergent adverse event.
Overall, TEAEs leading to dose modification were reported for 22 patients (52%; 16 due to regorafenib‐related TEAEs and six due to cetuximab‐related TEAEs; Table 3). Overall, TEAEs leading to discontinuation were reported in five patients (12%): two due to regorafenib‐related TEAEs (acute liver failure and disseminated intravascular coagulation in one patient, and upper gastrointestinal hemorrhage and vomiting in the other patient), one due to a cetuximab‐related TEAE (infusion‐related reaction) and two not related to treatment (urosepsis and sepsis, each n = 1).
During the run‐in period from Day −14 up to Day 1, 18 patients (43%) reported at least one TEAE, of which four (10%) were considered related to regorafenib and eight (19%) related to cetuximab. Four patients (10%) experienced grade ≥3 TEAEs (hypertension n = 2, hyponatremia n = 1 and hematemesis n = 1), none of which were considered drug related. One patient experienced a serious TEAE (hematemesis) which was considered unrelated to study drugs.
All patients with valid laboratory data (n = 39) experienced hematologic and biochemical abnormalities during the study period, the most common of which were grade 1 leukocytosis (100%) and grade 1–3 anemia (85%). Other laboratory abnormalities included hypomagnesemia (44%), hyponatremia (54%), hypocalcemia (64%) and hypokalemia (49%). Hypophosphatemia was the most common grade 3 biochemical abnormality (36%; Supporting Information Table S2). The grade 4 laboratory abnormalities were decreased lymphocyte count (n = 2), increased AST, increased lipase and decreased platelet count (each n = 1). A worsening by three grades was reported for hypophosphatemia (n = 14), lipase increased (n = 4), lymphocyte count decreased (n = 3), blood bilirubin increased (n = 2), alanine aminotransferase increased, gamma‐glutamyltransferase increased, hyperglycemia, hypokalemia and hypomagnesemia (each n = 1). A worsening by four grades was reported for lipase increased and platelet count decreased (each n = 1).
Pharmacokinetics
PK parameters were evaluated for regorafenib and its metabolites M‐2 and M‐5 after single dose (Day −14 and Day 1 Cycle 1) and multiple doses (Day 15 Cycle 1) of regorafenib in the absence (Day −14) and presence (Days 1 and 15, Cycle 1) of cetuximab (Table 5). Overall, 36 patients were evaluable for PK analyses of regorafenib and 33 patients for cetuximab. The single dose of regorafenib on Day −14 was received by 28 patients. After a single regorafenib dose (Day −14), regorafenib exposure was higher at the 160 mg dose than at the 120 mg dose (28.5 mg hr/l vs. 17.1 mg hr/l, respectively) with a high intersubject variability (geometric coefficient of variation [CV]%: 50.5 and 55.0%, respectively; Table 5). Regorafenib exposure was comparable after a single dose on Day −14 and in the presence of cetuximab on Day 1 (Supporting Information Fig. S2 and Table 5). The mean concentration–time profiles for regorafenib (160 mg QD) when administered alone (Day −14) and in combination with a single dose of cetuximab (Day 1) are shown in Supporting Information Fig. S3. Regorafenib exposure increased after multiple doses of regorafenib 160 mg and cetuximab on Day 15 (AUC(0–24)md 39.2 mg hr/l). A similar PK pattern was observed for M‐2 and M‐5 (Table 5). The exposure of cetuximab (AUC(0–26)) was similar across regorafenib dose levels (3,641, 3,657, 2,888 and 3,810 mg hr/l for regorafenib 60, 100, 120 and 160 mg, respectively).
Table 5.
Pharmacokinetic parameters of regorafenib alone (Day −14) or with cetuximab after a single regorafenib dose (Day 1); geometric mean (%CV)
| Day | Continuous regorafenib 60 mg plus cetuximab | Continuous regorafenib 100 mg plus cetuximab | Intermittent regorafenib 120 mg plus cetuximab | Intermittent regorafenib 160 mg plus cetuximab | |
|---|---|---|---|---|---|
| Regorafenib | |||||
| AUC(0–24) (mg hr/l) | Day −14 | (n = 5) 10.8 (88.6) | (n = 6) 20.8 (48.8) | (n = 8) 17.1 (55.0) | (n = 9) 28.5 (50.5) |
| C max (mg/l) | (n = 5) 0.7 (103) | (n = 6) 1.3 (46.9) | (n = 8) 1.2 (71.7) | (n = 9) 1.9 (58.3) | |
| T max 1 (hr) | (n = 5) 8.0 (4.0–24.0) | (n = 6) 8.0 (2.0–24.0) | (n = 8) 4.0 (2.0–7.5) | (n = 9) 2.3 (1.9–23.9) | |
| AUC(0–24) (mg hr/l) | Day 1 | (n = 4) 10.2 (114) | (n = 3) 21.7 (48.5) | (n = 2) NC | (n = 5) 30.1 (23.8) |
| C max (mg/l) | (n = 4) 0.8 (116) | (n = 4) 1.3 (98.3) | (n = 7) 1.1 (47.3) | (n = 9) 2.4 (44.3) | |
| T max 1 (hr) | (n = 4) 2.9 (2.0–23.6) | (n = 4) 7.9 (2.0–24.0) | (n = 7) 7.6 (2.0–22.0) | (n = 9) 2.0 (1.9–7.5) | |
| M‐2 | |||||
| AUC(0–last) (mg hr/l) | Day −14 | (n = 5) 2.6 (195) | (n = 6) 8.0 (96.1) | (n = 8) 4.8 (91.8) | (n = 9) 13.7 (85.8) |
| C max (mg/l) | (n = 5) 0.2 (208) | (n = 6) 0.5 (101) | (n = 8) 0.3 (100) | (n = 9) 0.8 (85.2) | |
| T max 1 (hr) | (n = 5) 8.0 (4.0–24.0) | (n = 6) 8.0 (8.0–24.0) | (n = 8) 7.5 (2.0–24.3) | (n = 9) 7.5 (3.9–23.8) | |
| AUC(0–last) (mg hr/l) | Day 1 | (n = 4) 3.7 (223) | (n = 4) 3.7 (158) | (n = 7) 2.8 (164) | (n = 9) 11.7 (100) |
| C max (mg/l) | (n = 4) 0.3 (168) | (n = 4) 0.3 (72.1) | (n = 7) 0.3 (57.9) | (n = 9) 1.3 (41.2) | |
| T max 1 (hr) | (n = 4) 3.9 (2.0–23.6) | (n = 4) 7.9 (2.0–24.0) | (n = 7) 7.6 (2.0–22.0) | (n = 9) 4.1 (2.0–8.0) | |
| M‐5 | |||||
| AUC(0–last) (mg hr/l) | Day −14 | (n = 3) 0.4 (28.5) | (n = 5) 0.8 (146) | (n = 7) 0.3 (126) | (n = 8) 1.4 (214) |
| C max (mg/l) | (n = 5) 0.01 (192) | (n = 6) 0.04 (221) | (n = 8) 0.02 (147) | (n = 9) 0.09 (180) | |
| T max 1 (hr) | (n = 5) 23.8 (7.5–24.0) | (n = 6) 24.0 (8.0–24.0) | (n = 8) 23.8 (22.6–24.5) | (n = 9) 23.8 (22.4–24.7) | |
| AUC(0–last) (mg hr/l) | Day 1 | (n = 3) 0.5 (214) | (n = 4) 0.3 (184) | (n = 6) 0.3 (259) | (n = 9) 1.3 (281) |
| C max (mg/l) | (n = 4) 0.02 (176) | (n = 4) 0.03 (72.7) | (n = 6) 0.03 (143) | (n = 9) 0.14 (127) | |
| T max 1 (hr) | (n = 4) 23.7 (23.5–24.5) | (n = 4) 23.9 (8.0–24.0) | (n = 6) 20.3 (7.6–23.7) | (n = 9) 8.0 (4.0–24.7) | |
Median (range).
Abbreviations: AUC, area under the concentration–time curve; C max, maximum observed drug concentration; CV, coefficient of variation; NC, not calculated; T max, time to reach maximum concentration in plasma.
Efficacy
Of the 24 patients included in the efficacy analysis, five patients achieved a partial response (PR; 21%), six had stable disease (25%) and 13 had progressive disease (54%; Fig. 1). The five patients with PR were diagnosed with anal squamous cell carcinoma, small cell lung cancer, adenoid cystic carcinoma, HCC and CRC. Two patients had durable (≥6 months) PR: one patient with CRC in the 160 mg dose group who responded from Cycles 10 to 20 and another in the 120 mg dose group with adenoid cystic carcinoma with lung metastasis who responded from Cycles 4 to 16.
Figure 1.
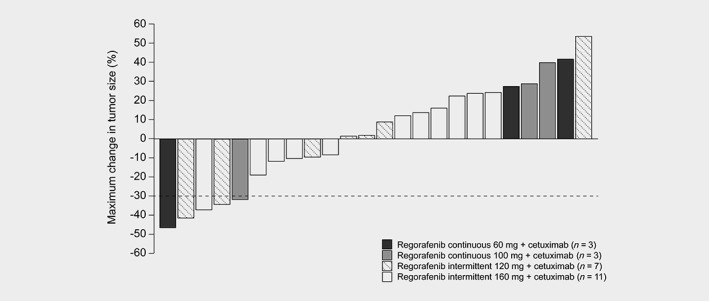
Best overall response by maximum change in tumor size (efficacy set, n = 24). Bars that reach the reference line (30% reduction in tumor size) meet the criterion for partial response.
Discussion
This phase 1 study evaluated the safety and tolerability of combining regorafenib with cetuximab for the treatment of advanced solid tumors. Both regorafenib and cetuximab have separately demonstrated activity in various advanced tumor types with preclinical data, suggesting possible additive clinical benefit in certain tumor types.6, 7, 8, 9, 14, 15, 17, 18, 19 Initially, both continuous and intermittent dosing schedules for regorafenib were evaluated; however, the continuous dose was subsequently terminated due to a substantially lower tolerable dose of 60 mg in the combination than the approved intermittent 160 mg dose. No DLTs were observed at the 160 mg dose level. Oral regorafenib 160 mg QD for 3 weeks on/1 week off in combination with the recommended dose of cetuximab was determined as the MTD.
A recently published phase 1 combination trial by Subbiah et al., of regorafenib plus cetuximab in 27 patients with advanced cancer refractory to several lines of therapy, described a considerably lower dose (regorafenib 80 mg QD plus cetuximab 200 mg/m2 loading dose, followed by cetuximab 150 mg/m2 weekly) as the MTD.16 At this dose, no DLTs were observed in the 19 evaluable patients; however, two DLTs (grade 3 thrombocytopenia in a patient with glioblastoma and grade 3 intra‐abdominal bleeding in a patient with CRC) were reported in five evaluable patients at the higher dose of regorafenib 120 mg QD plus cetuximab 200 mg/m2 loading dose followed by 150 mg/m2 weekly. The most common TEAEs were rash (n = 20, 74%) and fatigue (n = 7, 26%); the incidence of rash was therefore considerably higher than that observed in the intermittent arms in our study, in which just nine of 31 patients (29%) reported this TEAE. However, the incidence of fatigue is lower than in our study, where it was reported in 13 of 31 patients (42%) in the intermittent dosing arms. The reason for the different MTDs reported in the two trials is not clear, but may be partially related to variations in inclusion criteria (primary central nervous system malignancies were excluded in the current trial), DLT criteria and baseline disease heterogeneity.
Dermatologic toxicities have been frequently reported in clinical trials of cetuximab, in particular acneiform rash which occurred in 76–88% of 1,373 patients receiving cetuximab in the clinical trials leading to its approval, and was classed as severe in 1–17% of these patients.13 However, in the current combination trial, only 31% of patients with acneiform dermatitis of any grade were reported, with one grade 3. Whether this combination with regorafenib results in a lower incidence of dermatologic toxicities in patients receiving cetuximab is undetermined and requires confirmation in an adequately powered trial. The effect might be related to an immunomodulatory role of regorafenib, which has been previously described.2, 20 In addition, compared with data from the phase 3 CORRECT trial in patients with metastatic CRC, the incidence reported here for all‐grade regorafenib‐related HFSR was lower (47% vs. 17%, respectively).8 Notably, although a DLT of grade 3 thrombocytopenia was reported with regorafenib plus cetuximab in the trial by Subbiah et al.,16 only one case of grade ≥3 platelet count decrease was reported in our study, which was observed in the context of fatal liver failure and disseminated intravascular coagulation.
Cetuximab had no effect on the PK of regorafenib or its metabolites, and there was no clinically relevant effect of regorafenib on the PK of cetuximab. Consistent with the PK properties of regorafenib,10 some accumulation was observed after multiple dosing. The combination treatment showed promising signs of efficacy with five PRs (21%) observed, which is higher than that reported with regorafenib alone in mCRC (1% in CORRECT8 and 4% in CONCUR9). However, interpretation of efficacy should be carried out with caution because the study enrolled a highly heterogeneous group of patients, only a limited number of patients were evaluable for efficacy and efficacy was only evaluated as a secondary objective.
In conclusion, results from our study demonstrate that regorafenib at the MTD plus the standard dose of cetuximab was well tolerated with no unexpected toxicities and with promising initial signs of efficacy. In case of further development, the recommended phase 2 dose for the combination will be the standard dose for either drug.
Data availability statement
Availability of the data underlying this publication will be determined according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing.” This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the “Study sponsors section” of the portal. Data access will be granted to anonymized patient‐level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
Supporting information
Appendix S1: Supporting Information
Acknowledgements
We would like to thank Zuzana Jirakova Trnkova and Susanne Reschke for their contribution to the study. We would also like to thank the patients, their families and participating in study centers. Our study was sponsored by Bayer. Editorial assistance in the preparation of this article was provided by Alex Coulthard and Katrin Gudmundsdottir of OPEN Health Medical Communications (London, UK), with financial support from Bayer.
Conflict of interests: CW has received research funding from Bayer. JJL has received research funding from Merck and advisory board fees from AbbVie, Incyte and LOXO. IS is an employee of Bayer and owns Bayer stocks. AC is an employee of Bayer and owns stocks in Bayer, AstraZeneca and Pfizer. FH is an employee of Bayer and owns Bayer stocks. H‐JL has received honoraria from Bayer, Bristol‐Myers Squibb, Merck Serono and Roche, and advisory board and consulting fees from Bayer, Merck Serono and Roche. ACL has nothing to disclose.
References
- 1. Cook KM, Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin 2010;60:222–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abou‐Elkacem L, Arns S, Brix G, et al. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther 2013;12:1322–31. [DOI] [PubMed] [Google Scholar]
- 3. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73‐4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011;129:245–55. [DOI] [PubMed] [Google Scholar]
- 4. European Medicines Agency . Regorafenib (Stivarga) Summary of Product Characteristics. Canary Wharf, London: European Medicines Agency, 2018. [Google Scholar]
- 5. Food and Drug Administration . Regorafenib (Stivarga) Prescribing Information. Silver Spring, MD: FDA, 2018. [Google Scholar]
- 6. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;389:56–66. [DOI] [PubMed] [Google Scholar]
- 7. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:303–12. [DOI] [PubMed] [Google Scholar]
- 9. Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2015;16:619–29. [DOI] [PubMed] [Google Scholar]
- 10. Mross K, Frost A, Steinbild S, et al. A phase I dose‐escalation study of regorafenib (BAY 73‐4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 2012;18:2658–67. [DOI] [PubMed] [Google Scholar]
- 11. Shimizu T, Tolcher AW, Patnaik A, et al. Phase I dose‐escalation study of continuously administered regorafenib (BAY 73‐4506), an inhibitor of oncogenic and angiogenic kinases, in patients with advanced solid tumors. J Clin Oncol 2010;28:3035–5.20458031 [Google Scholar]
- 12. European Medicines Agency . Cetuximab (Erbitux) Summary of Product Characteristics. Canary Wharf, London: European Medicines Agency, 2018. [Google Scholar]
- 13. Food and Drug Administration . Cetuximab (Ertibux) Prescribing Information. Silver Spring, MD: FDA, 2018. [Google Scholar]
- 14. Napolitano S, Martini G, Rinaldi B, et al. Primary and acquired resistance of colorectal cancer to anti‐EGFR monoclonal antibody can be overcome by combined treatment of regorafenib with cetuximab. Clin Cancer Res 2015;21:2975–83. [DOI] [PubMed] [Google Scholar]
- 15. Shaheen RM, Ahmad SA, Liu W, et al. Inhibited growth of colon cancer carcinomatosis by antibodies to vascular endothelial and epidermal growth factor receptors. Br J Cancer 2001;85:584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subbiah V, Khawaja MR, Hong DS, et al. First‐in‐human trial of multikinase VEGF inhibitor regorafenib and anti‐EGFR antibody cetuximab in advanced cancer patients. JCI Insight 2017;2:e90380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408–17. [DOI] [PubMed] [Google Scholar]
- 18. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous‐cell carcinoma of the head and neck. N Engl J Med 2006;354:567–78. [DOI] [PubMed] [Google Scholar]
- 19. Vermorken JB, Mesia R, Rivera F, et al. Platinum‐based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359:1116–27. [DOI] [PubMed] [Google Scholar]
- 20. Hoff S, Grünewald S, Röse L, et al. 1198P: Immunomodulation by regorafenib alone and in combination with anti PD1 antibody on murine models of colorectal cancer. Ann Oncol 2017;28(suppl_5):423. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
Availability of the data underlying this publication will be determined according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing.” This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the “Study sponsors section” of the portal. Data access will be granted to anonymized patient‐level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.