

Abstract
Pre‐emptive pharmacogenetics (PGx) testing of a panel of germline genetic variants represents a new model for personalized medicine. Clinical impact of PGx testing is maximized when all variant alleles for which actionable clinical guidelines are available are included in the test panel. However, no such standardized panel has been presented to date, impeding adoption, exchange, and continuity of PGx testing. We, therefore, developed such a panel, hereafter called the PGx‐Passport, based on the actionable Dutch Pharmacogenetics Working Group (DPWG) guidelines. Germline‐variant alleles were systematically selected using predefined criteria regarding allele population frequencies, effect on protein functionality, and association with drug response. A PGx‐Passport of 58 germline variant alleles, located within 14 genes (CYP2B6,CYP2C9,CYP2C19,CYP2D6,CYP3A5,DPYD, F5,HLA‐A,HLA‐B,NUDT15,SLCO1B1,TPMT,UGT1A1, and VKORC1) was composed. This PGx‐Passport can be used in combination with the DPWG guidelines to optimize drug prescribing for 49 commonly prescribed drugs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Absence of a widely accepted pharmacogenetic (PGx) panel is impeding adoption, exchange, and continuity of panel‐based pre‐emptive PGx testing. Clinical impact of a PGx panel is optimized when it includes all variant alleles for which actionable clinical guidelines are available.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Here, we present the methods used and resulting selected variant alleles included in a proposed standardized panel, based on the actionable Dutch Pharmacogenetics Working Group guidelines; hereafter called the PGx‐Passport.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The resulting PGx‐Passport is a concise panel encompassing 58 germline clinically actionable variant alleles, located within 14 pharmacogenes (CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, F5, HLA‐A, HLA‐B, NUDT15, SLCO1B1, TPMT, UGT1A1, and VKORC1), which can be determined at low costs.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This PGx‐Passport can be used in combination with the DPWG guidelines to optimize drug prescribing for 49 commonly prescribed drugs and improve acceptance of PGx testing.
Pharmacogenetics (PGx)‐guided prescribing promises to personalize drug therapy by using an individual's germline genetic makeup.1, 2 This ameliorates the conventional “trial and error” approach of drug prescribing, thereby promising safer, more effective, and cost‐effective drug treatment.3 Several randomized controlled trials support the clinical utility of individual gene–drug pairs to either optimize dosing4, 5, 6, 7 or drug selection.8 Although there is extensive evidence supporting the utility of pre‐emptive PGx testing for individual gene–drug pairs, significant implementation barriers remain.9, 10, 11 One of the previously surmounted barriers is the development of clinical guidelines directing clinical application of PGx test results. In 2005, the Dutch Pharmacogenetics Working Group (DPWG) was established to devise pharmacotherapeutic recommendations based on systematic review of literature.12, 13 From 2005 onward, the DPWG has systematically reviewed 97 potential gene–drug interactions. Of these, 54 are actionable gene–drug interactions, providing a therapeutic recommendation for at least one interacting phenotype.12, 13 In parallel, the Clinical Pharmacogenetics Implementation Consortium (CPIC) has devised guidelines for over 40 drugs.14 The DPWG and CPIC guidelines have been formally compared, and efforts are ongoing to harmonize the two.15
Significant debate persists regarding the optimal timing and methodology of testing for delivering PGx testing in clinical care.16 Some support a pretherapeutic single gene–drug approach, in which a PGx test of a single relevant gene is ordered once a target drug is prescribed; whereas others advocate for a pre‐emptive panel‐based strategy in which multiple genes are tested simultaneously and saved for later use, in preparation of future prescriptions throughout a patient's lifetime.17 When combined with a clinical decision support system, the corresponding PGx guideline can be deployed by the clinical decision support system at the point of care, thereby providing clinicians with the necessary information to optimize drug prescribing when a target drug is prescribed. Patients will receive multiple drug prescriptions with potential gene–drug interactions within their lifetime.16, 18 It has been estimated that half of patients above 65 years will use at least one of the drugs for which PGx guidelines are available during a 4‐year period, and one fourth to one third will use two or more of these drugs.19 Logistics and cost‐effectiveness are, therefore, optimized when delivered in a pre‐emptive panel‐based approach; pharmacotherapy does not have to be delayed, awaiting single‐gene testing results and costs for genotyping are minimized, as marginal acquisition costs of testing and interpreting additional pharmacogenes is near‐zero.20 Although a sufficiently powered and well‐designed study assessing the (cost‐)effectiveness of pre‐emptive PGx testing is yet to be concluded,21 a number of small randomized observational studies indicate promising clinical utility of PGx panel testing.22, 23, 24, 25, 26 Another important challenge hampering adoption of pre‐emptive panel testing is the lack of standardization regarding variants included in such panels. Additionally, recommendations on which variants to test differ strikingly across the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) labels and also CPIC and DPWG recommendations.27 Standardization, however, would enable clinicians to understand PGx test results without extensive scrutiny of the alleles included in the panel. Despite the identification of standardization as a potential accelerator for PGx adoption, exchange, and continuity,28 there are currently no standards defining which variants must be tested.29, 30
Although some initiatives have developed standardized panels of relevant variants within individual genes,31 and other initiatives across multiple genes,32 a panel covering widely accepted genetic variants reflecting an entire set of guidelines is not yet available. Thus, in order to facilitate the clinical implementation of PGx testing, we here present such a panel based on actionable DPWG guidelines, hereafter called the PGx‐Passport. Clinical impact of such a PGx panel is maximized when all variant alleles for which actionable clinical guidelines are available are included. When implemented, it will maximize the incidence at which both an individual's predicted phenotype and the associated clinical guideline is available at the point of care, when a potential gene–drug interaction is encountered. In contrast, including variant alleles for which no clinical guidelines are available would not provide added clinical value, because results are not clinically actionable. This is an initiative of the Ubiquitous Pharmacogenomics Consortium (U‐PGx).21
Results
The PGx‐Passport represents the complete set of clinically actionable variant alleles for which the DPWG provides actionable recommendations. The selected genes and respective variant alleles are listed in Table 1. Overall, 58 variant alleles in 14 pharmacogenes complied with the selection criteria. Of these, 6 variant alleles are found in CYP2B6, 4 in CYP2C9, 9 in CYP2C19, 12 in CYP2D6, 3 in CYP3A5, 4 in DPYD, 1 in F5, 1 in HLA‐A, 4 in HLA‐B, 4 in NUDT15, 1 in SLCO1B1, 4 in TPMT, 4 in UGT1A1, and 1 in VKORC1. The panel can be used to optimize pharmacotherapy for 49 commonly prescribed drugs ranging in multiple therapeutic classes, including antidepressants (n = 10), immunosuppressants (n = 5), anticancer drugs (n = 5), anti‐infectives (n = 4), anticoagulants (n = 4), antiepileptics (n = 4), antipsychotics (n = 4), proton pump inhibitors (n = 3), anti‐arrhythmics (n = 2), analgesics (n = 2), antilipidemics (n = 2), an antihypertensive (n = 1), a psychostimulant (n = 1), treatment of Gaucher disease (n = 1), and anticontraceptives (n = 1).
Table 1.
Systematically selected clinically relevant variant alleles that reflect the complete set of actionable DPWG guidelines (58 variant alleles located in 14 pharmacogenes)
| Genes | Variant allele | Allele functional status | Drug for which actionable DPWG guideline is available |
|---|---|---|---|
| CYP2B6 | *6 | Decreased function or no function | Efavirenz |
| *9 | Decreased function or no function | ||
| *4 | Decreased function or no function | ||
| *16 | Decreased function or no function | ||
| *18 | Decreased function or no function | ||
| *5 | Decreased function or full function | ||
| CYP2C9 | *2 | Decreased function |
Phenytoin Warfarin |
| *3 | Decreased function | ||
| *5 | Decreased function | ||
| *11 | Decreased function | ||
| CYP2C19 | *2 | No function |
Clopidogrel Citalopram Escitalopram Sertraline Imipramine Lansoprazole Omeprazole Pantoprazole Voriconazole |
| *3 | No function | ||
| *4A/B | No function | ||
| *5 | No function | ||
| *6 | No function | ||
| *8 | Decreased function or no function | ||
| *9 | Decreased function | ||
| *10 | Decreased function | ||
| *17 | Increased function | ||
| CYP2D6 | *xN | Increased function |
Amitriptyline Aripiprazole Atomoxetine Clomipramine Codeine Doxepin Eliglustat Flecainide Haloperidol Imipramine Metoprolol Nortriptyline Paroxetine Pimozide Propafenone Tamoxifen Tramadol Venlafaxine Zuclopenthixol |
| *3 | No function | ||
| *4 | No function | ||
| *5 | No function | ||
| *6 | No function | ||
| *8 | No function | ||
| *9 | Decreased function | ||
| *10 | Decreased function | ||
| *14A | Decreased function | ||
| *14B | Decreased function | ||
| *17 | Decreased function | ||
| *41 | Decreased function | ||
| CYP3A5 | *3 | No function | Tacrolimus |
| *6 | No function | ||
| *7 | No function | ||
| DPYD | *2A | No function |
5‐Fluorouracil Capecitabine Tegafur |
| *13 | No function | ||
| 2846A>T | Decreased function | ||
| 1236G>A | Decreased function | ||
| F5 | 1691G>A | Decreased function | Estrogen contraceptive agents |
| HLA‐A | *31:01 | High‐risk allele | Carbamazepine |
| HLA‐B | *15:02 | High‐risk allele |
Carbamazepine Oxcarbazepine Phenytoin Lamotrigine |
| *15:11 | High‐risk allele | Carbamazepine | |
| *57:01 | High‐risk allele |
Abacavir Flucloxacillin |
|
| *58:01 | High‐risk allele | Allopurinol | |
| NUDT15 | *2 | Decreased function |
6‐Mercaptopurine Azathioprine Thioguanine |
| *3 | Decreased function | ||
| *6 | Decreased function | ||
| *9 | Decreased function | ||
| SLCO1B1 | *5/*15/*17 | Decreased function |
Atorvastatin Simvastatin |
| TPMT | *2 | No function |
6‐Mercaptopurine Azathioprine Thioguanine |
| *3A | No function | ||
| *3B | No function | ||
| *3C | No function | ||
| UGT1A1 | *6 | Decreased function | Irinotecan |
| *27 | Decreased function | ||
| *28 | Decreased function | ||
| *37 | Decreased function | ||
| VKORC1 |
−1639G>A; 1173 C>T |
Decreased expression |
Acenocoumarol Phenprocoumon Warfarin |
CYP, cytochrome P450; DPWG, Dutch Pharmacogenetics Working Group; DPYD, dihydropyrimidine dehydrogenase; F5, factor V Leiden; HLA, human leucocyte antigen; NUDT, nudix hydrolase; SLCO, solute carrier organic anion transporter; UGT, UDP‐glucuronosyltransferase; TPMT, thiopurine S‐methyltransferase; VKORC, vitamin K epoxide reductase complex.
Discussion
The presented PGx‐Passport encompasses 58 variant alleles within 14 pharmacogenes (CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, F5, HLA‐B, NUDT15, SLCO1B1, TPMT, UGT1A1, and VKORC1) and can be used to optimize pharmacotherapy for 49 commonly prescribed drugs throughout a patient's lifetime. Essentially, the PGx‐Passport represents the first curated summary of alleles across multiple genes for which, based on the consensus of the DPWG, adequate evidence is available to be applied in the clinic. A clear advantage of such curated summary is that all results translate into predicted phenotypes and clear clinical guidelines; avoiding report of clinically ambiguous results for which clinical guidelines are absent. Therefore, it can easily be implemented into the workflow of laboratories and clinicians worldwide. However, as with any curation process, deliberations and assumptions are made to justify simplification. Here, we present these deliberations in order to recognize the strengths and limitations of the PGx‐Passport.
A significant limitation, which is applicable not only to this variant selection but to PGx testing and interpretation as it is performed today, is that guidelines provide pharmacotherapeutic recommendations based on individual predicted phenotype categories rather than continuous scores. For example, for CYP2D6, patients are categorized into normal metabolizers, intermediate metabolizers, poor metabolizers, or ultrarapid metabolizers based upon their genotype. However, the actual CYP2D6 phenotype is likely normally distributed. Imposing categorization, as opposed to the interpretation of the actual genotype, therefore, sacrifices information in order to simplify clinical interpretation. In addition, we interpret the functionality of each allele individually and assume that the sum of these activity scores equals the total activity of the diplotype, thereby abstracting from potential compensatory effects. Furthermore, these categorizations are currently substrate invariant, even though the effects on metabolic capacity may differ between substrates.33 However, categorization is currently justified due to the lack of evidence to devise pharmacotherapeutic recommendations per diplotype or per substrate. For example, the CYP2D6 activity score is now set at 0.5 for CYP2D6*10 for all substrates. However, in reality, the effect on activity score may be different across substrates. As the field of PGx evolves, we foresee that phenotypes will be predicted substrate specifically on a continuous scale, and pharmacotherapeutic recommendations are provided for each value.
Even though multiple variants have been discovered within the selected actionable genes, we chose to restrict testing to a subset of these variants, based on their effect on protein functionality, minor allele frequency (MAF), and association with drug response. Restricting testing to individual variants disregards untested or undiscovered variants that may also influence the functionality of the gene product. However, despite progress in the computational interpretation of functional consequences of such uncharacterized variations,34 these variants are currently not clinically actionable. Significant debate persists regarding both the nature and strength of evidence required for clinical application of variant alleles. Fundamentally, the potential of a variant to accurately predict the genetic component of drug response is a function of both the predictability of a variant's effect on protein functionality and the extent to which the protein functionality is associated with clinical outcome. Because the strength of these functions differs across genes and gene–drug interactions, we do not foresee a one‐size‐fits‐all consensus regarding an evidence threshold across all gene–drug interactions but rather a different evidence threshold per individual gene–drug interaction based on the genetics and pharmacology of the interaction. For example, in the case of the TPMT–thiopurine interaction, the effect of TMPT variation on protein functionality has been firmly established because it exhibits behavior similar to monogenetic codominant traits.35 Therefore, identified variants in TPMT (*3A/*3B/*3D) are considered to have sufficient evidence to be applied in the clinic, even in the absence of studies specifically investigating clinical effects in patients carrying these particular variants. On the other hand, clinically relevant variant alleles in CYP2D6 are based on the pharmacology of the interaction. For example, the flecainide–CYP2D6 interaction is based on the associations between decreasing CYP2D6 activity leading to increasing flecainide plasma levels, which, in turn, leads to increased risk for flecainide intoxication. Therefore, all identified variants in CYP2D6 shown to have a significant effect on CYP2D6 enzyme activity are defined to have sufficient evidence to be applied in the clinic.
Here, we chose to limit variant selection to relatively common variant alleles. Therefore, we consider the PGx‐Passport a minimal list of clinically relevant variant alleles. An advantage of this approach is that the number of patients carrying actionable variants within their PGx‐Passport is maximized, while costs remain reasonable. On the other hand, a disadvantage is that the tested variants are unable to fully predict phenotype in patients carrying untested rare variants, which may indeed have an effect on protein functionality. In other words, including these very rare variants may strengthen the potential of the panel to predict drug response. However, because these are very rare variants, the absolute number of patients in which this is the case will be low. Still, a recent study has shown that indeed 30–40% of functional variability in pharmacogenes can be attributed to rare variants.36 On the contrary, the functional effect of many rare variants is yet unknown and may differ across substrates. Including these variants of unknown effect in the reported results would again provide clinically ambiguous results, and, therefore, we argue to exclude these until methods have been developed that enable accurate prediction of functional effects.37 Thus, until the effects of these variations on functional effect and subsequent drug response are validated, in silico,38 in vitro, or in vivo, we are unable to apply the results of testing for these variant alleles in clinical care. However, for some alleles for which the association with drug response is already well‐established, it may be useful to determine these alleles even though the frequency may be low. For example, the DPYD‐variant alleles, DPYD*2A (MAF < 1%), DPYD*13 (MAF < 1%), and DPYD c.2846A>T (MAF < 1%), were selected regardless of their MAF because their association with fluoropyrimidine‐induced toxicity has been well‐established and adopted clinically. Other examples include CYP2C19 *5, *6, *8, and *10.
In addition, many pharmacogenetic‐variant alleles have frequencies that vary across ethnicities.39 As self‐reported ethnicity is not always in agreement with genetic ethnicity,40 it is of clinical importance that the PGx‐Passport contains all variant alleles that are considered common in at least one defined ethnicity. For example, CYP2D6*6 has a global MAF < 1% but an MAF of 2% in Europeans and was, therefore, selected to be included in the panel. Determining this variant allele may be less relevant (but not irrelevant) in non‐European populations.
Importantly, we have selected‐variant alleles representing haplotype blocks, as opposed to defining variants, within the PGx‐Passport. Clinical evidence on associated drug response is commonly presented using variant alleles as opposed to defining variants. Therefore, the resulting pharmacotherapeutic recommendations and allele selection are also based on the *alleles. Nonetheless, in order to operationalize the PGx‐Passport, one must select defining variants representing variant alleles. Where sequencing platforms enable testing of the entire allele haplotype block without additional costs, it is much more economical to test a set of single nucleotide polymorphisms (SNPs) unique to haplotype blocks when using a genotyping platform. An example of an operationalized panel fit for genotyping platforms for a subset of genes in the PGx‐Passport, can be found in Table S1 . One must take special consideration when selecting and interpreting tagging SNPs for HLA genotyping because frequencies as linkage disequilibrium (LD) patterns vary across ethnicities. For example, HLA‐B*57:01 may be tested by using tagging SNP rs2395029(T>G). However, rs2395029(T>G) is in complete LD with HLA‐B*57:01 in Han Chinese, and LD is lower in Southeast Asians.41, 42, 43 Therefore, this result should be interpreted with caution in certain populations. Further examples are tagging SNPs for HLA‐A*31:01 and HLA‐B*15:02 in Asian populations, which cannot be interpreted in Europeans due to lower LD.44, 45
To support wide‐spread adoption of the PGx‐Passport, we recognize that evidence regarding clinical acceptance, clinical utility, and (cost‐)effectiveness is required by stakeholders. Clinical acceptance of a panel similar to the PGx‐Passport has been demonstrated among community pharmacists.46 Here, pharmacists requested a PGx panel test for 18% of eligible patients, indicating a relatively high level of acceptance. Additionally, clinical acceptance of PGx panel testing has also been shown by other initiatives.47 To appeal to the request for evidence demonstrating clinical utility, the collective clinical utility for a subset of genes in the PGx‐Passport ( Table S1 ) is being assessed in a cluster randomized controlled trial including 8,100 patients across healthcare institutions in 7 European countries.21 Several promising studies indicate the (cost‐)effectiveness of PGx panel‐based testing on healthcare utilization in psychiatry and polypharmacy,22, 23, 24, 26 where observed cost savings ranged from $21823 to $2,77848 per patient. Others have modeled the cost‐effectiveness of one‐time genetic testing to minimize a lifetime of adverse drug reactions, and concluded an incremental cost‐effectiveness ratio of $43,165 per additional life year and $53,680 per additional quality‐adjusted life year, and, therefore, cost‐effective.49 However, cost‐effectiveness may vary across ethnic populations as a result of varying in allele frequencies, the target population as a result of varying prescription patterns, and the healthcare setting as a result of varying healthcare costs and incremental cost‐effectiveness ratio thresholds.
The PGx‐Passport is a recommendation of alleles to be included in clinical laboratory assays, but it does not include information on genotype‐to‐phenotype translation or clinical interpretation of the PGx results. However, the correlation of genotypes to predicted phenotypes and recommendations for clinical actions based on these phenotypes are included in the clinical practice guidelines published by DPWG, CPIC, and other professional societies and regulatory bodies.
We recognize that as the field of pharmacogenetics continues to advance and novel associations between variant alleles and clinically relevant drug response are validated, new variant alleles will be added, and the PGx‐Passport panel will be updated. The DPWG continuously reviews literature and updates each guideline every 2 years. Additionally, the selected panel of variants also depends on the time point of selection; as available information on MAFs and allele functional status may change over time. An important example of this dynamic nature of the panel is the omission of CYP2C9*6 and *8 from the presented PGx‐Passport. At the time of variant selection, these variants did not comply with the selection criteria based on available information. At this time point, CYP2C9*6 was found to have an MAF < 1% in both global and selected populations50 and the allele functional status of CYP2C9*8 was defined to be increased function. Therefore, CYP2C9*6 did not comply to criterion 4 and CYP2C9*8 did not comply to criterion 1, because there was no DPWG guideline corresponding to the associated phenotype. However, based on current literature, these variants would be included in the panel. Therefore, the presented panel should not be perceived as a static entity, but rather a dynamic curated summary of clinically relevant variant alleles underlying the continuously updated guidelines. The updated PGx‐Passport will be published on the U‐PGx website (www.upgx.eu).
In summary, the selected variant alleles included in this panel fully cover the available, clinically actionable DPWG guidelines. This, now publicly available, panel can be used in combination with the DPWG guidelines to guide drug prescribing and dispensing of 49 commonly used drugs. The proposed PGx‐Passport is currently limited to the DPWG guidelines and common variants. As such, it can be considered a minimal list of clinically relevant variant alleles. We recommend commercial and hospital laboratories to incorporate these variant alleles in their clinical repertoire, thereby adopting a new model for personalized medicine in which dose and drug selection are personalized based upon an individual's PGx‐Passport.
Methods
Variant alleles included in the PGx‐Passport were systematically selected based on the five selection criteria shown in Figure 1. The DPWG guidelines were the starting point of the variant allele selection. At the time of initial selection (February 2017), these consisted of 90 gene–drug guidelines covering 81 drugs and 16 genes (see Table S2 ). After this initial selection, the panel was updated, because the DPWG released novel and updated guidelines. The update of the panel is a continuous process and is performed once an update is deemed necessary. The update was performed in January 2019 and based on 97 gene–drug guidelines covering 82 drugs and 19 genes (see Table S3 ). For the updated selection, actionable DPWG guidelines were compiled, consisting of 54 gene–drug guidelines covering 49 drugs and 14 genes (see Table S4 ). For the initial selection, variant alleles within 13 actionable genes reported within the DPWG, CPIC, PharmGKB, CYPAlleles, and other monographs were compiled (see Table S5 ). Second, a list of variant alleles of which the effect on protein functionality is established was compiled. Of these, all variant alleles with a global MAF ≥ 1% were included in the panel, as defined using 1000 Genomes project phase III allele frequencies. The global MAF is defined as the mean frequency across all populations. In addition, variant alleles that had a global MAF < 1%, but an MAF ≥ 1% among selected populations (European/Asian/African) was also included in the panel; again based on the 1000 Genomes project phase III allele frequencies for subpopulations. When variant alleles had both a global and selected population MAF of < 1%, they were excluded from the panel unless the association between a variant allele and drug response was well established. This included variants that were already tested for in routine clinical practice in one of the U‐PGx sites.
Figure 1.
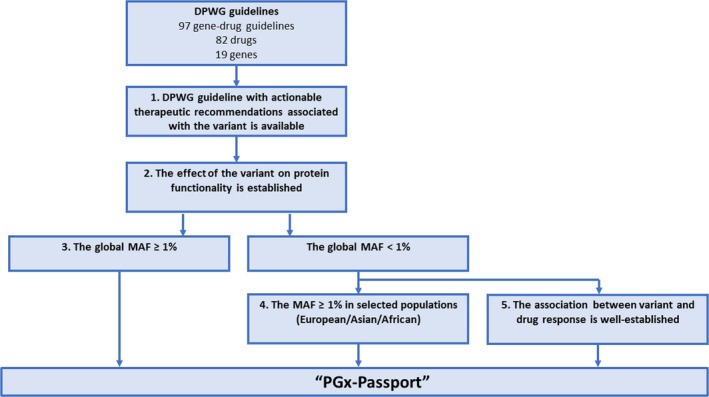
Decision tree to select relevant variant alleles to be included in the PGx‐Passport. DPWG, Dutch Pharmacogenetics Working Group; MAF, minor allele frequency, U‐PGx, Ubiquitous Pharmacogenomics Consortium. [Colour figure can be viewed at wileyonlinelibrary.com]
Funding
The research leading to these results has received funding from the European Community's Horizon 2020 Programme under grant agreement no. 668353 (U‐PGx). M.S. was, in part, supported by the Robert Bosch Stiftung, Stuttgart, Germany.
Conflicts of Interest
The authors declared no competing interests for this work.
Author Contributions
C.H.vdW., M.H.vR., J.J.S., H.‐J.G., W.O.M.J., M.I.‐S., V.M.L., L.K., and M.S. wrote the manuscript. C.H.vdW., M.H.vR., W.O.M.J., M.I.‐S., V.M.L., L.K., M.S., J.J.S., and H.‐J.G. designed the research. C.H.vdW., M.H.vR., W.O.M.J., M.I.‐S., V.M.L., L.K., M.S., J.J.S., and H.‐J.G. performed the research.
Supporting information
Table S1. An example of an operationalized panel fit for genotyping platforms, for a subset of genes in the PGx‐Passport.
Table S2. DPWG guidelines (n = 90): covering 81 drugs and 16 genes at the time of initial selection (February 13, 2017).
Table S3. DPWG guidelines (n = 97): covering 82 drugs and 19 genes, at the time of updated selection (January 25, 2019).
Table S4. DPWG guidelines which had an actionable therapeutic recommendation for at least one of the predicted phenotypes (n = 54): covering 49 drugs and 14 genes, at the time of updated selection (January 25, 2019).
Table S5. References used to compile variants in actionable pharmacogenes (n = 13).
References
- 1. Relling, M.V. & Evans, W.E. Pharmacogenomics in the clinic. Nature 526, 343–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinshilboum, R. & Wang, L. Pharmacogenomics: bench to bedside. Nat. Rev. Drug Discov. 3, 739–748 (2004). [DOI] [PubMed] [Google Scholar]
- 3. Pirmohamed, M. Personalized pharmacogenomics: predicting efficacy and adverse drug reactions. Annu. Rev. Genomics Hum. Genet. 15, 349–370 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Pirmohamed, M. et al A randomized trial of genotype‐guided dosing of warfarin. N. Engl. J. Med. 369, 2294–2303 (2013). [DOI] [PubMed] [Google Scholar]
- 5. Wu, A.H. Pharmacogenomic testing and response to warfarin. Lancet 385, 2231–2232 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Verhoef, T.I. et al A randomized trial of genotype‐guided dosing of acenocoumarol and phenprocoumon. N. Engl. J. Med. 369, 2304–2312 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Coenen, M.J. et al Identification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology 149, 907–917 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Mallal, S. et al HLA‐B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 358, 568–579 (2008). [DOI] [PubMed] [Google Scholar]
- 9. Abbasi, J. Getting pharmacogenomics into the clinic. JAMA 316, 1533–1535 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Haga, S.B. & Burke, W. Pharmacogenetic testing: not as simple as it seems. Genet. Med. 10, 391–395 (2008). [DOI] [PubMed] [Google Scholar]
- 11. Swen, J.J. et al Translating pharmacogenomics: challenges on the road to the clinic. PLoS Med. 4, e209 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swen, J.J. et al Pharmacogenetics: from bench to byte – an update of guidelines. Clin. Pharmacol. Ther. 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 13. Swen, J.J. et al Pharmacogenetics: from bench to byte. Clin. Pharmacol. Ther. 83, 781–787 (2008). [DOI] [PubMed] [Google Scholar]
- 14. Relling, M.V. & Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the pharmacogenomics research network. Clin. Pharmacol. Ther. 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bank, P. et al Comparison of the guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 103, 599–618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunnenberger, H.M. et al Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu. Rev. Pharmacol. Toxicol. 55, 89–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weitzel, K.W. , Cavallari, L.H. & Lesko, L.J. Preemptive panel‐based pharmacogenetic testing: the time is now. Pharm. Res. 34, 1551–1555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Driest, S.L. et al Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 95, 423–431 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samwald, M. et al Incidence of exposure of patients in the United States to multiple drugs for which pharmacogenomic guidelines are available. PLoS One 11, e0164972 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roden, D.M. et al Benefit of preemptive pharmacogenetic information on clinical outcome. Clin. Pharmacol. Ther. 103, 787–794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Wouden, C.H. et al Implementing pharmacogenomics in Europe: design and implementation strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 101, 341–358 (2017). [DOI] [PubMed] [Google Scholar]
- 22. Elliott, L.S. , Henderson, J.C. , Neradilek, M.B. , Moyer, N.A. , Ashcraft, K.C. & Thirumaran, R.K. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: a prospective pilot randomized controlled trial. PLoS One 12, e0170905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brixner, D. et al The effect of pharmacogenetic profiling with a clinical decision support tool on healthcare resource utilization and estimated costs in the elderly exposed to polypharmacy. J. Med. Econ. 19, 213–228 (2016). [DOI] [PubMed] [Google Scholar]
- 24. Pérez, V. et al Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: results of a randomized, double‐blind clinical trial. BMC Psychiatry 17, 250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walden, L.M. et al Genetic testing for CYP2D6 and CYP2C19 suggests improved outcome for antidepressant and antipsychotic medication. Psychiatry Res. 10.1016/j.psychres.2018.02.055. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 26. Espadaler, J. , Tuson, M. , Lopez‐Ibor, J.M. , Lopez‐Ibor, F. & Lopez‐Ibor, M.I. Pharmacogenetic testing for the guidance of psychiatric treatment: a multicenter retrospective analysis. CNS Spectr. 22, 315–324 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Lauschke, V.M. , Zhou, Y. & Ingelman‐Sundberg, M. Novel genetic an epigenetic factors of importance for inter‐individual differences in drug disposition, response and toxicity. Pharmacol. Ther. 10.1016/j.pharmthera.2019.01.002. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caudle, K.E. , Keeling, N.J. , Klein, T.E. , Whirl‐Carrillo, M. , Pratt, V.M. & Hoffman, J.M. Standardization can accelerate the adoption of pharmacogenomics: current status and the path forward. Pharmacogenomics 19, 847–860 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pratt, V.M. et al Characterization of 137 genomic DNA reference materials for 28 pharmacogenetic genes: a GeT‐RM Collaborative Project. J. Mol. Diagn. 18, 109–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pratt, V.M. et al Characterization of 107 genomic DNA reference materials for CYP2D6, CYP2C19, CYP2C9, VKORC1, and UGT1A1 A GeT‐RM and Association for Molecular Pathology Collaborative Project. J. Mol. Diagn. 12, 835–846 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pratt, V.M. et al Recommendations for clinical CYP2C19 genotyping allele selection: a report of the Association for Molecular Pathology. J. Mol. Diagn. 20, 269–276 (2018). [DOI] [PubMed] [Google Scholar]
- 32. Bush, W.S. et al Genetic variation among 82 pharmacogenes: the PGRNseq data from the eMERGE network. Clin. Pharmacol. Ther. 100, 160–169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hicks, J.K. , Swen, J.J. & Gaedigk, A. Challenges in CYP2D6 phenotype assignment from genotype data: a critical assessment and call for standardization. Curr. Drug Metab. 15, 218–232 (2014). [DOI] [PubMed] [Google Scholar]
- 34. Zhou, Y. , Mkrtchian, S. , Kumondai, M. , Hiratsuka, M. & Lauschke, V.M. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenomics J. 19, 115–126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weinshilboum, R.M. & Sladek, S.L. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am. J. Hum. Genet. 32, 651–662 (1980). [PMC free article] [PubMed] [Google Scholar]
- 36. Kozyra, M. , Ingelman‐Sundberg, M. & Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 19, 20–29 (2017). [DOI] [PubMed] [Google Scholar]
- 37. Drogemoller, B.I. , Wright, G.E. & Warnich, L. Considerations for rare variants in drug metabolism genes and the clinical implications. Expert Opin. Drug Metab. Toxicol. 10, 873–884 (2014). [DOI] [PubMed] [Google Scholar]
- 38. Li, B.In. et al silico comparative characterization of pharmacogenomic missense variants. BMC Genom. 15 (suppl. 4), S4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou, Y. , Ingelman–Sundberg, M. & Lauschke, V.M. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin. Pharmacol. Ther. 102, 688–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shraga, R. et al Evaluating genetic ancestry and self‐reported ethnicity in the context of carrier screening. BMC Genet. 18, 99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Melis, R. et al Copy number variation and incomplete linkage disequilibrium interfere with the HCP5 genotyping assay for abacavir hypersensitivity. Genet. Test Mol. Biomarkers 16, 1111–1114 (2012). [DOI] [PubMed] [Google Scholar]
- 42. Rodriguez‐Novoa, S. et al Use of the HCP5 single nucleotide polymorphism to predict hypersensitivity reactions to abacavir: correlation with HLA‐B*5701. J. Antimicrob. Chemother. 65, 1567–1569 (2010). [DOI] [PubMed] [Google Scholar]
- 43. Badulli, C. et al Tag SNPs of the ancestral haplotype 57.1 do not substitute HLA‐B* 57: 01 typing for eligibility to abacavir treatment in the Italian population. Pharmacogenomics 13, 247–249 (2012). [DOI] [PubMed] [Google Scholar]
- 44. de Bakker, P.I. et al A high‐resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Amstutz, U. et al HLA‐A 31:01 and HLA‐B 15:02 as genetic markers for carbamazepine hypersensitivity in children. Clin. Pharmacol. Ther. 94, 142–149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bank, P.C. , Swen, J.J. , Schaap, R.D. , Klootwijk, D.B. , Baak‐Pablo, R.F. & Guchelaar, H.J. Results of the implementation of pharmacogenomics into primary care project. Clin. Pharmacol. Ther. 101, S6‐S6 (2017). [Google Scholar]
- 47. Peterson, J.F. et al Attitudes of clinicians following large‐scale pharmacogenomics implementation. Pharmacogenomics J. 16, 393–398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winner, J.G. et al Combinatorial pharmacogenomic guidance for psychiatric medications reduces overall pharmacy costs in a 1 year prospective evaluation. Curr. Med. Res. Opin. 31, 1633–1643 (2015). [DOI] [PubMed] [Google Scholar]
- 49. Alagoz, O. , Durham, D. & Kasirajan, K. Cost‐effectiveness of one‐time genetic testing to minimize lifetime adverse drug reactions. Pharmacogenomics J. 16, 129–136 (2015). [DOI] [PubMed] [Google Scholar]
- 50. Kirchheiner, J. , Tsahuridu, M. , Jabrane, W. , Roots, I. & Brockmoller, J. The CYP2C9 polymorphism: from enzyme kinetics to clinical dose recommendations. Per. Med. 1, 63–84 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. An example of an operationalized panel fit for genotyping platforms, for a subset of genes in the PGx‐Passport.
Table S2. DPWG guidelines (n = 90): covering 81 drugs and 16 genes at the time of initial selection (February 13, 2017).
Table S3. DPWG guidelines (n = 97): covering 82 drugs and 19 genes, at the time of updated selection (January 25, 2019).
Table S4. DPWG guidelines which had an actionable therapeutic recommendation for at least one of the predicted phenotypes (n = 54): covering 49 drugs and 14 genes, at the time of updated selection (January 25, 2019).
Table S5. References used to compile variants in actionable pharmacogenes (n = 13).