

Abstract
Liver plays a major role in drug metabolism and is one of the main sites of drug adverse effects. Microphysiological systems (MPS), also known as organs‐on‐a‐chip, are a class of microfluidic platforms that recreate properties of tissue microenvironments. Among different properties, the liver microenvironment is three‐dimensional, fluid flows around its cells, and different cell types regulate its function. Liver MPS aim to recreate these properties and enable drug testing and measurement of functional endpoints. Tests with these systems have demonstrated their potential for predicting clinical drug effects. Properties of liver MPS that improve the physiology of cell culture are reviewed, specifically focusing on the importance of recreating a physiological microenvironment to evaluate and model drug effects. Advances in modeling hepatic function by leveraging MPS are addressed, noting the need for standardization in the use, quality control, and interpretation of data from these systems.
Microphysiological systems (MPS) are a class of microfabricated in vitro cellular platforms conceived with the intent of replicating specific physiological organ or tissue settings that define their function.1 MPS are currently being evaluated for different uses in drug development,2 disease modeling,3 basic research in human physiology,4, 5 and reducing the use of animal testing or the cost of clinical trials.6 According to the US National Institutes of Health, they are defined as microfluidic systems that enable the coculture of at least two types of human cells in three dimensions (3‐D)1 (Figure 1). In general, 3‐D microenvironments and cocultures of tissue‐specific cells mimic organ physiology better than monocultures of cells in traditional two‐dimensional (2‐D) plates/dishes.7 In accordance with this definition of MPS, the achievements, potential, and challenges of using hepatic/liver MPS in drug development are reviewed. Focus is placed on the opportunities to use this technology for filling gaps in the drug regulatory evaluation process, particularly for applications where human‐specific testing is required.
Figure 1.
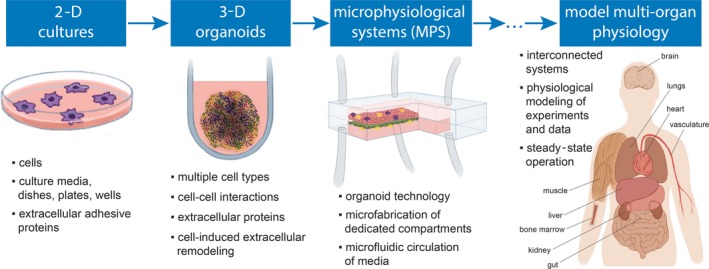
Microphysiological systems (MPS) as advanced platforms to model cellular properties in vitro. Petri dishes and similar two‐dimensional (2‐D) cell culture platforms have been used for almost a century as test beds for maintaining cell cultures and modeling the translation of their biological properties to clinical observations. More recently, spheroid technology has enabled the possibility of simultaneously coculturing different cell types that represent tissue‐specific cellular varieties in three‐dimensions (3‐D) configuration to recreate more physiologically relevant settings in vitro. For developing MPS, organoid‐like coculture approaches were integrated in microfluidic devices with the intent of further improving the physiological relevance of 3‐D cocultures, with the potential to also address several limitations related to lack of human‐specific properties of preclinical tests used in drug development. The field is fast evolving to the deployment of interconnected systems representing interorgan communications and the modeling of more physiologically relevant in vitro assays (63). Illustrations are inspired by different sources.89, 90 [Colour figure can be viewed at wileyonlinelibrary.com]
Other microfabricated cell culture platforms with characteristics that do not fit the definition of MPS can also enhance the physiological performance of hepatocytes. For example, micropatterning colonies of hepatocytes on a 2‐D surface and surrounding them with stromal cells has been shown to improve the lifetime and function of hepatocytes in culture,8 with demonstrated potential for drug development and for studying drug transport and drug‐drug interaction.9, 10
The initial motivation for developing MPS was to improve prediction of clinical drug effects early in drug development to improve its efficiency.1 This motivation primarily reflected the drug development burden resulting from the discordance in drug toxic effects between animals and humans.11, 12 Nine years after the first study reporting MPS was published,13 liver has become the most modeled organ, and different types of systems have been developed. Several publications have demonstrated advances in system improvement and reported different applications in recreating physiology and disease conditions, testing drug effects, toxicity screening, and capturing drug metabolism. Modeling liver function is important for drug development because liver is a key organ in the generation of toxic drug metabolites14 and drug metabolism, which has implications for drug clearance15 and drug–drug interactions.16 Data from such physiologically relevant hepatic setting (Figure 2 a) have the potential to improve predictions from physiologically‐based pharmacokinetic and pharmacodynamic models. Furthermore, drug‐induced liver injury accounts for approximately 30% of all drug retractions,17 and a physiologically relevant platform could improve the confidence of compound screening for hepatic toxicity early in drug development.
Figure 2.
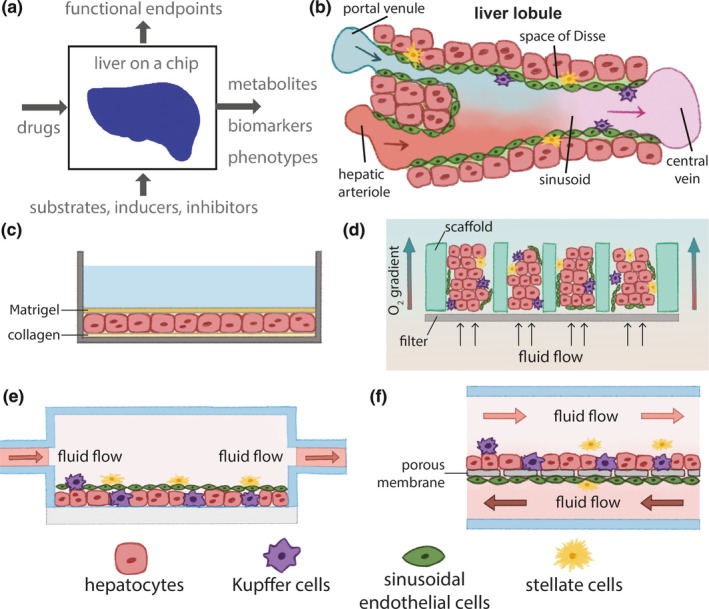
Drug development with liver MPS. Liver microphysiological systems (MPS) result from coculturing different cell types in microfluidic settings that are designed to set the physiology of cellular function. (a) MPS can model drug effects via analysis of metabolism, drug–drug interactions, biomarkers, and phenotypes, such as transport, structure, metabolites, and toxicity. (b) The microenvironment of the liver lobule is multicellular, three‐dimensional, under flow, and defines the sinusoid. (c) two‐dimensional sandwich culture of hepatocytes between a collagen‐coated surface and a layer of Matrigel preserves hepatic function. (d–f) Different designs of liver MPS. Microtissues attach to scaffolds and are exposed to media flow and oxygen gradients in d. Microfluidic chambers (e and f) can also maintain tissue function, under oxygen gradients.91 A porous membrane can separate endothelial cells from hepatocytes to mimic a barrier (f). Illustrations are inspired by different sources.42, 43 [Colour figure can be viewed at wileyonlinelibrary.com]
The abundance of published and commercialized liver systems, along with the potential impact that this technology may have on drug development, requires a focused assessment to determine how MPS may be utilized in contexts of use relevant to drug regulatory evaluations. This review describes the current capabilities of hepatic MPS that can improve drug development, defines future applications in toxicology and clinical pharmacology, and discusses the hurdles to employ these systems reliably and robustly.
Human Physiological Relevance In Vitro will Enable Clinical Predictions of Drug Effects
A principal challenge for developing in vitro cellular systems as drug development tools during the last decade has been the incorporation of relevant functional endpoints or toxicity output biomarkers that represent in vivo or clinical scenarios.12, 18 MPS have the potential to partially meet this challenge (Figure 2). This section focuses on defining and discussing potential physiological contexts of use for hepatic MPS. However, given the state of infancy of this field, these contexts will require deeper investigation, evaluation, and standardization to ensure the reliability and robustness of applied measurements.5, 19 In general, this level of standardization is required for cells as in vitro testing units of drug effects can be susceptible to change under varying cell isolation protocols and other noncontrollable experimental conditions because of their biological properties and functional performance.20, 21, 22
For cells from distinct isolation batches, differences in cellular function that do not relate to variations in the genetic background between donors can call into question the confidence of mechanistic predictions from experimental data. For this reason, quality control of isolated cell batches is pivotal for mechanistic evaluation of drug effects in vitro,23 and the experimental use of hepatocytes should consider quality control standards based on metabolism, transport, biomarker expression, and cell structure. Similar concerns exist in using cellular systems, where variability in results between systems can eventually arise from differences in the design of microfluidic circuits, flow profiles, cell–material interactions, and disparities between cell batches. Therefore, quality control assays and standards should be developed for cellular systems to enable confidence in their use. Cellular systems traditionally have low predictivity of drug candidates’ clinical hepatic toxicity,17 and enhanced physiological relevance of these systems and increased robustness and reliability in their use will improve their impact in drug development.
Overall, robustness and reliability of operation are key components for the evaluation of cell‐based platforms intended to predict clinical drug effects. For example, HepG2 cells belong to a class of standardized and robust human hepatocellular carcinoma immortalized cell lines, but they have impaired metabolic, transporter, and structural functions that limit their use in mechanistic studies that may better predict hepatic drug effects.24, 25, 26 That said, given their robustness, HepG2 cells can still predict hepatotoxicity of compounds with 80% sensitivity and 90% specificity.27 This shows great promise for early drug screening procedures that may precede more informative platforms for drug mechanisms of action, including MPS (Figure 2).
In contrast to immortalized cell lines, primary hepatocytes can enable mechanistic assays for metabolism, transport, and hepatic biology in general.28 However, animal and human hepatocytes differ in metabolism, transport, and sensitivity to many toxicants.29, 30, 31 In addition, the low availability, short time of duration in culture, and high interisolation variability of human hepatocytes28, 32 limit the standardization of their use. Recently discovered hepatocytes differentiated from human induced pluripotent stem cells (iPSCs) represent a promising alternative for mechanistic evaluation of drug effects,5, 33 but improvements are still required in differentiation and maturation protocols to develop cells that fully replicate mature hepatocyte‐like functions.34
Hepatocytes differentiated from iPSCs tend to have fetal‐like properties when compared with primary cells, such as lower metabolic activity, lower albumin secretion, and higher expression of fetal markers such as alpha‐fetoprotein, glutathione‐S‐transferase, and heat shock protein 47.35 However, these cells have the potential to model hepatic biology better than immortalized cell lines. In general, different types of hepatic cells for in vitro use present limitations that relate to biological variability, impaired hepatic properties, fetal‐like profile, or lack of human‐specific properties. Opportunities to solve hurdles in drug development and change regulatory paradigms can arise from solving these limitations, which include the following: (i) better predictability of clinical drug toxicity,36 (ii) physiological performance in drug uptake and efflux, metabolism, and interaction with other drugs,37 (iii) mechanistic information of drug effects,38 and (iv) assay genetic predispositions for differential drug effects among populations.39
Of considerable interest to the MPS field is that many of the limitations inherent to the use of hepatic cells can be addressed by culturing them in 3‐D under media flow or in the presence of other hepatic cell types.5, 40, 41, 42, 43 For example, by prolonging the lifetime and stability of cellular cultures, more accurate physiological microenvironments improve the hepatic properties of HepG2 cells and iPSC‐hepatocytes.5, 44 This improvement in hepatic function when iPSC‐hepatocytes are cultured in MPS suggests that the cellular microenvironment within such advanced in vitro models can improve differentiation of maturation.5 This potential should be the object of future research focusing on dissecting the biological regulatory pathways in iPSC‐hepatocytes that are activated under more physiological microenvironments.45, 46 For primary hepatocytes, enzymatic function and albumin production can be prolonged for more than one month after isolation or plating when cells are maintained in physiological microenvironments, such as 3‐D spheroids or MPS.47, 48 In contrast, in 2‐D sandwich cultures, the hepatic function of primary hepatocytes is lost within 3–9 days of culture following a dedifferentiation process.32 In conclusion, liver MPS have the potential to overcome the current limitations inherent to the use of hepatocytes or hepatocyte‐like cells.
The Supplementary Material associated with this paper focuses on the physiological relevance of each characteristic of liver MPS that distinguishes these systems from traditional 2‐D culture platforms, i.e., 3‐D organization, interstitial media flow, and co‐culture with different cell types (Figure 1). With regard to media flow, the importance of controlling oxygen concentration in MPS to mimic liver physiology as a function of oxygen tension is also described (Figure 3). Other cellular functional properties vary along the hepatic sinusoid, a topic reviewed elsewhere.49, 50, 51 In addition, the relevance of including different cell types for regulatory applications is described, where the presence of specific cell types dictates the ability to replicate physiological settings in vitro.
Figure 3.
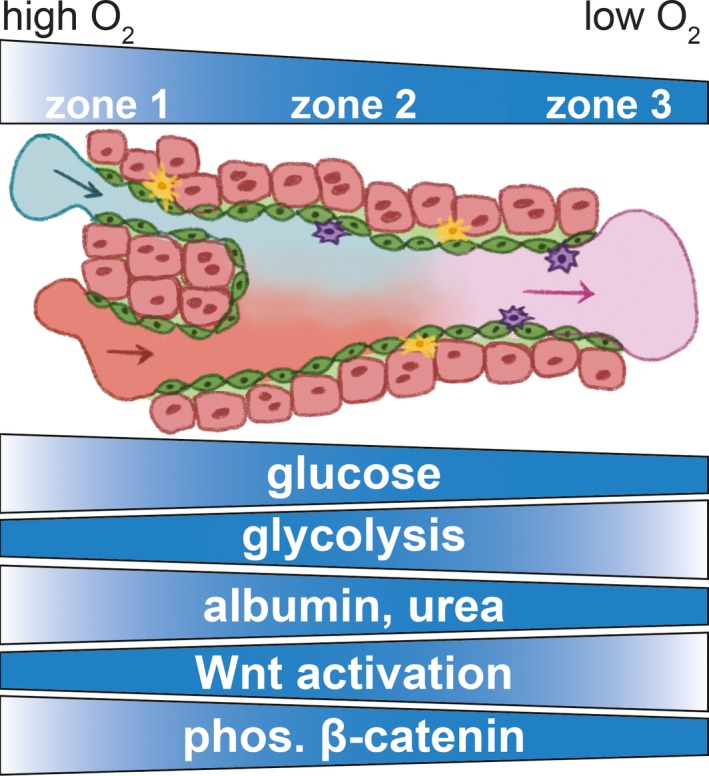
Physiological zonation and cellular activity in the sinusoid unit of the liver. In a healthy and mature liver, oxygen (O2) tension decreases as blood flows along the sinusoid unit (as detailed in Figure 2), from the portal triad (zone 1) to the central vein (zone 3). Different types of cell‐based molecular activities relate to this oxygen tension gradient. Glucose concentration, glycolytic activity, albumin and urea production, Wnt signaling and phosphorylation (phos.) of β‐catenin vary as noted in relation to oxygen concentration. The illustration is inspired by different sources.43, 50 Other cellular functions vary along the hepatic sinusoid, a topic reviewed elsewhere.49, 50, 51 [Colour figure can be viewed at wileyonlinelibrary.com]
Hepatic MPS Can Improve the Predictive Ability of Metabolism Studies
MPS have the capability to solve several hurdles in modeling clinical pharmacology52 for the following reasons: (i) their function is more physiologically relevant, (ii) their biological activity lasts longer in culture than traditional culture systems, (iii) they enable quantification of the intracellular and extracellular drug and metabolite concentrations as in vitro proxies for a physiological tissue. Furthermore, MPS can be exposed to different drug treatments and can contain different genetic backgrounds from distinct cell donors that with further research, may have the potential to translate to clinical doses and regimens and hence to patient‐specific effects.39, 53, 54
The discipline of clinical pharmacology studies the use, effects, and mechanisms of action of human drugs to predict the safety and efficacy of different dosages and regimens. Modeling drug actions requires the combination of data from different fields of investigation, including pharmacokinetics, pharmacodynamics, mechanisms of therapeutic efficacy, and adverse effects (Figure 4). The roles of clinical pharmacology and the contributions of its different branches for improving drug development are reviewed elsewhere.55, 56, 57
Figure 4.
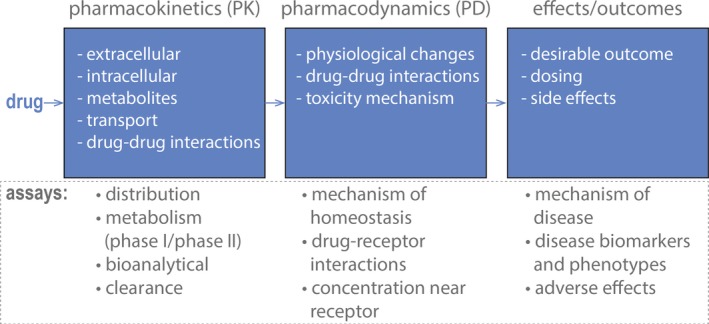
Applications of liver microphysiological systems (MPS) in the field of clinical pharmacology. To understand clinical drug action, studies in clinical pharmacology aim to characterize drug pharmacokinetics, pharmacodynamics, and their relationship to drug effects. For this purpose, different assays can be developed or adapted to liver MPS, and these systems can be used simultaneously to measure, for example, therapeutic and toxic concentrations, characterize metabolism and transporter effects, and evaluate drug–drug interactions. The potential to multiplex different types of measurements in MPS can provide data to support pharmacology models to inform the clinical study design. Pharmacokinetic studies with MPS are also useful for understanding mechanisms that affect drug (and metabolite) concentrations in plasma and liver. [Colour figure can be viewed at wileyonlinelibrary.com]
A comprehensive assessment of drugs’ metabolic fate and evaluation of the effect of various intrinsic and extrinsic factors is difficult to obtain solely based on clinical data and should be complemented with in vitro assays and animal models.52 Due to their human‐specific properties, MPS can complement current in vitro and animal models and may serve as a link between preclinical and clinical studies. Following this use, the physiological relevance of MPS may also support better‐designed clinical trials that minimize the number of participants.
As the liver is an organ involved in drug metabolism, the use of hepatic MPS can address needs in the fields of clinical pharmacology and toxicity assessment that involve evaluation of the following: (i) drug–drug interactions,53, 58 (ii) transport and intracellular drug accumulation and binding,59, 60 and (iii) prediction of drug hepatotoxicity due to chronic exposure.61, 62 The usefulness of MPS to comprehensively model the interaction between drugs and organs can rely on how cell culture media are exposed to cells (Figure 5).63 Some types of hepatic MPS have been designed to recirculate cell culture medium and require it to be changed every 2–3 days to provide fresh nutrients to cells and remove cell waste (Figure 5 a). Media changes abruptly alter the concentration of soluble components of the cellular microenvironment that may affect cell metabolism and physiology.63 Therefore, use of these types of systems may be challenging in some settings where the evolution of effects on drug metabolism are slow or depend upon repeated exposures. To better mimic such situations, modeling should take into consideration media changes and determine how they can impact the clinical translation of results from these systems.63
Figure 5.
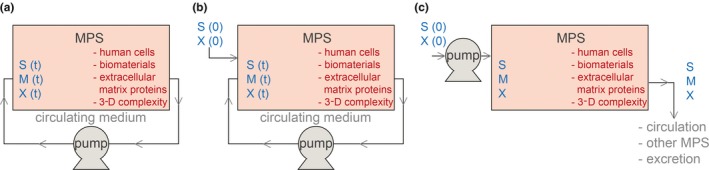
Different possible modes of operation of liver microphysiological systems (MPS) based on circulation of cell culture medium. Developed systems can differ from each other on operational levels that relate to the way cell culture medium is supplied to the cell culture unit. These operational levels have a strong impact in the kinetics of cellular metabolism and energetics due to differences in cellular exposure to nutrients, metabolites, and drugs, which eventually affect system response to drugs.63 (a) Medium can be recirculated and completely changed every 2–3 days throughout the operation time.53 (b) Soluble medium components or compounds can be supplied between media changes.39 (c) Fresh medium can be continuously perfused into the cell culture unit.61 S represents drug substrates, M represents drug metabolites, and X represents carbon sources or other soluble components that set cellular function or physiology. 3‐D, three‐dimensional. [Colour figure can be viewed at wileyonlinelibrary.com]
Other designs can incorporate the possibility of perfusing media into the cell culture chamber of the system (Figure 5 b) or maintain homeostasis of soluble components within the cellular microenvironment by continuously adding and removing cell culture medium (Figure 5 c). The effects of different designs for handling cell culture medium in drug evaluation should be considered for modeling purposes. The ability to connect different MPS that represent different types of organs further increases the potential of this technology for use in clinical pharmacology by evaluating drug effects that rely on interactions between different organs.64
Connecting Hepatic Systems to Other Organ Systems Increases the Physiological Relevance of Drug Testing
A major goal in the field of MPS is simulation of physiological multiorgan interactions involved in drug effects and bioprocessing via the interconnection of different organ systems.4 Interconnected MPS can facilitate the following: (i) detection of drug effects that depend on multiorgan interactions and (ii) set physiological states that are regulated by multiorgan interactions. The consistent use of liver MPS in different versions of multiorgan platforms42, 54, 64, 65, 66, 67 is necessary because of their roles in drug metabolism.
Coupling cardiac to liver MPS has the potential to improve predictions of clinical cardiac drug side effects, a main cause of drug attrition.68 As one example, terfenadine can cause cardiac toxicity depending on liver function:69 work with multiorgan systems has already demonstrated the ability of these platforms to detect variable terfenadine toxicity as a function of liver metabolism.42 Research has further demonstrated the ability to interconnect other organs susceptible to toxicity, such as kidney, skin, lung, gastrointestinal tract, testis, and brain.64, 70, 71
The main hurdles for using multiorgans in drug evaluation are the high cost and practical difficulties associated with maintaining several interconnected systems.72 However, this cost may be reduced if data from preliminary experiments with cheaper platforms, including computational tools, can already inform researchers concerning an affordable number of experimental conditions to consider. For example, pre‐experimental in silico modeling for recreating clinically relevant levels of exposure in MPS may decrease the number of experimental conditions required for testing, and minimize the cost of using these systems for drug evaluation.54, 73 Furthermore, modeling data in silico from experiments with MPS will ultimately improve predictions of drug pharmacokinetic and pharmacodynamic profiles utilized in the planning of clinical trials.72, 73 Modeling pharmacokinetics and pharmacodynamics from experiments with MPS representing different organs and with interconnected MPS is reviewed elsewhere.72
Multiorgan MPS use requires data from liver metabolism and transport experiments to determine several pharmacokinetic parameters. Pharmacokinetic parameters that can be derived from bioanalytical experiments with liver MPS, and used for physiologically‐based pharmacokinetic modeling, include liver intrinsic clearance and the partition coefficient from circulation into liver.72 In combination with other in vitro information and/or clinical data, parameters derived from MPS can be used to extrapolate clinical results from modeling drug pharmacokinetics.72, 74 A direct application of in vivo extrapolations from MPS data is the possibility of predicting ranges of clinical drug regimens that may maximize efficacy while also minimizing toxicity and predicting risk.
Interconnection of various MPS representing different organs can also predict drug effects that depend on multiorgan interactions. Techniques to interconnect up to 10 organ systems in one platform have been successfully developed.64 However, 10 organs and the in vitro interactions between them is likely still an underrepresentation of various multitissue interactions that regulate drug response physiology.75 For example, endocrine regulation of organ function by secretion of hormones or peptides into the bloodstream is missing from the 10‐MPS platform. The incorporation of a “missing organ” in interconnected MPS platforms for release of paracrine factors involved in specific physiological settings has been proposed as a solution to further improve the physiological relevance of multiorgan MPS.75
Setting the Quality and Functional Characteristics of Liver MPS
Cell culture conditions, cell types, media flow, being 3‐D, co‐culture of different cells, circulation of cell culture medium, interconnection with other organ systems, and modeling of experiments and experimental results are discussed in detail in this section to motivate the use of liver MPS in drug evaluation.
Standards regarding fabrication of MPS are also of key importance to ensure the robustness and reliability of their use in regulatory applications, as they generally determine the robustness and reliability of microdevices, and are reviewed elsewhere in the biomicrofabrication and microfluidics literature.76, 77 For example, polydimethylsiloxane (PDMS) is a material often used by MPS developers because of its optimal properties for micromolding.78 However, PDMS is highly hydrophobic, has unstable surface properties, and easily adsorbs small molecules; these considerations make it unsuitable for testing several types of small molecules.79 Therefore, fabrication and material properties should be given equal attention as standardization processes discussed for other MPS properties.
In closed compartmentalized systems, for example, cells are cultured in microfluidic chambers (Figure 2 e,f), as opposed to open systems (Figure 2 d). Closed systems have the potential to expose cells to controlled media volumes within the range of physiological volumes of fluids surrounding cells in tissues.1 Such conditions can potentially better replicate physiological compositions of the cellular microenvironment that rely on homeostatic states of cell metabolism, absorption, and secretion. To evaluate this potential, further research should test the effects of the dimensions of microfluidic enclosures and media exchange rates on cell function to enable a comprehensive comparison of data from different MPS. Altogether, several developers are commercializing distinct versions of liver MPS, and additional academic laboratories have also published detailed protocols and designs of novel systems.
Other challenges in pharmacokinetic applications aiming to predict clinical drug effects are the ability to scale volumes from experiments to clinical settings and the extrapolation to human‐specific physiology.80 In silico modeling is particularly important in addressing this challenge, and efforts are underway to extrapolate clinically relevant information from experiments.54 Ideally, various types of liver MPS that use similar cells should lead to similar results with similar drug treatments, while accounting for their differences in volume and media flow (Figures 2 and 5) in models for clinical data extrapolation.
Given the variety in available systems, the field lacks a comprehensive evaluation of various potential uses that may both differentiate MPS from various developers and establish similarities between them. Independent and publicly funded academic testing centers have already initiated such evaluation to address concerns regarding variability of results between laboratories using the same MPS.81 Testing centers primarily focus on site‐to‐site variability of MPS assembly and use,82 but their work has raised questions about the quality and origin of cellular material, biointerfaces between microfluidic materials and media or cells, modeling of experimental results, and clinical translation of data.83 For example, adsorption of small molecules to MPS materials that replace PDMS and approaches to account for drug‐material non‐specific interactions in pharmacokinetic simulations have been proposed for standardization purposes.39, 84 This example concerning the characterization and standardization of MPS operation illustrates the need for standardization. In summary, standardization is required in:
MPS properties: cell origin, cell quality, media, extracellular components, preparation protocols, operation quality control properties, and tissue function
MPS use in drug development: drug treatment schedules, measurement schedules, toxicity, biomarkers, mechanistic endpoints, pharmacokinetics, pharmacodynamics, efficacy, and experiment and data modeling
Multiple donors with distinct genetic backgrounds: isolated cellular material85 or differentiated iPSC‐hepatocytes86 from different individuals with functional properties that reliably represent clinical settings
For liver MPS, a focused standardization effort should consider metabolism, transport, drug intracellular accumulation, drug–drug interactions, inflammation, and interactions between different cell types that regulate drug effects on liver physiology. Robustly capturing variability in liver function between different groups of donors87 and differentiating donor‐specific results from the effects of cell handling procedures should also be considered for ensuring reliability of results with liver MPS. The execution of this effort should not fall solely on academic research but should also involve regulatory agencies, the pharmaceutical industry, commercial developers of MPS, and other drug development stakeholders.
Concluding Comments and Future Work
Different studies have validated the suitability of liver MPS to predict drug effects that traditional culture systems fail to predict because of the loss of cellular hepatic properties after a few days of culture or because cell lines or iPSC‐derived cells have poorly developed metabolism or transport. In general, different versions of liver MPS (Figure 2 d–f) increase the physiological relevance of cultured cells’ hepatic function and can enable clinical drug effect prediction with higher reliability. In addition, under physiological conditions, liver MPS can provide more clinically relevant data on pharmacokinetics, which can improve models in the domain of clinical pharmacology (Figure 4). Different properties of MPS that set their physiological relevance, and the importance of these properties for designing and analyzing assays in the context of drug development, have been discussed.
Different liver MPS (Figure 2 d–f) can vary in the design of microfluidic devices, pumping systems, profile of media circulation (Figure 5), coculture of varied cell types, and their connectivity to other MPS that represent different organs. Given the complexity of MPS and the relative infancy of this field, the main consideration for their use in drug development relates to their reliability and robustness.88 Therefore, engendering confidence in this technology requires a concerted standardization effort that builds upon the work of the tissue‐chip testing centers and focuses on different contexts of use. Such an effort should involve multiple stakeholders within drug development. Such standardization efforts will also require comprehensive studies of the operational effects of system materials, cellular properties, and fluidic design that yield informed interpretations of results to ensure assay quality.
Funding
The authors acknowledge support from the Defense Advanced Research Projects Agency via an Interagency Agreement with the FDA.
Conflict of Interest
The authors declared no competing interests for this work.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the views or policies of the FDA.
Supporting information
Supplementary Material
Acknowledgments
The authors would like to thank the following members of the US Food and Drug Administration's (FDA) Division of Applied Regulatory Science and Office of Clinical Pharmacology for valuable discussions: Donna Volpe, Neil Hartman, Kristina Howard, Hisham Qosa, Jim Weaver, and Shiew Mei Huang. The authors would also like to thank J. Rick Turner, PhD, DSc (DRT Strategies, Inc.) for editorial assistance.
The copyright line for this article was changed on 18 July 2019 after original online publication.
References
- 1. Wikswo, J.P. The relevance and potential roles of microphysiological systems in biology and medicine. Exp. Biol. Med. (Maywood) 239, 1061–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Low, L.A. & Tagle, D.A. Tissue chips ‐ innovative tools for drug development and disease modeling. Lab Chip 17, 3026–3036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Low, L.A. & Tagle, D.A. Microphysiological systems (tissue chips) and their utility for rare disease research. Adv. Exp. Med. Biol. 1031, 405–415 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Wikswo, J.P. et al Scaling and systems biology for integrating multiple organs‐on‐a‐chip. Lab Chip 13, 3496–3511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raasch, M. , Fritsche, E. , Kurtz, A. , Bauer, M. & Mosig, A.S. Microphysiological systems meet hiPSC technology ‐ New tools for disease modeling of liver infections in basic research and drug development. Adv. Drug Deliv. Rev. (2018) 10.1016/j.addr.2018.06.008. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6. Marx, U. et al Biology‐inspired microphysiological system approaches to solve the prediction dilemma of substance testing. Altex 33, 272–321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duval, K. et al Modeling physiological events in 2D vs 3D cell culture. Physiology (Bethesda) 32, 266–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 8. March, S. et al Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat. Protoc. 10, 2027–2053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin, C. & Khetani, S.R. Micropatterned co‐cultures of human hepatocytes and stromal cells for the assessment of drug clearance and drug‐drug interactions. Curr. Protoc. Toxicol. 72, 14.7.1–14.7.23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davidson, M.D. , Kukla, D.A. & Khetani, S.R. Microengineered cultures containing human hepatic stellate cells and hepatocytes for drug development. Integr. Biol. (Camb) 9, 662–677 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Olson, H. et al Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67 (2000). [DOI] [PubMed] [Google Scholar]
- 12. Sistare, F.D. & DeGeorge, J.J. Preclinical predictors of clinical safety: opportunities for improvement. Clin. Pharmacol. Ther. 82, 210–214 (2007). [DOI] [PubMed] [Google Scholar]
- 13. Huh, D. , Matthews, B.D. , Mammoto, A. , Montoya‐Zavala, M. , Hsin, H.Y. & Ingber, D.E. Reconstituting organ‐level lung functions on a chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park, B.K. et al Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug. Discov. 10, 292–306 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Pond, S.M. & Tozer, T.N. First‐pass elimination, Basic concepts and clinical consequences. Clin. Pharmacokinet. 9, 1–25 (1984). [DOI] [PubMed] [Google Scholar]
- 16. Orhan, H. & Vermeulen, N.P. Conventional and novel approaches in generating and characterization of reactive intermediates from drugs/drug candidates. Curr. Drug Metab. 12, 383–394 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Tralau, T. & Luch, A. Drug‐mediated toxicity: illuminating the ‘bad’ in the test tube by means of cellular assays? Trends Pharmacol. Sci. 33, 353–364 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Muller, P.Y. & Dieterle, F. Tissue‐specific, non‐invasive toxicity biomarkers: translation from preclinical safety assessment to clinical safety monitoring. Expert Opin. Drug Me Toxicol. 5, 1023–1038 (2009). [DOI] [PubMed] [Google Scholar]
- 19. Dehne, E.M. , Hasenberg, T. & Marx, U. The ascendance of microphysiological systems to solve the drug testing dilemma. Future Sci. OA. 3, FSO185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guantes, R. , Diaz‐Colunga, J. & Iborra, F.J. Mitochondria and the non‐genetic origins of cell‐to‐cell variability: more is different. BioEssays 38, 64–76 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Paladino, F.V. , Peixoto‐Cruz, J.S. , Santacruz‐Perez, C. & Goldberg, A.C. Comparison between isolation protocols highlights intrinsic variability of human umbilical cord mesenchymal cells. Cell Tissue Bank 17, 123–136 (2016). [DOI] [PubMed] [Google Scholar]
- 22. McGillicuddy, N. , Floris, P. , Albrecht, S. & Bones, J. Examining the sources of variability in cell culture media used for biopharmaceutical production. Biotechnol. Lett. 40, 5–21 (2018). [DOI] [PubMed] [Google Scholar]
- 23. Knobeloch, D. et al Human hepatocytes: isolation, culture, and quality procedures. Methods Mol. Biol. 806, 99–120 (2012). [DOI] [PubMed] [Google Scholar]
- 24. Gerets, H.H.J. et al Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 28, 69–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gissen, P. & Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 63, 1023–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Louisa, M. , Suyatna, F.D. , Wanandi, S.I. , Asih, P.B.S. & Syafruddin, D. Differential expression of several drug transporter genes in HepG2 and Huh‐7 cell lines. Adv. Biomed. Res. 5, 104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Brien, P.J. et al High concordance of drug‐induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell‐based model using high content screening. Arch. Toxicol. 80, 580–604 (2006). [DOI] [PubMed] [Google Scholar]
- 28. Zeilinger, K. , Freyer, N. , Damm, G. , Seehofer, D. & Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. (Maywood) 241, 1684–1698 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merrill, J.C. , Beck, D.J. , Kaminski, D.A. & Li, A.P. Polybrominated biphenyl induction of cytochrome P450 mixed function oxidase activity in primary rat and human hepatocytes. Toxicology 99, 147–152 (1995). [DOI] [PubMed] [Google Scholar]
- 30. Richert, L. et al Species differences in the response of liver drug‐metabolizing enzymes to (S)‐4‐O‐tolylsulfanyl‐2‐(4‐trifluormethyl‐phenoxy)‐butyric acid (EMD 392949) in vivo and in vitro. Drug Me Dispos. 36, 702–714 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Li, M. et al Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. Eur. J. Pharm. Sci. 35, 114–126 (2008). [DOI] [PubMed] [Google Scholar]
- 32. Heslop, J.A. et al Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 91, 439–452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mallanna, S.K. & Duncan, S.A. Differentiation of hepatocytes from pluripotent stem cells. Curr. Protoc. Stem. Cell Biol. 26, Unit 1G 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palakkan, A.A. , Nanda, J. & Ross, J.A. Pluripotent stem cells to hepatocytes, the journey so far. Biomed. Rep. 6, 367–373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baxter, M. et al Phenotypic and functional analyses show stem cell‐derived hepatocyte‐like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 62, 581–589 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKim, J.M. Jr . Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb. Chem. High Throughput Screen. 13, 188–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cherkaoui‐Rbati, M.H. , Paine, S.W. , Littlewood, P. & Rauch, C. A quantitative systems pharmacology approach, incorporating a novel liver model, for predicting pharmacokinetic drug‐drug interactions. PLoS ONE 12, e0183794 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leist, M. et al Adverse outcome pathways: opportunities, limitations and open questions. Arch. Toxicol. 91, 3477–3505 (2017). [DOI] [PubMed] [Google Scholar]
- 39. Tsamandouras, N. , Kostrzewski, T. , Stokes, C.L. , Griffith, L.G. , Hughes, D.J. & Cirit, M. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three‐dimensional human liver microphysiological system. J. Pharmacol. Exp. Ther. 360, 95–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Messner, S. , Agarkova, I. , Moritz, W. & Kelm, J.M. Multi‐cell type human liver microtissues for hepatotoxicity testing. Arch. Toxicol. 87, 209–213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nguyen, D.G. et al Bioprinted 3D primary liver tissues allow assessment of organ‐level response to clinical drug induced toxicity in vitro. PLoS ONE 11, e0158674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vernetti, L. et al Functional coupling of human microphysiology systems: intestine, liver, kidney proximal tubule, blood‐brain barrier and skeletal muscle. Sci. Rep. 7, 42296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beckwitt, C.H. et al Liver ‘organ on a chip’. Exp. Cell Res. 363, 15–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramaiahgari, S.C. et al A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver‐like properties for repeated dose high‐throughput toxicity studies. Arch. Toxicol. 88, 1083–1095 (2014). [DOI] [PubMed] [Google Scholar]
- 45. Ma, X. et al Deterministically patterned biomimetic human iPSC‐derived hepatic model via rapid 3D bioprinting. Proc. Natl Acad. Sci. USA 113, 2206–2211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang, B. et al Functional maturation of induced pluripotent stem cell hepatocytes in extracellular matrix‐a comparative analysis of bioartificial liver microenvironments. Stem Cells Transl. Med. 5, 1257–1267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bell, C.C. et al Characterization of primary human hepatocyte spheroids as a model system for drug‐induced liver injury, liver function and disease. Sci. Rep. 6, 25187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kostrzewski, T. et al Three‐dimensional perfused human in vitro model of non‐alcoholic fatty liver disease. World J. Gastroenterol. 23, 204–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gebhardt, R. & Matz‐Soja, M. Liver zonation: novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 20, 8491–8504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soto‐Gutierrez, A. , Gough, A. , Vernetti, L.A. , Taylor, D.L. & Monga, S.P. Pre‐clinical and clinical investigations of metabolic zonation in liver diseases: the potential of microphysiology systems. Exp. Biol. Med. (Maywood) 242, 1605–1616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kietzmann, T. Metabolic zonation of the liver: the oxygen gradient revisited. Redox. Biol. 11, 622–630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo, Y. et al Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Long, T.J. et al Modeling therapeutic antibody‐small molecule drug‐drug interactions using a three‐dimensional perfusable human liver coculture platform. Drug Me Dispos. 44, 1940–1948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maass, C. , Stokes, C.L. , Griffith, L.G. & Cirit, M. Multi‐functional scaling methodology for translational pharmacokinetic and pharmacodynamic applications using integrated microphysiological systems (MPS). Integr. Biol. (Camb) 9, 290–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holford, N. Clinical pharmacology = disease progression + drug action. Br. J. Clin. Pharmacol. 79, 18–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mehrotra, N. et al Role of quantitative clinical pharmacology in pediatric approval and labeling. Drug Me Dispos. 44, 924–933 (2016). [DOI] [PubMed] [Google Scholar]
- 57. Reichel, A. & Lienau, P. Pharmacokinetics in drug discovery: an exposure‐centred approach to optimising and predicting drug efficacy and safety. Handb. Exp. Pharmacol. 232, 235–260 (2016). [DOI] [PubMed] [Google Scholar]
- 58. Cascorbi, I. Drug interactions–principles, examples and clinical consequences. Dtsch. Arztebl. Int. 109, 546–555; quiz 56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chu, X. et al Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin. Pharmacol. Ther. 94, 126–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chang, S.Y. , Weber, E.J. , Ness, K.V. , Eaton, D.L. & Kelly, E.J. Liver and kidney on chips: microphysiological models to understand transporter function. Clin. Pharmacol. Ther. 100, 464–478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vernetti, L.A. et al A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp. Biol. Med. (Maywood) 241, 101–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bell, L.N. & Chalasani, N. Epidemiology of idiosyncratic drug‐induced liver injury. Semin. Liver Dis. 29, 337–347 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maass, C. et al Establishing quasi‐steady state operations of microphysiological systems (MPS) using tissue‐specific metabolic dependencies. Sci. Rep. 8, 8015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Edington, C.D. et al Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep. 8, 4530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Skardal, A. et al Multi‐tissue interactions in an integrated three‐tissue organ‐on‐a‐chip platform. Sci. Rep. 7, 8837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen, W.L.K. et al Integrated gut/liver microphysiological systems elucidates inflammatory inter‐tissue crosstalk. Biotechnol. Bioeng. 114, 2648–2659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maschmeyer, I. et al A four‐organ‐chip for interconnected long‐term co‐culture of human intestine, liver, skin and kidney equivalents. Lab Chip 15, 2688–2699 (2015). [DOI] [PubMed] [Google Scholar]
- 68. Ferri, N. , Siegl, P. , Corsini, A. , Herrmann, J. , Lerman, A. & Benghozi, R. Drug attrition during pre‐clinical and clinical development: understanding and managing drug‐induced cardiotoxicity. Pharmacol. Ther. 138, 470–484 (2013). [DOI] [PubMed] [Google Scholar]
- 69. Yap, Y.G. & Camm, A.J. Potential cardiac toxicity of H1‐antihistamines. Clin. Allergy. Immunol. 17, 389–419 (2002). [PubMed] [Google Scholar]
- 70. Heylman, C. , Sobrino, A. , Shirure, V.S. , Hughes, C.C. & George, S.C. A strategy for integrating essential three‐dimensional microphysiological systems of human organs for realistic anticancer drug screening. Exp. Biol. Med. (Maywood) 239, 1240–1254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Truskey, G.A. Human microphysiological systems and organoids as in vitro models for toxicological studies. Front. Public Health 6, 185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Prantil‐Baun, R. , Novak, R. , Das, D. , Somayaji, M.R. , Przekwas, A. & Ingber, D.E. Physiologically based pharmacokinetic and pharmacodynamic analysis enabled by microfluidically linked organs‐on‐chips. Annu. Rev. Pharmacol. Toxicol. 58, 37–64 (2018). [DOI] [PubMed] [Google Scholar]
- 73. Yu, J. et al Quantitative systems pharmacology approaches applied to microphysiological systems (MPS): data interpretation and multi‐MPS integration. CPT. Pharmacometrics Syst. Pharmacol. 4, 585–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pharmacokinetics: the dynamics of drug absorption, distribution, metabolism, and elimination In Goodman and Gilman's Manual of Pharmacology and Therapeutics 2nd edn. (eds. Hilal‐Dandan R. & Brunton L.L.) 12‐26 (McGraw‐Hill Education, New York, NY, 2014). [Google Scholar]
- 75. Wikswo, J.P. Looking to the future of organs‐on‐chips: interview with Professor John Wikswo. Future Sci. OA. 3, FSO163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mark, D. , Haeberle, S. , Roth, G. , von Stetten, F. & Zengerle, R. Microfluidic lab‐on‐a‐chip platforms: requirements, characteristics and applications. Chem. Soc. Rev. 39, 1153–1182 (2010). [DOI] [PubMed] [Google Scholar]
- 77. Sosa‐Hernández, J.E. et al Organs‐on‐a‐chip module: a review from the development and applications perspective. Micromachines (Basel) 9, pii: E536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Duffy, D.C. , McDonald, J.C. , Schueller, O.J. & Whitesides, G.M. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem. 70, 4974–4984 (1998). [DOI] [PubMed] [Google Scholar]
- 79. Toepke, M.W. & Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 6, 1484–1486 (2006). [DOI] [PubMed] [Google Scholar]
- 80. Mahmood, I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv. Drug Deliv. Rev. 59, 1177–1192 (2007). [DOI] [PubMed] [Google Scholar]
- 81. Low, L.A. & Tagle, D.A. Microphysiological systems (“organs‐on‐chips”) for drug efficacy and toxicity testing. Clin. Transl. Sci. 10, 237–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sakolish, C. et al Technology transfer of the microphysiological systems: a case study of the human proximal tubule tissue chip. Sci. Rep. 8, 14882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cirit, M. & Stokes, C.L. Maximizing the impact of microphysiological systems with in vitro‐in vivo translation. Lab Chip 18, 1831–1837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kanamori, T. , Sugiura, S. & Sakai, Y. Technical aspects of microphysiological systems (MPS) as a promising wet human‐in‐vivo simulator. Drug Metab. Pharmacokinet. 33, 40–42 (2018). [DOI] [PubMed] [Google Scholar]
- 85. Green, C.J. , Charlton, C.A. , Wang, L.M. , Silva, M. , Morten, K.J. & Hodson, L. The isolation of primary hepatocytes from human tissue: optimising the use of small non‐encapsulated liver resection surplus. Cell Tissue Bank 18, 597–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cayo, M.A. et al A drug screen using human iPSC‐derived hepatocyte‐like cells reveals cardiac glycosides as a potential treatment for hypercholesterolemia. Cell Stem Cell 20, 478–489 e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Parkinson, A. , Mudra, D.R. , Johnson, C. , Dwyer, A. & Carroll, K.M. The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol. Appl. Pharmacol. 199, 193–209 (2004). [DOI] [PubMed] [Google Scholar]
- 88. Dehne, E.M. , Hasenberg, T. & Marx, U. The ascendance of microphysiological systems to solve the drug testing dilemma. Future Sci. OA. 3, FSO185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ranga, A. , Gjorevski, N. & Lutolf, M.P. Drug discovery through stem cell‐based organoid models. Adv. Drug Deliv. Rev. 69–70, 19–28 (2014). [DOI] [PubMed] [Google Scholar]
- 90. Schepers, A. , Li, C. , Chhabra, A. , Seney, B.T. & Bhatia, S. Engineering a perfusable 3D human liver platform from iPS cells. Lab Chip 16, 2644–2653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li, X. , George, S.M. , Vernetti, L. , Gough, A.H. & Taylor, D.L. A glass‐based, continuously zonated and vascularized human liver acinus microphysiological system (vLAMPS) designed for experimental modeling of diseases and ADME/TOX. Lab Chip 18, 2614–2631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material