

Abstract
Mast cells are implicated in the innate proinflammatory immune defence against bacterial insult, but the mechanisms through which mast cells respond to bacterial encounter are poorly defined. Here, we addressed this issue and show that mast cells respond vividly to wild type Streptococcus equi by up‐regulating a panel of proinflammatory genes and by secreting proinflammatory cytokines. However, this response was completely abrogated when the bacteria lacked expression of sagA, whereas the lack of a range of other potential virulence genes (seeH, seeI, seeL, seeM, hasA, seM, aroB, pyrC, and recA) had no effect on the amplitude of the mast cell responses. The sagA gene encodes streptolysin S, a lytic toxin, and we next showed that the wild type strain but not a sagA‐deficient mutant induced lysis of mast cells. To investigate whether host cell membrane perturbation per se could play a role in the activation of the proinflammatory response, we evaluated the effects of detergent‐ and pneumolysin‐dependent lysis on mast cells. Indeed, exposure of mast cells to sublytic concentrations of all these agents resulted in cytokine responses of similar amplitudes as those caused by wild type streptococci. This suggests that sublytic membrane perturbation is sufficient to trigger full‐blown proinflammatory signalling in mast cells. Subsequent analysis showed that the p38 and Erk1/2 signalling pathways had central roles in the proinflammatory response of mast cells challenged by either sagA‐expressing streptococci or detergent. Altogether, these findings suggest that sagA‐dependent mast cell membrane perturbation is a mechanism capable of activating the innate immune response upon bacterial challenge.
Keywords: mast cells, streptococci, toxins
Abbreviations
- BMMC
bone marrow‐derived mast cell
- IL
interleukin
- MCP
monocyte chemoattractant protein
- PCMC
peritoneal cell‐derived mast cell
- PI
propidium iodide
- SLS
streptolysin S
- TLR
toll‐like receptor
- TNF
tumour necrosis factor
- WT
wild type
1. INTRODUCTION
When pathogens encounter the host, they face physical barriers of the innate immune system, for example, skin or mucosae, which also contain a large array of cell types that sense and react to the foreign invader (Yatim & Lakkis, 2015). Mast cells, derived from the haematopoietic lineage, are strategically localised at sites close to the exterior and have therefore been strongly implicated in the first line host defence against infection by bacterial and other pathogens (Dawicki & Marshall, 2007; Marshall, 2004). A main characteristic of mast cells is their high content of secretory granules, filled with a plethora of preformed inflammatory mediators, including proteases, chemokines, cytokines, proteoglycans, and histamine (Wernersson & Pejler, 2014). In addition to these stored components, which can be released into the environment in minutes, mast cells can de novo synthesise inter alia cytokines, prostaglandins, and growth factors to communicate and interact with their environment and other immune cells (Wernersson & Pejler, 2014).
Mast cells are classically known for their crucial role in IgE‐mediated allergic responses, triggered by antigen crosslinking of IgE molecules bound to their high affinity receptor, FcεRI. However, it has also been demonstrated that mast cells express a panel of pattern recognition receptors such as toll‐like receptors (TLRs), which has led to the notion that mast cells could also directly respond bacterial infection (reviewed in Abraham & St John, 2010; Johnzon, Rönnberg, & Pejler, 2016; Marshall, 2004). Indeed, animals lacking mast cells or individual mast cell components have been shown to respond differentially to bacterial infection in comparison with wild type (WT) controls (Chan, St John, & Abraham, 2013; Dahdah et al., 2014; Di Nardo, Yamasaki, Dorschner, Lai, & Gallo, 2008; Echtenacher, Mannel, & Hultner, 1996; Malaviya, Ikeda, Ross, & Abraham, 1996; Piliponsky et al., 2010; Piliponsky et al., 2012; Thakurdas et al., 2007; van den Boogaard et al., 2014). A role for mast cells in regulating the host defence against bacterial infection has therefore emerged (Abraham & St John, 2010; Johnzon, Rönnberg, & Pejler, 2016; Marshall, 2004).
Intriguingly, previous studies have revealed that mast cells can respond in fundamentally different ways to bacterial challenge, depending on the particular bacterial species and even strains thereof (Johnzon, Rönnberg, & Pejler, 2016). In fact, whereas most studies suggest a beneficial role of mast cells, there are other studies pointing to a detrimental (or even dispensable) role of mast cells under different settings of bacterial challenge (reviewed in Johnzon, Rönnberg, & Pejler, 2016). In line with this notion, the direct response of mast cells varies substantially depending on the nature of the bacterium. For example, mast cells degranulate when they encounter Escherichia coli (E. coli) (Malaviya, Ross, Jakschik, & Abraham, 1994) or Staphylococcus aureus (Abel et al., 2011) but not in response to Mycobacterium tuberculosis (Garcia‐Rodriguez, Goenka, Alonso‐Rasgado, Hernandez‐Pando, & Bulfone‐Paus, 2017) or Streptococcus equi (Rönnberg, Guss, & Pejler, 2010). It has also been demonstrated that, whereas Streptococcus faecium and E. coli are phagocytosed by mast cells (Arock et al., 1998), Listeria monocytogenes is not (Dietrich et al., 2010). Further, Sutherland et al. demonstrated that mast cells induce killing of intracellular Klebsiella pneumoniae (Sutherland, Olsen, McKinstry, Villalta, & Wolters, 2008). Finally, there are indications that mast cells can produce extracellular traps as a means of combating inter alia bacterial pathogens such as Streptococcus pyogenes (von Kockritz‐Blickwede et al., 2008) or L. monocytogenes (Campillo‐Navarro et al., 2017).
Altogether, from previous research efforts, a complex picture emerges in which mast cells can respond in multiple ways to bacterial infection and differentially impact on the host responses depending on the nature of the bacterial insult and the particular setting. However, several central issues remain to be fully addressed. One important, and partly unanswered, question regards the mechanism through which mast cells are activated by bacterial encounter. Considering that mast cells express various pattern recognition receptors, a likely scenario would be that engagement of these by classical cell wall‐derived pathogen‐associated molecular patterns (PAMPs) trigger proinflammatory signalling. Indeed, several studies have indicated that mast cells can be activated through such mechanisms (Marshall, 2004). However, we showed previously that, whereas primary mast cells respond vividly to live group C streptococci, they were completely refractory to heat‐inactivated bacteria in which cell wall‐associated PAMPs are intact (Rönnberg et al., 2010). Moreover, whereas mast cells responded to live S. aureus by robust vascular endothelial growth factor release, they were unresponsive to stimulation by peptidoglycan, lipopolysaccharide, Pam3CSK4, and lipoteichoic acid (Johnzon, Ronnberg, Guss, & Pejler, 2016). It has also been demonstrated that the activation of human mast cells by E. coli is independent of TLR2 and TLR4 (Kramer et al., 2008). A generic mechanism for bacterial activation of proinflammatory responses in mast cells has therefore remained elusive. Here, we addressed this issue with the specific aim of identifying the bacterial component(s) of group C streptococci that activate proinflammatory responses in mast cells. To this end, we challenged primary mast cells with live Streptococcus equi subspecies equi (S. equi) or with S. equi mutants lacking a panel of potential virulence genes, followed by an evaluation of downstream proinflammatory responses. Our findings reveal that sagA (encoding streptolysin S [SLS])‐mediated perturbation of mast cell membranes represents the major mechanism of mast cell activation in response to the bacterial challenge. Strikingly, mast cells responded with equal amplitude to low concentrations of pore‐forming detergents or pneumolysin, thus demonstrating that sublytic membrane perturbation per se is a generic trigger of proinflammatory signalling in mast cells.
2. RESULTS
2.1. Mast cell activation in response to S. equi mutants lacking multiple virulence genes
To search for potential virulence factors involved in mast cell activation in response to group C streptococci, we developed mouse bone marrow‐derived mast cells (BMMCs) and cocultured these with live S. equi. In the absence of bacteria, no detectable release of proinflammatory interleukin 6 (IL‐6), tumour necrosis factor (TNF), or monocyte chemoattractant protein 1 (MCP‐1) from mast cells was detected. However, upon challenge with live WT S. equi, a robust release of all of these cytokines/chemokines was observed (Figure 1a–c), this being in concordance with our previous observations (Rönnberg et al., 2010). In further agreement, challenge with live bacteria also induced expression of the genes coding for IL‐6, TNF, and Nr4a3 (Figure 1d–f), the latter representing a nuclear receptor that was previously shown to be strongly induced by various mast cell activators (Lundequist, Calounova, Wensman, Ronnberg, & Pejler, 2011; Rönnberg et al., 2010). To start identifying the bacterial factor(s) responsible for the strong mast cell response, we cocultured the BMMCs with two S. equi mutant strains, each lacking a panel of potential virulence factors (Table 1). In the HILM strain, genes encoding four bacterial superantigens (seeH, seeI, seeL, and seeM) are lacking, whereas the strain denoted Multi lacks hasA, seM, pyrC, sagA, and aroB. After incubation of the BMMCs with the HILM strain, mast cell responses were indistinguishable from those seen after incubation with WT bacteria (Figure 1a–f), thus demonstrating that mast cell activation was independent of the bacterial superantigens. In contrast, the mast cell responses were completely abolished when BMMCs were challenged with the Multi strain, indicating that mast cell activation was due to the effects of either (or a combination) of the genes lacking in this strain (Figure 1a–f).
Figure 1.
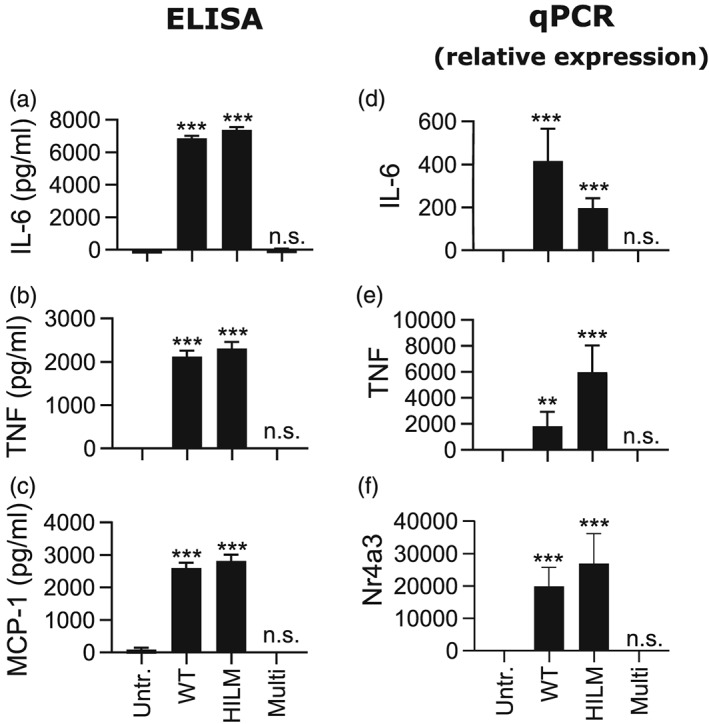
Activation of mast cells by wild type and multiple‐gene‐mutant Streptococcus equi strains. 1 × 106 BMMCs/ml were cultured either alone (untreated; Untr.) or in the presence of wild type (4047; WT) S. equi or S. equi mutant strains lacking multiple genes (seeH/seeI/seeL/seeM [HILM] or hasA/sagA/seM/aroB/pyrC [Multi]; multiplicity of infection = 10). (a–c) After 4 hr, conditioned media were harvested and analysed for IL‐6, TNF, and MCP‐1 by ELISA. (d–f) Cells were recovered after 4‐hr incubation of mast cells with bacteria and were analysed by qPCR for expression of the IL6, TNF, and Nr4a3 genes. Expression relative to the housekeeping gene (HPRT) is presented. Data are given as mean values ± standard error of the mean, pooled from four biological replicates; ** p ≤ .01, *** p ≤ .001; n.s., not significant
Table 1.
Wild type and mutant strains of Streptococcus equi used in this study
| Designation | Mutated genes | Description |
|---|---|---|
| S. equi 4047 | Wild type S. equi 4047 | |
| HILM | seeH, seeI, seeL, and seeM | Lacks genes encoding four bacterial superantigens |
| Multi | hasA, seM, pyrC, sagA, and aroB | Lacks five genes: hasA, seM, pyrC, sagA, and aroB |
| ΔhasA | hasA | Lacks hasA: encoding hyaluronan synthase |
| ΔseM | seM | Lacks seM: encoding an antiphagocytic M‐protein |
| ΔpyrC | pyrC | Lacks pyrC: encoding a dihydroorotase involved in DNA biosynthesis |
| ΔsagA | sagA | Lacks sagA: encoding a haemolytic toxin; streptolysin S |
| ΔaroB | aroB | Lacks aroB: encoding a 3‐dehydroquinate synthase involved in aromatic amino acid biosynthesis |
| ΔrecA | recA | Lacks recA: encoding a recombinase involved in homologous recombination of DNA |
2.2. sagA is the single mast cell‐activating virulence gene of S. equi
To gain a deeper understanding of which gene(s) of S. equi are responsible for mast cell activation, we next cocultured BMMCs with single gene knockout strains deficient in individual genes included in the Multi knockout strain. We also evaluated a strain lacking recA. As seen in Figure 2a–f, the strains lacking seM, recA, pyrC, and aroB were all capable of inducing a robust cytokine response and triggering of inflammatory gene expression in mast cells, although the amplitude of activation showed some variability between the knockout strains. By contrast, the mutant strain lacking sagA (Molloy, Cotter, Hill, Mitchell, & Ross, 2011) was completely unable to induce any detectable response in mast cells (Figure 2a–f), suggesting that the effects of sagA alone are responsible for the strong proinflammatory response of mast cells challenged with the live bacteria. To assess whether the blunted response to sagA‐deficient bacteria was related to differential bacterial growth rate, we followed the growth rate of WT versus ΔsagA mutant S. equi. However, the growth characteristics of these strains were similar in the medium used for the bacteria : mast cell cocultures, arguing against such a scenario (Figure S1A). Further, both strains showed equal growth and morphology in the presence of mast cells (Figure S1B,C). A time course experiment showed that the release of IL‐6, TNF, and MCP‐1 was first detected 4 hr after initiation of the coculture of BMMCs with S. equi, with a further increase in cytokine output at 24 hr (Figure 2g–i). In agreement with the data above, secretion of IL‐6, TNF, or MCP‐1 was undetectable at all time points following coculture of BMMCs with ΔsagA mutant bacteria (Figure 2g–i). Together, these findings demonstrate that sagA is the main bacterial virulence gene capable of activating mast cells.
Figure 2.
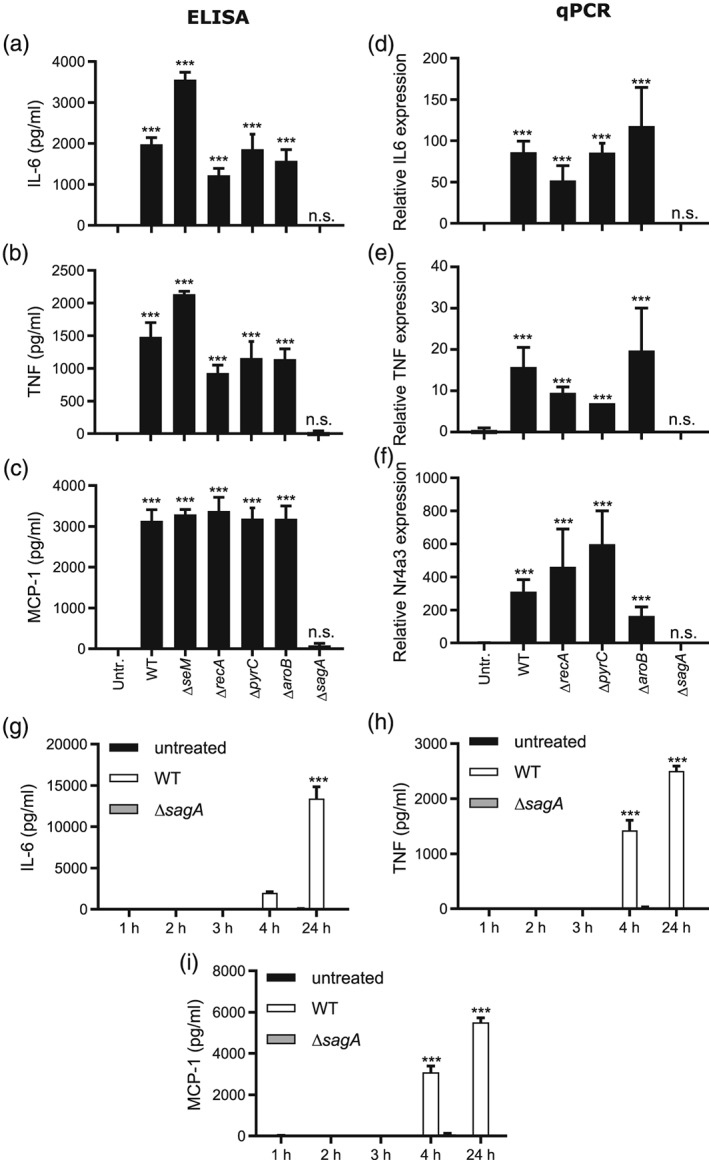
Streptococcus equi mutants lacking sagA fail to elicit a mast cell response. (a–c) 1 × 106 BMMCs/ml were cultured either alone (Untr.) or in the presence of WT S. equi (4047; WT) or knockout strains lacking seM, recA, pyrC, aroB, or sagA as indicated (multiplicity of infection = 10). After 4 hr, conditioned media were harvested and analysed for IL‐6, TNF, and MCP‐1 by ELISA. (d–f) Cells were recovered after 4‐hr incubation of mast cells with bacteria and were analysed by qPCR for expression of the IL6, TNF, and Nr4a3 genes. Expression relative to the housekeeping gene (HPRT) is presented. (g–i) Time course experiment showing the release of IL‐6, TNF, and MCP‐1, measured by ELISA. Data are given as mean values ± standard error of the mean, pooled from at least 4 biological replicates (1‐3 hr, n = 5; 4 hr, n = 6; 24 hr, n = 10). Two‐factor analysis of variance was performed with Tukey's post hoc test. *** p ≤ .001; n.s., not significant
The sagA (streptolysin associated gene cluster A) gene encodes SLS, a lytic toxin. To confirm that WT S. equi has lytic capacity and that this is absent in the sagA mutants, we performed experiments with blood agar. As seen in Figure S2, WT S. equi indeed displayed strong haemolytic activity, which was completely abrogated by the sagA mutation. Further, all other mutants tested, that is, those except the sagA and Multi mutants, displayed lytic activity that was indistinguishable from that of WT bacteria. Hence, the haemolytic capacity of S. equi is dependent on sagA.
2.3. sagA is the main factor causing activation of peritoneal cell‐derived mast cells
BMMCs represent mature primary mast cell populations with features that are in most parts compatible with those seen in in vivo‐derived mast cells and are therefore commonly used to study mast cell function. In addition, mast cells can be derived from the expansion of the peritoneal mast cell population, that is, peritoneal cell‐derived mast cells (PCMCs). Similar to BMMCs, PCMCs represent a mature MC population, but with a somewhat altered morphology and phenotype as regards granule composition (Figure 3a,b; Malbec et al., 2007). To study if the mast cell‐activating mechanism(s) seen in BMMCs is also operative in PCMCs, we cocultured PCMCs with either WT or ΔsagA mutant bacteria, followed by the assessment of mast cell responses. As seen in Figure 3c–e, PCMCs, similar to BMMCs, responded to WT S. equi by a robust release of IL‐6 and TNF, whereas the release of these cytokines was undetectable if the bacteria lacked sagA expression. PCMCs also released low amounts of MCP‐1 in response to WT S. equi, and this release was blunted if the bacteria lacked sagA expression. At the mRNA level, WT bacteria induced the expression of the IL‐6 and Nr4a3 genes, but these responses were not induced if the bacteria lacked sagA expression (Figure 3f,h). However, despite the release of TNF protein, no detectable induction of expression of the TNF gene could be observed after coculture of PCMCs with WT bacteria (Figure 3g). Possibly, this finding could reflect that the encounter of PCMCs with bacteria causes release of TNF from preformed stores within the mast cell granules (Gordon & Galli, 1990), without triggering TNF gene expression.
Figure 3.
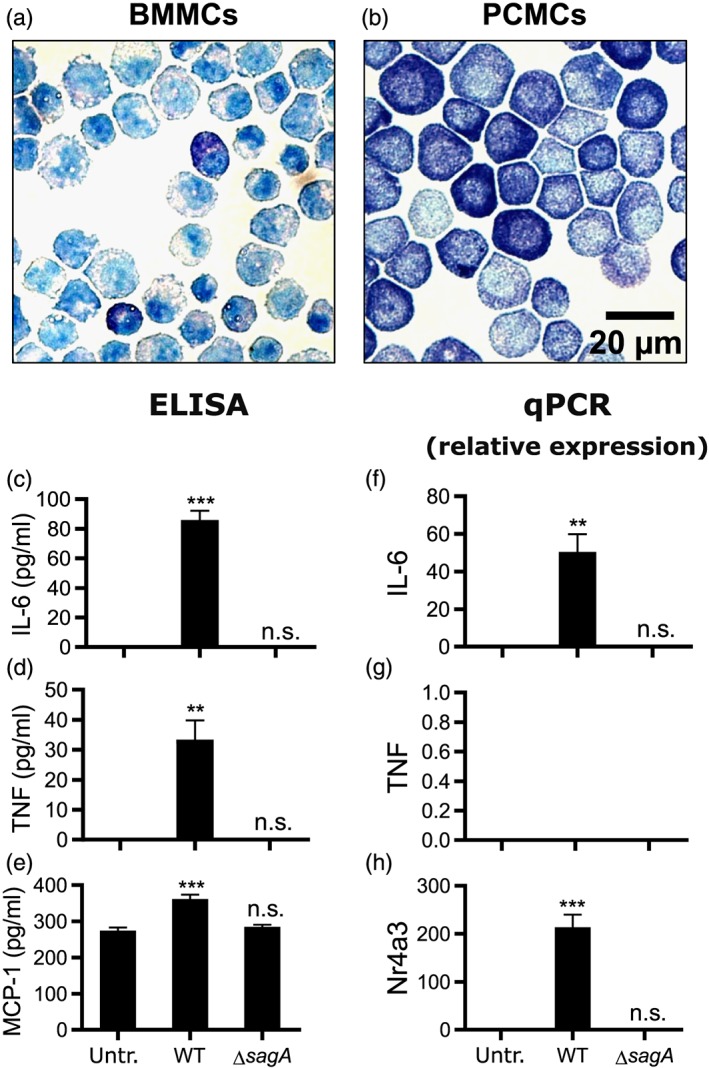
The sagA gene is required to elicit a proinflammatory response in peritoneal cell‐derived mast cells (PCMCs). (a–b) BMMCs (a) and PCMCs (b) depicted after staining with Toluidine blue. (c–e) 1 × 106 PCMCs/ml were cultured either alone (Untr.) or in the presence of WT (4047) or ΔsagA mutant Streptococcus equi (multiplicity of infection = 10). After 4 hr, conditioned media were harvested and analysed for IL‐6, TNF, and MCP‐1 by ELISA. (f–h) Cells were recovered after 4‐hr incubation of mast cells with bacteria and were analysed by qPCR for expression of the IL6, TNF, and Nr4a3 genes. Expression relative to the housekeeping gene (HPRT) is presented. Data are given as mean values ± standard error of the mean, pooled from three biological replicates. ** p ≤ .01, *** p ≤ .001; n.s., not significant
2.4. sagA accounts for mast cell lysis but not degranulation, and mast cell activation is not triggered by components released during mast cell lysis
Since the sagA gene codes for the precursor of the cytolytic toxin SLS (Molloy et al., 2011), we hypothesised that WT, but not ΔsagA mutant bacteria, could cause lysis of mast cells. Indeed, WT S. equi lysed mast cells as evaluated by lactate dehydrogenase (LDH) release, with partial lysis seen at 4 hr and with the majority of the cells being lysed after 24‐hr coculture (Figure 4a). In contrast, ΔsagA mutant bacteria did not cause any detectable increase in the lysis of mast cells in comparison with cells cultured without bacteria (Figure 4a). Knowing that sagA accounts for lysis of mast cells, we next hypothesised that products released from lysed mast cells, such as alarmins, could serve as triggers to activate nonlysed surrounding mast cells to secrete proinflammatory mediators. To address this possibility, we first evaluated whether incubation of mast cells with bacteria caused the release of the alarmins IL‐33 or HMGB1. However, we were not able to detect any of these alarmins in the culture supernatant after incubation of mast cells with WT S. equi (Figure S3A,B). We also assessed supernatants from activated mast cells for IL‐1β but did not see elevated release of this cytokine in response to live bacteria (Figure S3C). Hence, mast cell activation by live S. equi does not cause profound inflammasome activation. To further address whether mast cell activation is triggered by components released from lysed mast cells, we lysed mast cells by repeated freeze–thaw cycles, incubated the lysates together with viable mast cells, and then measured IL‐6 release. However, we were not able to detect IL‐6 release from intact mast cells in response to the mast cell lysates, arguing against also this scenario (Figure S3D). To ask whether mast cell‐activating components are released during the bacteria : mast cell coculture conditions, we incubated bacteria together with mast cells for 4 and 24 hr, sterile filtered the supernatants, and incubated mast cells for 24 hr with these. However, no additional IL‐6 besides that present at baseline in the conditioned supernatants could be detected after the incubation (Figure S3E). We also incubated mast cells with supernatants from the bacterial cultures. As seen in Figure 4b, bacterial supernatants were not able to trigger any detectable IL‐6 release from the mast cells. This suggests that cell–cell contact is needed for mast cell activation mediated by bacteria‐derived SLS.
Figure 4.
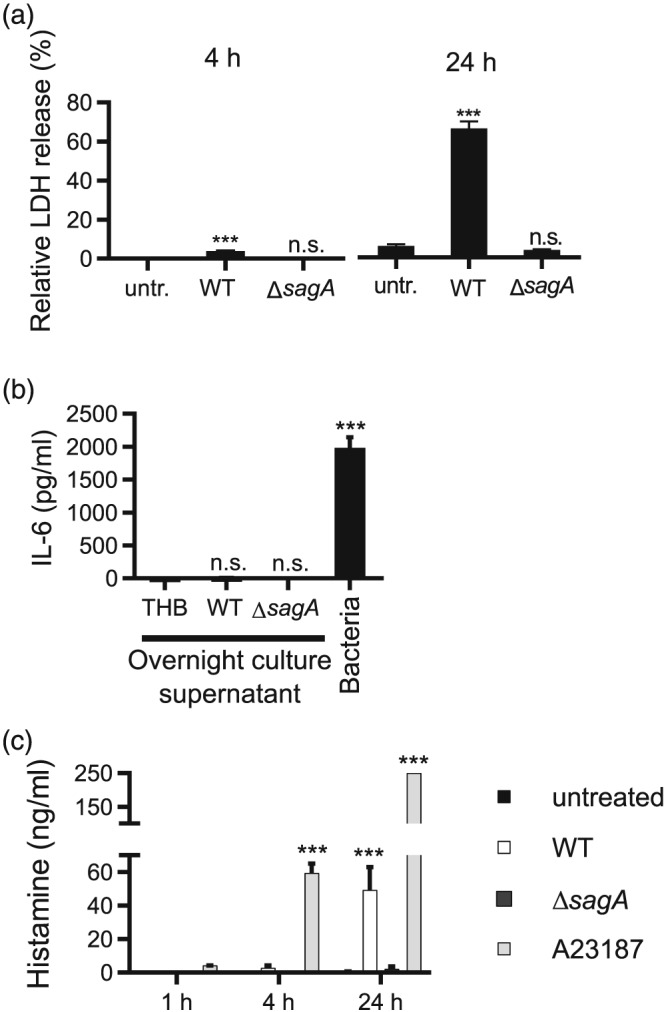
sagA causes lysis of mast cells, cell–cell contact dependent activation of mast cells, and limited release of histamine. (a) Mast cells (1 × 106 BMMCs/ml) were cultured either alone (untr.) or in the presence of WT (4047) or ΔsagA mutant bacteria (multiplicity of infection = 10). Conditioned media were collected at 4 and 24 hr and were assayed for % release of lactate dehydrogenase (LDH). LDH activity was normalised to total LDH in lysed BMMCs. (b) Conditioned media (12 μl) from overnight cultures of Streptococcus equi in Todd Hewitt Broth (centrifuged 10 min, 4,000× g) or control Todd Hewitt Broth medium was added to 1 × 106 BMMCs (in 1 ml). After 4 hr, culture supernatants were harvested and analysed for IL‐6 by ELISA. (c) Mast cells (1 × 106 BMMCs/ml) were cultured either alone (untreated), treated with 1‐μM calcium ionophore A23187, or in the presence of WT (4047) or ΔsagA mutant S. equi (multiplicity of infection = 10). At the time points indicated, conditioned media were harvested and analysed for histamine by ELISA. Data are given as mean values ± standard error of the mean, pooled from at least three biological replicates. Two‐factor analysis of variance was performed with Tukey's post hoc test. *** p ≤ .001; n.s., not significant
To address whether encounter of mast cells with the live bacteria causes mast cell degranulation, we measured the levels of histamine in the culture supernatants taken after incubation of mast cells with S. equi. As a positive control, we used the calcium ionophore A23187. As shown in Figure 4c, only slow release of histamine was seen in response to live bacteria, and the total amount of histamine release was relatively low. In contrast, calcium ionophore, as expected, triggered a rapid and more profound histamine release. This indicates that the live bacteria do not induce substantial mast cell degranulation.
Altogether, our findings indicate that mast cell activation co‐occurs with cellular lysis but is not triggered by mast cell products released during lysis or degranulation.
2.5. Mast cell activation precedes cellular lysis
To achieve a more precise view of the interaction between mast cells and streptococci, we utilised live microscopy using propidium iodide (PI) to stain lysed mast cells. As seen in Figure 5a and Videos S1 and S2, incubation of BMMCs with WT bacteria resulted in strong PI positivity, indicating cell lysis and cell death. By contrast, and in agreement with the LDH release assay, the ΔsagA mutant bacteria did not cause detectable lysis of the mast cells (Figure 5a; Video S3). Notably though, the incubation of mast cells with ΔsagA mutant bacteria caused aggregation of the mast cells (Figure 5a, upper panels; Video S3). Mast cell aggregation was also seen in cocultures of mast cells with WT bacteria, although to a lesser extent (Figure 5a, upper panels; Video S2). Conceivably, the mast cell aggregation seen could be an effect of interactions between bacteria and mast cells, that is, that the bacteria interact persistently with mast cells and thereby cause mast cell clumping. To address this possibility, we performed high resolution live imaging analysis of the mast cell : bacterial cocultures. However, although transient contacts between mast cells and bacteria could be seen; no signs of persistent contacts were observed (Figure S1C and Video S4).
Figure 5.
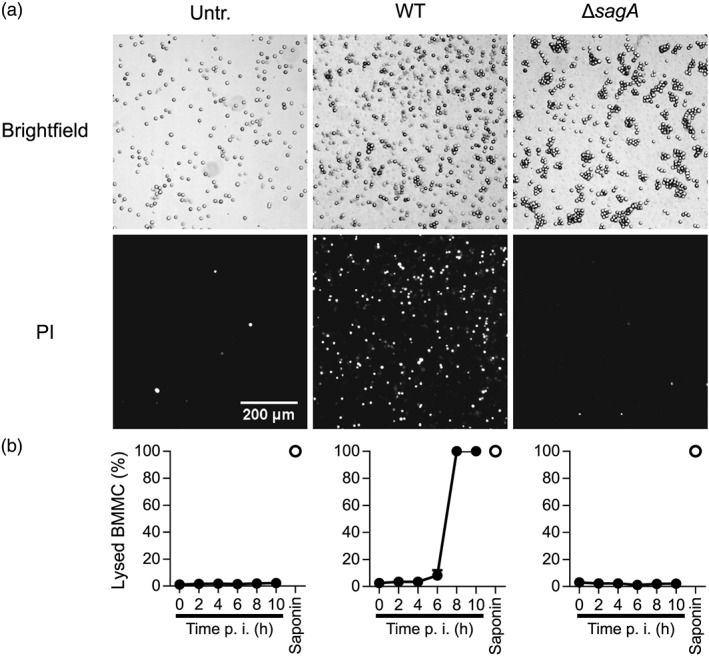
Live imaging kinetic analysis of mast cell lysis in response WT or ΔsagA mutant Streptococcus equi. Mast cells (1 × 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. After 1 hr, cells were infected with WT (4047) S. equi or ΔsagA mutant S. equi (multiplicity of infection = 10); BMMCs without bacteria (Untr.) served as control. Every 5 min, images of every well were taken. At least 150 cells were present in each image. (a) Representative brightfield and PI images, 7‐hr postinfection. (b) Percent PI+ (or previously PI+ and subsequently desintegrated) cells over the time course postinfection (p.i.), counted automatically with manual verification. The percent lysis did not change after 10 hr. As a positive control for complete lysis, saponin (final concentration 0.5%) was added 22‐hr postinfection, and images were generated after 10 min. Data are given as mean values ± standard error of the mean, of four fields of view representative of four independent experiments (error bars for most time points are hidden by the symbols)
By using the live imaging approach, we were also able to quantify the extent of PI positivity over time, thus enabling a kinetic assessment of mast cell lysis. As seen in Figure 5b and Videos S2 and S3, no detectable mast cell lysis was seen up to 4 hr of coculture of mast cells with WT S. equi, and only limited cell lysis was seen at 6‐hr incubation. Thereafter, rapid cell lysis occurred with complete cell lysis noted at 8 hr of incubation (saponin was used as positive control for complete lysis). Again, no lysis was seen after coculture of mast cells with ΔsagA mutant bacteria. Importantly, these findings show that the onset of mast cell lysis (8 hr and onwards) occurs substantially later than the onset of proinflammatory events, as detected by cytokine release (see Figure 2g–i). Hence, mast cell activation is an event that precedes mast cell lysis.
2.6. Sublytic membrane perturbation mimics the effect of sagA
The results above indicate that bacterial sagA causes lytic cell death in mast cells and that the effects of sagA are sufficient to trigger full mast cell activation in terms of inflammatory gene expression. We next asked whether the membrane perturbation in itself could constitute a trigger for mast cell activation. To address this possibility, we first incubated mast cells with increasing concentrations of saponin, followed by measurement of cell lysis and release of IL‐6. Saponin is a gentle detergent that, similar to SLS, is known to introduce small pores in a target membrane (Seeman, 1967). As depicted in Figure 6a, saponin concentrations up to 0.015% did not cause any detectable cell lysis, whereas 0.018–0.025% saponin resulted in partial lysis. Saponin concentrations of 0.03% and above caused complete mast cell lysis. Intriguingly, incubation of BMMCs with low concentrations of saponin induced expression of IL‐6, at levels similar to those seen after incubation of mast cells with live WT S. equi (see Figure 1). It is also notable that IL‐6 release was seen at saponin concentrations starting at 0.012–0.015%, that is, at clearly sublytic concentrations, whereas the cytokine response was abrogated at higher (lytic) concentrations of saponin. Low concentrations of saponin also triggered proinflammatory gene expression in mast cells (Figure 6b–d). These findings suggest that mast cell activation by S. equi SLS can be fully phenocopied by low concentrations of the membrane‐integrating detergent saponin. This reveals a general mechanism of mast cell activation through subtle perturbation of the plasma membrane.
Figure 6.
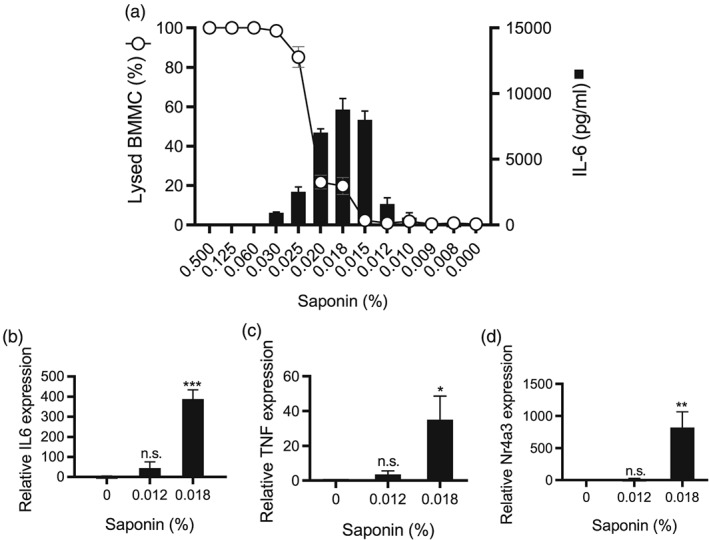
Sublytic concentrations of saponin activate mast cells. Mast cells (1 × 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. Saponin was added to the final concentrations indicated. After 15 min, PI+ (lysed) cells as percentage of the total number of cells was quantified. Mean values ± standard error of the mean of four fields of view are shown, representative of four independent experiments. Conditioned media were analysed in parallel for IL‐6 by ELISA. Mean values ± standard error of the mean are shown for three to four biological replicates. (b–d) After incubation of mast cells with the indicated concentrations of saponin, cells were analysed by qPCR for expression of the IL‐6, TNF, and Nr4a3 genes, relative to housekeeping gene (HPRT). Data are given as mean values ± standard error of the mean, based on three to four biological replicate experiments; * p ≤ .05, ** p ≤ .01, *** p ≤ .001; n.s., not significant
To ask whether lytic mechanisms beyond those represented by sagA and saponin cause similar mast cell activation responses, we also evaluated the detergent digitonin and pneumolysin, the latter a lytic toxin expressed by Streptococcus pneumoniae. As shown in Figure S4A,B, digitonin caused a profound activation of mast cells in terms of IL‐6 secretion. Similar to saponin and sagA, mast cell activation was seen at digitonin concentrations below those causing overt mast cell lysis. Pneumolysin also caused profound mast cell activation, as measured by IL‐6 secretion. Similar to digitonin, saponin, and sagA, effects on IL‐6 secretion were seen at pneumolysin concentrations below those causing substantial mast cell lysis (Figure S4C,D). Altogether, these findings suggest that multiple lytic agents/mechanisms have strong activating effects on mast cells. Further, our data indicate that maximal mast cell activation in response to all of these agents/mechanisms is seen at sublytic stages.
2.7. Mast cell activation by sagA and saponin is dependent on the MAP kinases p38 and Erk1/2 but not on JNK
Next, we aimed to identify signalling pathways operative in sagA/saponin‐triggered proinflammatory signalling in mast cells. MAP kinase signalling has been implicated in responses downstream of bacterial encounter (McGuire & Arthur, 2015). Therefore, we hypothesised that sagA‐induced mast cell activation could be MAP kinase dependent. To address this, we incubated mast cells with inhibitors of Erk1/2, p38, and c‐JunNH2‐terminal kinase (JNK) and then challenged the mast cells with either live bacteria or sublytic concentrations of saponin. Mast cell activation was monitored by measuring the secretion of IL‐6. As depicted in Figure 7a,b, inhibition of the p38 or Erk1/2 pathways almost completely abolished the IL‐6 release in response to live bacteria and caused a significant reduction in IL‐6 release in response to saponin exposure. By contrast, JNK inhibition had no significant effect. As a control, neither of these inhibitors caused IL‐6 release in mast cells cultured in the absence of bacteria/saponin (Figure 7c). Hence, these findings indicate that the p38 and Erk1/2 pathways play crucial roles in SLS/saponin‐dependent activation of mast cells. To verify that these pathways are activated after bacterial challenge, we performed western blot analysis to monitor the phosphorylation status of the respective signalling molecules. Indeed, stimulation of mast cells with WT but not ΔsagA mutant S. equi induced phosphorylation of p38 and Erk1/2, and this was blocked by inhibitors of the respective signalling molecules (Figure 7d,e).
Figure 7.
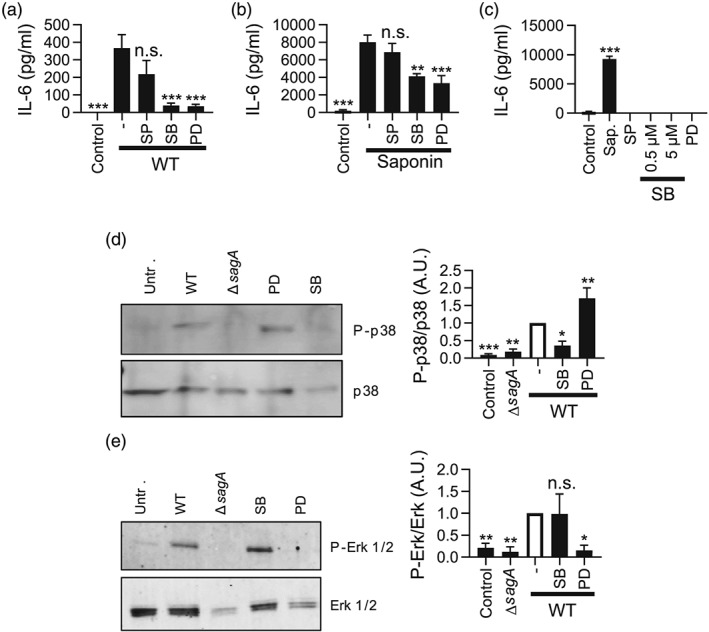
Mast cell responses to Streptococcus equi are dependent on the p38 and Erk1/2 MAP kinases. (a) Mast cells (1 × 106 BMMCs/ml) were cultured alone or with WT (4047) S. equi (multiplicity of infection = 10), either in the absence or presence of MAP kinase inhibitors: 1‐μM SP600125 (SP), 5‐μM SB203580 (SB), or 50‐μM PD98059 (PD) for inhibition of the JNK, p38, or Erk1/2 pathways, respectively. After 4 hr, conditioned media were harvested and analysed for IL‐6 by ELISA. (b) IL‐6 release in response to saponin (0.018% final concentration) ± MAP kinase inhibitors (as in (a)). (c) IL‐6 release in response to MAP kinase inhibitors alone, with 0.018% saponin as positive control. Data are given as mean values ± standard error of the mean, based on at least three biological replicate experiments. (d, e) Mast cells (1 × 106 BMMCs/ml) were cultured with either WT (4047) or ΔsagA mutant bacteria (multiplicity of infection = 10); or WT bacteria ± MAP kinase inhibitors as in (a). Cell pellets were recovered and evaluated by western blot analysis for phosphorylation of p38 (P‐p38) and Erk1/2 (P‐Erk‐1/2), with nonphosphorylated proteins as controls. Quantification of the western blot data is shown to the right, based on three to five independent experiments. * p ≤ .05, ** p ≤ .01, *** p ≤ .001; n.s., not significant
3. DISCUSSION
In previous studies, we have shown that mast cells mount a powerful proinflammatory response after encountering S. equi. After stimulation with S. equi, mast cells were shown to up‐regulate a large panel of proinflammatory genes, many of which were up‐regulated several thousand‐fold, hence revealing that mast cells are remarkably sensitive to these types of bacterial pathogens (Rönnberg et al., 2010). However, the bacterial virulence factor(s) responsible for mast cell activation in this setting has not been identified prior to this investigation. Here, we addressed this issue and identify sagA (encoding SLS) as the main virulence gene of S. equi causing activation of proinflammatory responses in mast cells. Notably, in the absence of sagA/SLS, the mast cell responses were completely abrogated, suggesting that other candidate mast cell‐activating compounds contribute minimally to the proinflammatory effects of S. equi. Such candidate mast cell‐activating factors include cell wall compounds, for example, peptidoglycan and lipoteichoic acids, which are typically resistant to heat inactivation. However, although we earlier noted a modest impact of TLR2 or TLR4 on the mast cell response to live bacteria, mast cells were completely refractory to stimulation by heat‐inactivated S. equi (Rönnberg et al., 2010). In combination with the present data, this suggests that classical cell wall‐associated PAMPs have minimal roles in activating mast cells in response to this group of bacteria.
The sagA gene codes for the precursor of the cytolytic toxin SLS and our data thus suggest that mast cell lysis is triggered by sagA‐encoded SLS. The mechanism by which sagA causes mast cell activation is however intriguing. Initially, we hypothesised that lysis of mast cells could result in the release of alarmins such as IL‐33 and HMGB1 that are capable of activating surrounding viable mast cells (Tung, Plunkett, Huang, & Zhou, 2014). However, we were not able to detect the release of alarmins after coculture of mast cells with WT S. equi. Moreover, we did not see any mast cell‐activating effect of lysed mast cells (containing a large spectrum of compounds with potential alarmin activities). We therefore regard this possibility as unlikely. Another possibility would be that sagA‐dependent cell membrane perturbation per se has proinflammatory effects on mast cells. Indeed, we showed that exposure of mast cells to lytic detergents (i.e., saponin or digitonin) mirrors the effects of WT S. equi exposure. This suggests that interference with the mast cell membrane itself results in mast cell activation.
An intriguing finding was that mast cell activation by S. equi, as assessed by induced expression of proinflammatory genes and secretion of proinflammatory cytokines, occurred several hours before the time point when cell lysis was first detected. Furthermore, we noted that the activation of mast cells by saponin or digitonin occurred at concentrations that were clearly below those required to cause overt mast cell lysis, that is, sublytic concentrations. These findings indicate that mast cell activation is triggered at the stage before actual lysis occurs, that is, at a sublytic stage. At present, we cannot with certainty explain how this response is initiated. However, it has been hypothesised that bacterial lysins from sources other than S. equi could potentially trigger cellular responses by causing osmotic stress (Carr, Sledjeski, Podbielski, Boyle, & Kreikemeyer, 2001; Flaherty, Puricelli, Higashi, Park, & Lee, 2015). The proinflammatory effects of sagA‐encoded SLS on mast cells could thus potentially be explained by such a scenario (Figure 8). It is also notable that mast cell activation occurred without profound degranulation, as indicated by only limited histamine release. Hence, the signalling mechanisms triggered by membrane perturbation do not appear to crosstalk with pathways leading to mast degranulation. Intriguingly, it has been shown that, in contrast to SLS, the cholesterol‐dependent cytolysins streptolysin O from S. pyogenes and pneumolysin can cause mast cell degranulation (Fritscher et al., 2018; Stassen et al., 2003), hence suggesting that the lytic toxins expressed by these three streptococcal species have differential activating impact on mast cells depending on the class of toxin. This is further supported by the observation of mast cell activation by another cholesterol‐dependent cytolysin, listeriolysin O from L. monocytogenes (Gekara et al., 2007).
Figure 8.
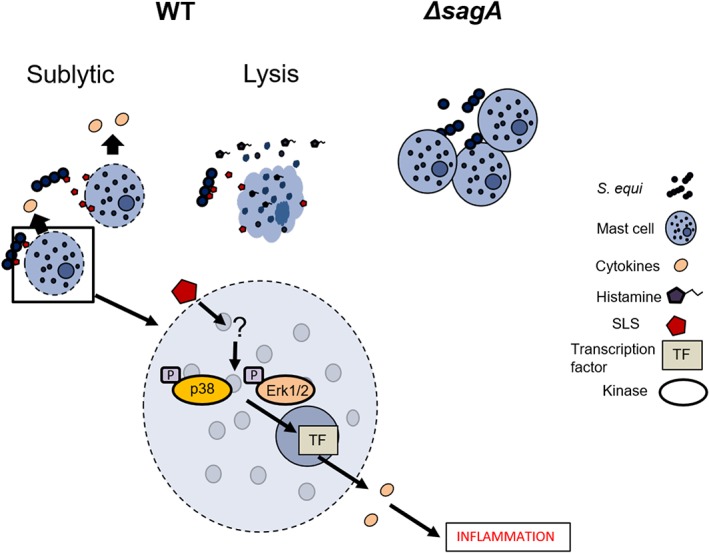
Model depicting how sagA can activate proinflammatory responses in mast cells
In a previous study, we showed that the responses of mast cells to live S. equi required cell–cell contact, as judged by transwell experiments (Rönnberg et al., 2010). In line with this, we show here that neither conditioned medium from S. equi cultures nor conditioned media from mast cell: S. equi cocultures had the ability to cause mast cell activation. This reinforces that mast cells need to be in physical contact with S. equi to mount a proinflammatory response, which is consistent with previous studies revealing that SLS‐like toxins are only active in the context of presentation on the bacterial surface or on carrier structures (Molloy et al., 2011). In further support, we showed by live imaging that mast cells aggregate when cocultured with S. equi. Most likely, the mechanism of mast cell activation thus involves a first aggregation step, followed by lysis caused by SLS. It was also notable that the aggregation was more pronounced when mast cells were cocultured with ΔsagA mutant versus WT bacteria. A plausible explanation for these findings could be that the aggregation is resolved after SLS‐dependent mast cell lysis and thus that the aggregation of mast cells will persist if sagA is absent. Further, this scenario implies that sagA‐encoded SLS is anchored to the cell wall or, alternatively, that SLS released by S. equi is an effective lysin for mast cells only if released in proximity to the target cell (Figure 8). In contrast to this observation, Flaherty et al. showed that SLS expressed by Streptococcus pyogenes (Molloy et al., 2011) was released into the medium and that SLS‐dependent effects on keratinocytes did not require cell–cell contact (Flaherty et al., 2015).
Mast cells have been known for decades to interact and/or become activated by bacteria and other pathogens (Johnzon, Rönnberg, & Pejler, 2016). Although many bacteria are believed to activate mast cells via TLR signalling (Sandig & Bulfone‐Paus, 2012), this study together with a limited number of previous studies indicate that mast cell activation can be caused by cell membrane‐altering cytolytic toxins (Fritscher et al., 2018; Kramer et al., 2008; Stassen et al., 2003). As judged by the complete abrogation of mast cell responses in the absence of sagA, we propose that membrane‐perturbing toxins, rather than classical PAMPs, represent the dominating bacterial factors driving mast cell activation. On a more general level, bacterial lysins have been shown to activate a range of other cell types, such as keratinocytes (Flaherty et al., 2015), neutrophils (Ma, Chang, Zhang, Zhou, & Yu, 2012), epithelial cells (Ratner et al., 2006), and myoblasts (Kennedy, Smith, Lyras, Chakravorty, & Rood, 2009). Importantly, the exact mechanism for how these toxins activate immune (and other) cells remains poorly understood. In several studies, it has been shown that lysin‐dependent effects on target cells cause an up‐regulation of specific signalling pathways including p38 MAP kinase, Akt, and NF‐κB and that the effect of bacterial lysis ultimately results in cell death (Autheman et al., 2013; Flaherty et al., 2015; Kao et al., 2011; Kennedy et al., 2009; Ma et al., 2012; Ratner et al., 2006; Wiles, Dhakal, Eto, & Mulvey, 2008). In line with such findings, we found that mast cell activation in response to sagA was strongly dependent on MAP kinase signalling through the p38 and Erk1/2 pathways and also that the bacteria eventually caused lytic cell death of the mast cells. However, the exact mechanism linking the sublytic effects of bacterial lysins to p38/Erk1/2‐activation and the downstream initiation of proinflammatory signalling events in mast cells remains to be fully elucidated.
Altogether, the present study identifies bacteria‐derived sagA as a major stimulus for mast cell activation and reveals that sagA and low levels of detergents and pneumolysin activate mast cells by a sublytic mechanism, leading to proinflammatory gene expression and cytokine release (Figure 8).
4. EXPERIMENTAL PROCEDURES
4.1. Cultured primary mast cells
BMMCs were derived from C57BL/6J mice and were cultured as described (Rönnberg & Pejler, 2012). PCMCs were generated and cultured as described (Malbec et al., 2007; Waern et al., 2012). Cell viability was determined by Trypan blue staining, quantified by an automated cell counter (Countess™ II FL; Life Technologies, Bothell, WA).
4.2. Bacteria
WT Streptococcus equi subsp. equi (strain 4047) and mutant strains were as described (Paillot et al., 2010; Robinson et al., 2015; Table 1). Each strain was streaked from glycerol stocks onto horse or cow blood agar plates (SVA, Uppsala, Sweden) and incubated overnight at 37°C (5% CO2); 14.5‐ml aliquots of Todd Hewitt Broth (CM0189 medium [Oxoid, Malmö, Sweden] containing 10 g of infusion from 450‐g fat‐free minced meat, 20‐g tryptone, 2‐g glucose, 2‐g sodium bicarbonate, 2‐g sodium chloride, and 0.4‐g disodium phosphate in 1‐L H2O (pH 7.8 at 25°C) were inoculated with four colonies respectively and incubated statically overnight at 37°C in a humidified atmosphere containing 5% CO2.
4.3. In vitro coculture of BMMCs/PCMCs and bacteria
BMMCs or PCMCs were cultured at 1 × 106 cells/ml; 0.5–1 ml of these cultures were added to wells of 24‐well plates. Bacteria from overnight cultures were washed in Tyrode's buffer (130‐mM NaCl, 5‐mM KCl, 1.4‐mM CaCl2, 1‐mM MgCl2, 5.6‐mM glucose, 10‐mM HEPES, and 0.1% BSA, pH 7.4; washed by centrifugation for 7 min at 3,000× g) and added to the BMMC or PCMC cultures at a multiplicity of infection of 10 (adjusted by optical density; 12 μl of culture with optical density = 0.5). At selected time points, supernatants (conditioned media) and cell pellets were harvested (10‐min centrifugation, 300× g) and stored at −20°C. Bacterial counts were determined by colony forming units using horse or cow blood agar plates. For inhibition of MAP kinases JNK, p38, or Erk1/2, 1‐μM SP600125 (Santa Cruz Biotechnology, Dallas, TX), 5‐μM SB203580 (MedChem Express, Sollentuna, Sweden), or 50‐μM PD98059 (InvivoGen, San Diego, CA) were added respectively, 1 hr prior to addition of bacteria.
4.4. Live microscopy
250‐μl BMMCs (1 × 106 cells/ml) in medium containing 1 μg/ml PI (ThermoFisher Scientific, Uppsala, Sweden) were added to 8‐well chamber slides (Nunc® Lab‐Tek® II—CC2™ Chamber Slide™; ThermoFisher Scientific). Slides were incubated at 37°C (5% CO2) during microscopy, and cells were equilibrated in the microscope chamber 1 hr prior to the start of the experiment. Images were acquired every 5 min in brightfield and red fluorescence (PI) channels with a Prime 95B (Photometrics) camera through a 10×/0.45 NA Plan Apochromatic objective (Nikon). Micromanager was used for controlling the microscope (μManager plugin; Edelstein, Amodaj, Hoover, Vale, & Stuurman, 2010), and ImageJ was used for analysing images (Schindelin et al., 2012).
4.5. ELISA
IL‐6, IL‐33, TNF, MCP‐1 (CCL2; ThermoFisher Scientific), HMGB1 (Aviva Systems Biology, San Diego, CA), IL‐1β (PeproTech, Stockholm, Sweden), and histamine (Histamine ELISA Fast Track, Genway Biotech Inc., San Diego, CA, and ImmuSmol, Pessac, France) were quantified by ELISA following the manufacturer's protocols and using high protein binding ELISA plates (Sarstedt). Plates were measured with an Emax® plate reader (Molecular Devices, San Jose, CA).
4.6. Quantitative real time RT‐PCR (qPCR)
Total RNA from cell pellets was extracted following the manufacturer's protocol (NucleoSpin® RNA, Machery‐Nagel, Germany) but excluding the viscosity filtering step (6.1 step 3 described in the manual). Equal concentrations of RNA (NanoDrop) were converted to cDNA (iScript™ cDNA Synthesis Kit, Bio‐Rad) and stored at −20°C; 1 μl of each cDNA was added to 9 μl of master mix: 5‐μl iTaq™ Universal SYBR® (Bio‐Rad, Hercules, CA), 3.6‐μl H2O, and 0.2‐ to 0.4‐μl primer mix (10‐μl forward primer and 10‐μl reverse primer, diluted in 80‐μl H2O, frozen and stored at −20°C; final concentration in master mix: 200–400 nM for each primer; sequences shown in Table S1). Samples were analysed in duplicates on 384‐well microtiter plates (Sarstedt, Nümbrecht, Germany) with centrifugation 5 min (2,000× g) prior to PCR. qPCR (CFX384 Touch™, Bio‐Rad) was performed with 1: 95°C, 30 s; 2: 95°C, 5 s; 3: 60°C, 30 s; steps 2–3 40× + dissociation stage. Data are shown in the form of 2−ΔCq. If no signal was detected, a Cq of 40 was assigned (see McCall, McMurray, Land, & Almudevar, 2014).
4.7. Western blot
Pellets from 1 × 106 BMMCs were lysed in SDS‐PAGE sample buffer (2.5% 2‐mercaptoethanol [Sigma], 2% SDS, 10% glycerol, and 0.002% μl bromphenol blue), boiled for 5 min at 99°C in a heating block and applied to gels (4–20%); 5‐μl ladder was added and SDS‐PAGE was run at 100 V. We used GLASS gels (WSHT, Shanghai, China), ClearPage™ HEPES‐Running buffer (WSHT) and PageRuler™ Plus Prestained Protein Ladder (Fermentas International Inc., ON, Canada). Wet transfer was performed with PVDF‐FL membranes (Millipore, Bedford, MA) activated in methanol. All components were soaked in transfer buffer (20‐mM Tris, 150‐mM glycine, 20% methanol, 0.5% SDS) before assembling the transfer box (Li‐Cor, Lincoln, NE) with ice‐block. After covering with transfer buffer, transfer was run for 1.5–2 hr at 100 V. Blocking was performed by dilution of the blocking buffer (Li‐Cor) 1:1 in TBS (10‐mM Tris, 150‐mM NaCl, pH 7.4), and membranes were placed for 1 hr on a rocking table. Primary antibodies (to p38, P‐p38, Erk1/2, P‐Erk1/2; all from Cell Signalling; Cat no. #8690, #4511, #9102, #9101, respectively) were diluted 1:1,000 in blocking buffer and incubated with membranes overnight at 4°C. After washing 3 × 10 min in TBS‐T (TBS + 0.1% Tween‐20) and 5 min in TBS, secondary antibodies (IRDye 800 CW Donkey Anti‐Rabbit IgG polyclonal [Li‐cor]) diluted 1:10,000 in TBS were added, and the filters were incubated for 60 min in the dark. Washing was performed in black boxes (2 × 10 min TBS‐T and 1 × 10 min in TBS) before scanning using an Odyssey Infrared Imager.
4.8. LDH assay
LDH release was measured by the Pierce™ LDH Cytotoxicity Assay Kit (ThermoFisher Scientific), following the manufacturer's instructions. Lysed BMMCs (using supplied lysis buffer) were used as positive control for total LDH content. For these assays, mast cell : bacterial cocultures were performed in phenol red‐free DMEM (ThermoFisher Scientific).
4.9. Flow cytometry
Cell lysis was determined by flow cytometry using DRAQ7 staining (Biostatus, Shepshed, UK), utilising a BD Accuri™ C6 Plus instrument (BD Biosciences, San Jose, CA). Digitonin was purchased from Sigma‐Aldrich (Steinheim, Germany), and pneumolysin was provided by MyBioSource (MBS1141054, expressed in E. coli, LPS free; San Diego, CA).
4.10. Haemolysis
Haemolytic activity was determined by identifying a zone of β haemolysis surrounding colonies of the mutant strains of S. equi following overnight growth on COBA streptococcal selective agar at 37°C in a humidified atmosphere containing 5% CO2.
4.11. Statistical analysis
Data are presented generally as mean ± standard error of the mean. Microsoft Excel 2017 and Graphpad Prism 7 (plotting and statistical testing) were used to analyse the data by one‐way analysis of variance with the Dunnett's post hoc test for comparison with controls if not otherwise indicated in the figures. Significance levels: *** p < .001, ** p < .01, * p < .05, or n.s.: not significant. For expression data, ΔCq values were analysed for significance and transformed to 2−ΔCq. For all statistical tests, data from all cocultures from all mice inside an experiment were pooled.
AUTHOR CONTRIBUTIONS
C. v. B. performed most of the experiments, planned the experiments, and wrote the paper; I. W. planned and performed experiments and contributed to the writing of the paper; J. E. planned and performed experiments; F. R. M. contributed to the experiments; C. R. generated essential reagents and contributed to the writing of the paper; A. S. W. generated essential reagents and contributed to the writing of the paper; M. E. S. contributed to the planning of the study and experiments and to the writing of the paper; B. G. conceived the study and contributed to the experiments and to the writing of the paper; G. P. conceived the study, planned the experiments, and wrote the paper.
CONFLICT OF INTERESTS
The authors report no conflict of interest in relation to this study.
Supporting information
Table S1. Primers used for RT‐PCR.
Video S1. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. Images were taken every 5 min. At least 200 cells were present in each picture. The video is representative of four independent experiments.
Video S2. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. After 1h, cells were infected with WT (4047) S. equi (MOI = 10). Images were taken every 5 min. At least 300 cells were present in each picture. The video is representative of four independent experiments.
Video S3. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. After 1h, cells were infected with ΔsagA mutant S. equi (MOI = 10). Images were taken every 5 min. At least 300 cells were present in each picture. The video is representative of four independent experiments.
Video S4. Mast cells (1 x 106 BMMCs/ml) were added to chamber slides and cells were infected with ΔsagA mutant S. equi (MOI = 10). After 24h, 1 μg/ml PI were added and images were taken every 1 min. The video corresponds to Suppl. Fig. 1C.
Fig. S1. Wild‐type and ΔsagA mutant S. equi have a similar growth kinetics. (A) Overnight cultures of WT and ΔsagA mutant S. equi and were adjusted to an OD 600 nm of 0.05 and growth in the medium used for mast cell‐bacteria co‐cultures was monitored once per hour. (B) CFUs for both strains after 24h of co‐cultivation with BMMCs. 50 μl from the co‐cultures were plated on horse blood agar in appropriate dilutions and plates were incubated overnight. Data are given as means ± SEM of three biological replicates. (C) 100x (NA 1.45) images of mast cell: S. equi co‐cultures after 24h. 1 x 106 BMMCs/ml were added to chamber slides and infected with WT (4047) S. equi, or ΔsagA mutant S. equi (MOI = 10); BMMCs without bacteria (Untr.) served as control. Representative brightfield images are shown for three biological replicates.
Fig. S2. Mutation of sagA abolishes the hemolytic activity of S. equi. Colony phenotypes of S. equi mutants grown overnight at 37 °C (5% CO2) on COBA streptococcal selective agar. Zones of hemolysis can be seen as clear areas around the colonies and where present, are indicated by arrows. Note the absence of hemolysis for the ΔsagA mutant.
Fig. S3. Mast cells are not acitvated by mast cell lysis, and mast cell activation by S. equi does not cause release of alarmins or IL‐1β. (A‐C) Mast cells (1 x 106 BMMCs/ml) were cultured either alone (untreated) or in the presence of WT (4047) or ΔsagA mutant S. equi (MOI = 10). At the time points indicated, conditioned media were harvested and analyzed for IL‐33 (A) or HMGB1 (B) by ELISA. (C) 1 x 106 BMMCs/ml were cultured either alone (Contr.) or in presence of WT (4047) or ΔsagA mutant S. equi ± SB203580 (SB) or SP600125 (SP) as indicated (MOI = 10). After 4h, conditioned media were harvested and analyzed for IL‐1β by ELISA. (D) Mast cells (1 x 106 BMMCs/ml) were lysed by three repeated freeze‐thaw cycles and the lysate (diluted 1:1 with growth medium) was added to BMMCs. As a control, the same amount of untreated BMMCs was used. After 4h incubation, conditioned medium was harvested and analyzed for IL‐6 by ELISA. (E) BMMCs were co‐cultured with WT or ΔsagA mutant S. equi as described above for 4 or 24h. At the time points indicated, conditioned media were harvested, sterile filtered (absence of bacteria confirmed by CFU) and added in a 1:1 ratio with normal growth medium to non‐treated BMMCs (1 x 106 BMMCs/ml). After 24h, culture supernatants were recovered and analyzed for IL‐6 by ELISA. N.s., not significant.
Fig. S4. Digitonin and pneumolysin activate mast cells by a sublytic mechanism. (A) 1 x 106 BMMCs/ml were treated with digitonin at the indicated concentrations for 4h. Supernatants were analyzed for IL‐6 and the percent lysis was determined by flow cytometry. (B) Flow cytometry showing recording of viable (healthy) cells. Note that BMMCs were mostly viable in cultures without digitonin, that a modest reduction of viable cells was seen at 3 μg/ml digitonin and that few viable cells remained after treatment with 10 μg/ml digitonin. (C) 1 x 106 BMMCs/ml were treated with the indicated concentrations of recombinant pneumolysin for 4h. Supernatants were analyzed for IL‐6 and the % lysed cells was determined by flow cytometry using DRAQ7 staining. (D) Strategy used for detection of cell lysis. Cell pellets were resuspended in PBS containing 1.5 μM DRAQ7 as a marker for cellular lysis. The data represent mean values ± SEM, representative of 2‐4 biological replicates.
ACKNOWLEDGEMENTS
This study was supported by grants from The Swedish Research Council (G. P.), from the SciLifeLab fellows program (M. E. S.), and from the Swedish Foundation for Strategic Research (M. E. S.; Grant ICA16‐0031).
von Beek C, Waern I, Eriksson J, et al. Streptococcal sagA activates a proinflammatory response in mast cells by a sublytic mechanism. Cellular Microbiology. 2019;21:e13064 10.1111/cmi.13064
Christopher von Beek and Ida Waern have equal contributions.
REFERENCES
- Abel, J. , Goldmann, O. , Ziegler, C. , Holtje, C. , Smeltzer, M. S. , Cheung, A. L. , … Medina, E. (2011). Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. Journal of Innate Immunity, 3, 495–507. 10.1159/000327714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham, S. N. , & St John, A. L. (2010). Mast cell‐orchestrated immunity to pathogens. Nature Reviews, 10, 440–452. 10.1038/nri2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arock, M. , Ross, E. , Lai‐Kuen, R. , Averlant, G. , Gao, Z. , & Abraham, S. N. (1998). Phagocytic and tumor necrosis factor α response of human mast cells following exposure to Gram‐negative and Gram‐positive bacteria. Infection and Immunity, 66, 6030–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autheman, D. , Wyder, M. , Popoff, M. , D'Herde, K. , Christen, S. , & Posthaus, H. (2013). Clostridium perfringens β‐toxin induces necrostatin‐inhibitable, calpain‐dependent necrosis in primary porcine endothelial cells. PLoS ONE, 8, e64644 10.1371/journal.pone.0064644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillo‐Navarro, M. , Leyva‐Paredes, K. , Donis‐Maturano, L. , Gonzalez‐Jimenez, M. , Paredes‐Vivas, Y. , Cerbulo‐Vazquez, A. , … Pérez‐Tapia, S. M. (2017). Listeria monocytogenes induces mast cell extracellular traps. Immunobiology, 222, 432–439. 10.1016/j.imbio.2016.08.006 [DOI] [PubMed] [Google Scholar]
- Carr, A. , Sledjeski, D. D. , Podbielski, A. , Boyle, M. D. , & Kreikemeyer, B. (2001). Similarities between complement‐mediated and streptolysin S‐mediated hemolysis. The Journal of Biological Chemistry, 276, 41790–41796. 10.1074/jbc.M107401200 [DOI] [PubMed] [Google Scholar]
- Chan, C. Y. , St John, A. L. , & Abraham, S. N. (2013). Mast cell interleukin‐10 drives localized tolerance in chronic bladder infection. Immunity, 38, 349–359. 10.1016/j.immuni.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahdah, A. , Gautier, G. , Attout, T. , Fiore, F. , Lebourdais, E. , Msallam, R. , … Launay, P. (2014). Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. The Journal of Clinical Investigation, 124, 4577–4589. 10.1172/JCI75212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawicki, W. , & Marshall, J. S. (2007). New and emerging roles for mast cells in host defence. Current Opinion in Immunology, 19, 31–38. 10.1016/j.coi.2006.11.006 [DOI] [PubMed] [Google Scholar]
- Di Nardo, A. , Yamasaki, K. , Dorschner, R. A. , Lai, Y. , & Gallo, R. L. (2008). Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. Journal of Immunology, 180, 7565–7573. 10.4049/jimmunol.180.11.7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, N. , Rohde, M. , Geffers, R. , Kroger, A. , Hauser, H. , Weiss, S. , & Gekara, N. O. (2010). Mast cells elicit proinflammatory but not type I interferon responses upon activation of TLRs by bacteria. Proceedings of the National Academy of Sciences of the United States of America, 107, 8748–8753. 10.1073/pnas.0912551107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtenacher, B. , Mannel, D. N. , & Hultner, L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis. Nature, 381, 75–77. 10.1038/381075a0 [DOI] [PubMed] [Google Scholar]
- Edelstein, A. , Amodaj, N. , Hoover, K. , Vale, R. , & Stuurman, N. (2010) Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14:Unit14 20 [DOI] [PMC free article] [PubMed]
- Flaherty, R. A. , Puricelli, J. M. , Higashi, D. L. , Park, C. J. , & Lee, S. W. (2015). Streptolysin S promotes programmed cell death and enhances inflammatory signaling in epithelial keratinocytes during group A streptococcus infection. Infection and Immunity, 83, 4118–4133. 10.1128/IAI.00611-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritscher, J. , Amberger, D. , Dyckhoff, S. , Bewersdorf, J. P. , Masouris, I. , Voelk, S. , … Koedel, U. (2018). Mast cells are activated by Streptococcus pneumoniae in vitro but dispensable for the host defense against pneumococcal central nervous system infection in vivo. Frontiers in Immunology, 9, 550 10.3389/fimmu.2018.00550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Rodriguez, K. M. , Goenka, A. , Alonso‐Rasgado, M. T. , Hernandez‐Pando, R. , & Bulfone‐Paus, S. (2017). The role of mast cells in tuberculosis: Orchestrating innate immune crosstalk? Frontiers in Immunology, 8, 1290 10.3389/fimmu.2017.01290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekara, N. O. , Westphal, K. , Ma, B. , Rohde, M. , Groebe, L. , & Weiss, S. (2007). The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol‐dependent cytolysin of Listeria monocytogenes . Cellular Microbiology, 9, 2008–2021. 10.1111/j.1462-5822.2007.00932.x [DOI] [PubMed] [Google Scholar]
- Gordon, J. R. , & Galli, S. J. (1990). Mast cells as a source of both preformed and immunologically inducible TNF‐α/cachectin. Nature, 346, 274–276. 10.1038/346274a0 [DOI] [PubMed] [Google Scholar]
- Johnzon, C. F. , Ronnberg, E. , Guss, B. , & Pejler, G. (2016). Live Staphylococcus aureus induces expression and release of vascular endothelial growth factor in terminally differentiated mouse mast cells. Frontiers in Immunology, 7, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnzon, C. F. , Rönnberg, E. , & Pejler, G. (2016). The role of mast cells in bacterial infection. The American Journal of Pathology, 186, 4–14. 10.1016/j.ajpath.2015.06.024 [DOI] [PubMed] [Google Scholar]
- Kao, C. Y. , Los, F. C. , Huffman, D. L. , Wachi, S. , Kloft, N. , Husmann, M. , … Sagong, Y. (2011). Global functional analyses of cellular responses to pore‐forming toxins. PLoS Pathogens, 7, e1001314 10.1371/journal.ppat.1001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, C. L. , Smith, D. J. , Lyras, D. , Chakravorty, A. , & Rood, J. I. (2009). Programmed cellular necrosis mediated by the pore‐forming α‐toxin from Clostridium septicum . PLoS Pathogens, 5, e1000516 10.1371/journal.ppat.1000516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, S. , Sellge, G. , Lorentz, A. , Krueger, D. , Schemann, M. , Feilhauer, K. , … Bischoff, S. C. (2008). Selective activation of human intestinal mast cells by Escherichia coli hemolysin. Journal of Immunology, 181, 1438–1445. 10.4049/jimmunol.181.2.1438 [DOI] [PubMed] [Google Scholar]
- Lundequist, A. , Calounova, G. , Wensman, H. , Ronnberg, E. , & Pejler, G. (2011). Differential regulation of Nr4a subfamily nuclear receptors following mast cell activation. Molecular Immunology, 48, 1753–1761. 10.1016/j.molimm.2011.04.017 [DOI] [PubMed] [Google Scholar]
- Ma, X. , Chang, W. , Zhang, C. , Zhou, X. , & Yu, F. (2012). Staphylococcal Panton‐Valentine leukocidin induces pro‐inflammatory cytokine production and nuclear factor‐κB activation in neutrophils. PLoS ONE, 7, e34970 10.1371/journal.pone.0034970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya, R. , Ikeda, T. , Ross, E. , & Abraham, S. N. (1996). Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF‐α [see comments. Nature, 381, 77–80. 10.1038/381077a0 [DOI] [PubMed] [Google Scholar]
- Malaviya, R. , Ross, E. , Jakschik, B. A. , & Abraham, S. N. (1994). Mast cell degranulation induced by type 1 fimbriated Escherichia coli in mice. The Journal of Clinical Investigation, 93, 1645–1653. 10.1172/JCI117146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malbec, O. , Roget, K. , Schiffer, C. , Iannascoli, B. , Dumas, A. R. , Arock, M. , & Daeron, M. (2007). Peritoneal cell‐derived mast cells: An in vitro model of mature serosal‐type mouse mast cells. Journal of Immunology, 178, 6465–6475. 10.4049/jimmunol.178.10.6465 [DOI] [PubMed] [Google Scholar]
- Marshall, J. S. (2004). Mast‐cell responses to pathogens. Nature Reviews, 4, 787–799. 10.1038/nri1460 [DOI] [PubMed] [Google Scholar]
- McCall, M. N. , McMurray, H. R. , Land, H. , & Almudevar, A. (2014). On non‐detects in qPCR data. Bioinformatics (Oxford, England), 30, 2310–2316. 10.1093/bioinformatics/btu239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire, V. A. , & Arthur, J. S. (2015). Subverting toll‐like receptor signaling by bacterial pathogens. Frontiers in Immunology, 6, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy, E. M. , Cotter, P. D. , Hill, C. , Mitchell, D. A. , & Ross, R. P. (2011). Streptolysin S‐like virulence factors: The continuing sagA. Nature Reviews. Microbiology, 9, 670–681. 10.1038/nrmicro2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillot, R. , Robinson, C. , Steward, K. , Wright, N. , Jourdan, T. , Butcher, N. , … Waller, A. S. (2010). Contribution of each of four superantigens to Streptococcus equi‐induced mitogenicity, γ interferon synthesis, and immunity. Infection and Immunity, 78, 1728–1739. 10.1128/IAI.01079-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piliponsky, A. M. , Chen, C. C. , Grimbaldeston, M. A. , Burns‐Guydish, S. M. , Hardy, J. , Kalesnikoff, J. , … Galli, S. J. (2010). Mast cell‐derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6‐KitW‐sh/W‐sh mice. The American Journal of Pathology, 176, 926–938. 10.2353/ajpath.2010.090342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piliponsky, A. M. , Chen, C. C. , Rios, E. J. , Treuting, P. M. , Lahiri, A. , Abrink, M. , … Galli, S. J. (2012). The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. The American Journal of Pathology, 181, 875–886. 10.1016/j.ajpath.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner, A. J. , Hippe, K. R. , Aguilar, J. L. , Bender, M. H. , Nelson, A. L. , & Weiser, J. N. (2006). Epithelial cells are sensitive detectors of bacterial pore‐forming toxins. The Journal of Biological Chemistry, 281, 12994–12998. 10.1074/jbc.M511431200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, C. , Heather, Z. , Slater, J. , Potts, N. , Steward, K. F. , Maskell, D. J. , … Waller, A. S. (2015). Vaccination with a live multi‐gene deletion strain protects horses against virulent challenge with Streptococcus equi . Vaccine, 33, 1160–1167. 10.1016/j.vaccine.2015.01.019 [DOI] [PubMed] [Google Scholar]
- Rönnberg, E. , Guss, B. , & Pejler, G. (2010). Infection of mast cells with live streptococci causes a toll‐like receptor 2‐ and cell‐cell contact‐dependent cytokine and chemokine response. Infection and Immunity, 78, 854–864. 10.1128/IAI.01004-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönnberg, E. , & Pejler, G. (2012). Serglycin: The master of the mast cell. Methods in Molecular Biology, 836, 201–217. 10.1007/978-1-61779-498-8_14 [DOI] [PubMed] [Google Scholar]
- Sandig, H. , & Bulfone‐Paus, S. (2012). TLR signaling in mast cells: Common and unique features. Frontiers in Immunology, 3, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , … Cardona, A. (2012). Fiji: An open‐source platform for biological‐image analysis. Nature Methods, 9, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman, P. (1967). Transient holes in the erythrocyte membrane during hypotonic hemolysis and stable holes in the membrane after lysis by saponin and lysolecithin. The Journal of Cell Biology, 32, 55–70. 10.1083/jcb.32.1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassen, M. , Muller, C. , Richter, C. , Neudorfl, C. , Hultner, L. , Bhakdi, S. , … Schmitt, E. (2003). The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor α by p38 mitogen‐activated protein kinase‐ and protein kinase C‐dependent pathways. Infection and Immunity, 71, 6171–6177. 10.1128/IAI.71.11.6171-6177.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, R. E. , Olsen, J. S. , McKinstry, A. , Villalta, S. A. , & Wolters, P. J. (2008). Mast cell IL‐6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. Journal of Immunology, 181, 5598–5605. 10.4049/jimmunol.181.8.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakurdas, S. M. , Melicoff, E. , Sansores‐Garcia, L. , Moreira, D. C. , Petrova, Y. , Stevens, R. L. , & Adachi, R. (2007). The mast cell‐restricted tryptase mMCP‐6 has a critical immunoprotective role in bacterial infections. The Journal of Biological Chemistry, 282, 20809–20815. 10.1074/jbc.M611842200 [DOI] [PubMed] [Google Scholar]
- Tung, H. Y. , Plunkett, B. , Huang, S. K. , & Zhou, Y. (2014). Murine mast cells secrete and respond to interleukin‐33. Journal of Interferon & Cytokine Research, 34, 141–147. 10.1089/jir.2012.0066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard, F. E. , Brands, X. , Roelofs, J. J. , de Beer, R. , de Boer, O. J. , van't Veer, C. , & van der Poll, T. (2014). Mast cells impair host defense during murine Streptococcus pneumoniae pneumonia. The Journal of Infectious Diseases, 210, 1376–1384. 10.1093/infdis/jiu285 [DOI] [PubMed] [Google Scholar]
- von Kockritz‐Blickwede, M. , Goldmann, O. , Thulin, P. , Heinemann, K. , Norrby‐Teglund, A. , Rohde, M. , & Medina, E. (2008). Phagocytosis‐independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood, 111, 3070–3080. 10.1182/blood-2007-07-104018 [DOI] [PubMed] [Google Scholar]
- Waern, I. , Karlsson, I. , Thorpe, M. , Schlenner, S. M. , Feyerabend, T. B. , Rodewald, H. R. , … Wernersson, S. (2012). Mast cells limit extracellular levels of IL‐13 via a serglycin proteoglycan‐serine protease axis. Biological Chemistry, 393, 1555–1567. 10.1515/hsz-2012-0189 [DOI] [PubMed] [Google Scholar]
- Wernersson, S. , & Pejler, G. (2014). Mast cell granules: Armed for battle. Nature Reviews, 14, 478–494. 10.1038/nri3690 [DOI] [PubMed] [Google Scholar]
- Wiles, T. J. , Dhakal, B. K. , Eto, D. S. , & Mulvey, M. A. (2008). Inactivation of host Akt/protein kinase B signaling by bacterial pore‐forming toxins. Molecular Biology of the Cell, 19, 1427–1438. 10.1091/mbc.e07-07-0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim, K. M. , & Lakkis, F. G. (2015). A brief journey through the immune system. Clinical Journal of the American Society of Nephrology, 10, 1274–1281. 10.2215/CJN.10031014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used for RT‐PCR.
Video S1. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. Images were taken every 5 min. At least 200 cells were present in each picture. The video is representative of four independent experiments.
Video S2. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. After 1h, cells were infected with WT (4047) S. equi (MOI = 10). Images were taken every 5 min. At least 300 cells were present in each picture. The video is representative of four independent experiments.
Video S3. Mast cells (1 x 106 BMMCs/ml) in medium containing 1 μg/ml PI were added to chamber slides. After 1h, cells were infected with ΔsagA mutant S. equi (MOI = 10). Images were taken every 5 min. At least 300 cells were present in each picture. The video is representative of four independent experiments.
Video S4. Mast cells (1 x 106 BMMCs/ml) were added to chamber slides and cells were infected with ΔsagA mutant S. equi (MOI = 10). After 24h, 1 μg/ml PI were added and images were taken every 1 min. The video corresponds to Suppl. Fig. 1C.
Fig. S1. Wild‐type and ΔsagA mutant S. equi have a similar growth kinetics. (A) Overnight cultures of WT and ΔsagA mutant S. equi and were adjusted to an OD 600 nm of 0.05 and growth in the medium used for mast cell‐bacteria co‐cultures was monitored once per hour. (B) CFUs for both strains after 24h of co‐cultivation with BMMCs. 50 μl from the co‐cultures were plated on horse blood agar in appropriate dilutions and plates were incubated overnight. Data are given as means ± SEM of three biological replicates. (C) 100x (NA 1.45) images of mast cell: S. equi co‐cultures after 24h. 1 x 106 BMMCs/ml were added to chamber slides and infected with WT (4047) S. equi, or ΔsagA mutant S. equi (MOI = 10); BMMCs without bacteria (Untr.) served as control. Representative brightfield images are shown for three biological replicates.
Fig. S2. Mutation of sagA abolishes the hemolytic activity of S. equi. Colony phenotypes of S. equi mutants grown overnight at 37 °C (5% CO2) on COBA streptococcal selective agar. Zones of hemolysis can be seen as clear areas around the colonies and where present, are indicated by arrows. Note the absence of hemolysis for the ΔsagA mutant.
Fig. S3. Mast cells are not acitvated by mast cell lysis, and mast cell activation by S. equi does not cause release of alarmins or IL‐1β. (A‐C) Mast cells (1 x 106 BMMCs/ml) were cultured either alone (untreated) or in the presence of WT (4047) or ΔsagA mutant S. equi (MOI = 10). At the time points indicated, conditioned media were harvested and analyzed for IL‐33 (A) or HMGB1 (B) by ELISA. (C) 1 x 106 BMMCs/ml were cultured either alone (Contr.) or in presence of WT (4047) or ΔsagA mutant S. equi ± SB203580 (SB) or SP600125 (SP) as indicated (MOI = 10). After 4h, conditioned media were harvested and analyzed for IL‐1β by ELISA. (D) Mast cells (1 x 106 BMMCs/ml) were lysed by three repeated freeze‐thaw cycles and the lysate (diluted 1:1 with growth medium) was added to BMMCs. As a control, the same amount of untreated BMMCs was used. After 4h incubation, conditioned medium was harvested and analyzed for IL‐6 by ELISA. (E) BMMCs were co‐cultured with WT or ΔsagA mutant S. equi as described above for 4 or 24h. At the time points indicated, conditioned media were harvested, sterile filtered (absence of bacteria confirmed by CFU) and added in a 1:1 ratio with normal growth medium to non‐treated BMMCs (1 x 106 BMMCs/ml). After 24h, culture supernatants were recovered and analyzed for IL‐6 by ELISA. N.s., not significant.
Fig. S4. Digitonin and pneumolysin activate mast cells by a sublytic mechanism. (A) 1 x 106 BMMCs/ml were treated with digitonin at the indicated concentrations for 4h. Supernatants were analyzed for IL‐6 and the percent lysis was determined by flow cytometry. (B) Flow cytometry showing recording of viable (healthy) cells. Note that BMMCs were mostly viable in cultures without digitonin, that a modest reduction of viable cells was seen at 3 μg/ml digitonin and that few viable cells remained after treatment with 10 μg/ml digitonin. (C) 1 x 106 BMMCs/ml were treated with the indicated concentrations of recombinant pneumolysin for 4h. Supernatants were analyzed for IL‐6 and the % lysed cells was determined by flow cytometry using DRAQ7 staining. (D) Strategy used for detection of cell lysis. Cell pellets were resuspended in PBS containing 1.5 μM DRAQ7 as a marker for cellular lysis. The data represent mean values ± SEM, representative of 2‐4 biological replicates.