

Abstract
Here, we report the catalytic effect of vibrational strong coupling (VSC) on the solvolysis of para‐nitrophenyl acetate (PNPA), which increases the reaction rate by an order of magnitude. This is observed when the microfluidic Fabry–Perot cavity in which the VSC is generated is tuned to the C=O vibrational stretching mode of both the reactant and solvent molecules. Thermodynamic experiments confirm the catalytic nature of VSC in the system. The change in the reaction rate follows an exponential relation with respect to the coupling strength of the solvent, indicating a cooperative effect between the solvent molecules and the reactant. Furthermore, the study of the solvent kinetic isotope effect clearly shows that the vibrational overlap of the C=O vibrational bands of the reactant and the strongly coupled solvent molecules is critical for the catalysis in this reaction. The combination of cooperative effects and cavity catalysis confirms the potential of VSC as a new frontier in chemistry.
Keywords: cavity catalysis, cooperativity, polaritonic chemistry, vibrational strong coupling
Strong light–matter coupling offers a unique way to control internal reaction coordinates by coupling molecular transitions to the vacuum field (zero‐point energy) of a cavity mode.1, 2, 3, 4, 5, 6, 7 VSC allows the selective coupling of a vibrational mode,8, 9 which is easily achieved in the liquid state in a microfluidic Fabry–Perot (FP) cavity.10 Thomas et al. observed the modification of the rate of a C−Si bond‐breaking process and, more recently, demonstrated changes to the branching ratio of the products by selective VSC of silane derivatives.3, 6 Hiura et al. recently reported the possibility of accelerating reactions by vibrational ultra‐strong coupling of water molecules involved in hydrolysis reactions.7 In both cases, reactants were directly coupled to cavity modes and the reaction rates could be controlled by moving the physical distance between the mirrors of a FP cavity, that is, by tuning the coupling to a given vibrational resonance. Recent theoretical studies confirm that potential energy surfaces and reactivity can be modified by coupling to the vacuum field, although the critical role of the solvent is hard to simulate.11, 12, 13, 14, 15, 16
VSC conditions can be achieved by injecting a molecular liquid into a FP cavity in which the vibrational transition interacts with the cavity mode (Figure 1 a) to generate two new eigenstates, called vibro‐polaritonic states (P+ and P−). The interaction energy (Rabi energy; ) is proportional to the transition dipole moment d and the electric field ( ) of the electromagnetic mode of the cavity:
| (1) |
Figure 1.
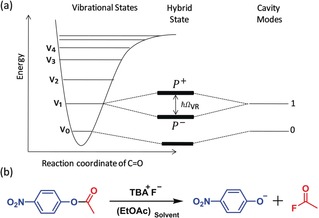
a) Schematic representation of vibro‐polaritonic states formed from a molecular vibrational state and a FP cavity mode. b) Hydrolysis of PNPA in ethyl acetate (EtOAc).
where is the vacuum permittivity, is the vibrational frequency, is the mode volume of the cavity, and the number of photons involved in the coupling process. When goes to zero, a residual Rabi splitting energy remains even in the dark due to the coupling to the zero‐point energy of the cavity. Importantly, is proportional to , where N is the number and C the concentration of the coupled molecules interacting strongly with the cavity mode.1, 2, 3
In this Communication, we report the solvolysis of PNPA catalyzed by VSC under a cooperative strong coupling effect between the reactant and the solvent molecules. In other words, when the solvent and the reactant molecules have overlapping vibrational bands, but only the solvent is at a sufficiently high concentration to be strongly coupled to the cavity mode, it increases the reaction rate more than one order of magnitude at room temperature.
The FP cavities used in the present experiments were prepared with Au mirrors sputtered onto IR‐transparent BaF2 windows by following a reported procedure.10 The Au mirrors were protected with 100 nm of sputtered SiO2. The spacing between the mirrors used for the current study is set to 18 μm to couple the C=O stretching mode of the PNPA molecules (Figure 2 a). Such FP cavities can hold up to 3 μL of a reaction solution. The non‐cavity experiments were carried out in a microfluidic cell made of the same windows without the Au mirrors (see the Supporting Information for further details). The PNPA solvolysis17, 18, 19 in this study was carried out under mild basic conditions provided by tetrabutylammonium fluoride (TBAF), as schematically shown in Figure 1 b. TBAF in ethyl acetate (EtOAc) normally facilitates a base‐catalysed acyl (BAc2) bond breaking mechanism (see Supporting Information and Scheme S1). EtOAc was chosen as the solvent because its carbonyl stretching transition overlaps with that of the reactant PNPA (Figure 2 b; red traces). Both PNPA and TBAF solutions were prepared separately, mixed outside and injected into the FP cavity.
Figure 2.
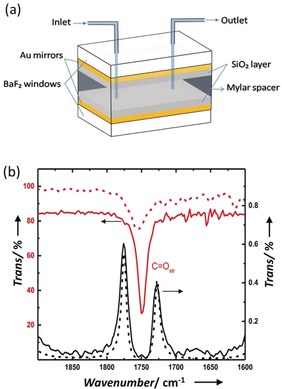
a) Parts of a flow‐cell micro‐cavity QED reactor. b) IR transmission spectra of 10 % EtOAc (red trace) and 0.1 m PNPA (dotted red trace; magnified by factor 100) in hexane. Vibro‐polaritonic states P+ and P− formed by coupling to the tenth cavity mode (black trace; path length is approx. 18 μm) with a TMM simulation (dotted black trace).
Strong coupling of the C=O stretching mode of neat 12EtOAc to the tenth mode of the FP cavity gives a Rabi splitting of 155 cm−1, which is equivalent to a 4.4 % change in the actual transition energy with P+ and P− at 1840 cm−1 and 1685 cm−1, respectively (Figures 2 b and S2). In the reactive mixture, the PNPA represents only 0.1 wt % of the mixture and is thus unable to undergo strong coupling to the FP cavity because of the low concentration.1 One of the questions we address here is whether VSC of 12EtOAc can nevertheless influence the reactivity of PNPA under these conditions even though it is not a reactant.
The major product of the PNPA solvolysis is the para‐nitro phenoxide (PNP−), which has an intense absorption peak at 407 nm. Since the transparency region of BaF2 windows extends well into the UV range (ca. 200 nm), the progress of the reaction can be easily monitored by following the temporal rise of the PNP− absorption peak as shown in Figure 3 a (see also Figure S6). In other words, while the FP cavity is set to couple the C=O vibrational band in the IR, the influence of strong coupling on the reaction rate is being monitored in the UV/Vis region. This condition helps to avoid complications such as filter effects and other issues related to the FP cavity configuration. By having a tenfold excess of PNPA compared to TBAF (0.1 m vs. 0.01 m, respectively), a pseudo‐first‐order kinetics is observed. Apparent rate constants (k app) were determined by linear regression (Figure 3 b).17 In the normal non‐cavity, the apparent rate constant (k app nc) is 0.2×10−2 s−1 (Figure 3 b). To prepare an on‐resonance cavity, the empty cavity is slowly tuned to the desired path length and kept undisturbed for 30 min (see Supporting Information). Afterwards, the reaction mixture is injected into the cavity and the kinetic traces are recorded. Under the on‐resonance condition, that is, when the cavity is tuned to the 12C=O stretching mode of ethylacetate (12EtOAc), the k app cav increased by more than one order of magnitude (2.5×10−2 s−1) compared to the non‐cavity, as shown in Figure 3 b. k app cav levels off to normal values (k app nc) when the cavity is detuned from the C=O mode of 12EtOAc (the off‐resonance measurements), as shown in Figure 3 d. Please note that the non‐cavity and the cavity conditions are exactly the same except for the presence of Au mirrors in the cavity, and k app nc is unaffected by small variation in the spacer thickness (blue empty circle in the Figure 3 d). Interestingly, the increase in k app cav follows the envelope of the C=O stretching mode of the 12EtOAc and PNPA molecules (FWHM; 25 cm−1) which are coupled to the cavity mode (Figure 3 c,d). This suggests a cooperative effect between the strongly coupled solvent and the reactant.
Figure 3.
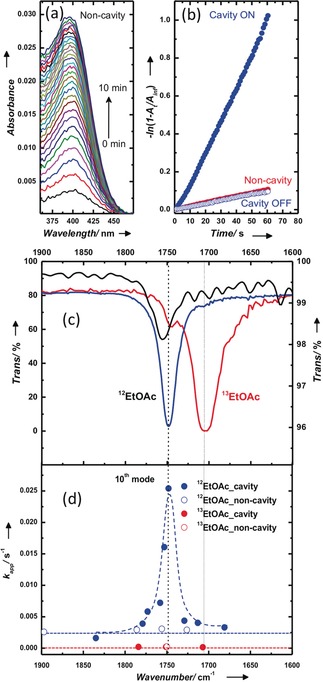
a) Absorption spectra showing the evolution of PNP− species during ester hydrolysis. b) Pseudo‐first‐order kinetic traces measured at 407 nm for cavity on‐resonance (blue circle; 1.6×10−2), cavity off‐resonance (blue hollow circle; 0.16×10−2), and non‐cavity (red circle; 0.18×10−2) for 12EtOAc. c) Carbonyl stretching modes of pure PNPA (ATR spectrum; black trace); FTIR spectra of 12EtOAc (blue trace) and 13EtOAc (red trace) at 10 % in hexane. d) Corresponding kinetic traces measured by tuning the cavity (12EtOAc: blue filled circles; 13EtOAc: red filled circles) and non‐cavity (12EtOAc blue empty circles; 13EtOAc red empty circles); tenth mode of the cavity overlapping with the carbonyl stretching mode of 12EtOAc and PNPA. The dashed curves are guides to the eye.
To confirm this cooperative effect, we have conducted kinetic isotope effect (KIE) experiments using the 12C=O (12EtOAc) and 13C=O (13EtOAc) isotopes with carbonyl vibrational resonances at 1750 and 1706 cm−1, respectively (Figure 3 c). Outside the cavity, the use of 13EtOAc gives an apparent rate that is one order of magnitude lower (13 k app=2.4×10−4 s−1) than the corresponding 12EtOAc isotope rate. This large change in the reaction rate might be caused by a secondary isotope effect in which the 12EtOAc is coupled to the PNPA carbonyl vibrational state, facilitating external vibrational energy transfer in the system.20 Please note that the rate‐determining step for the BAc2 mechanism is the attack of the nucleophile on the electron‐deficient carbonyl carbon atom.17, 18, 19 Interestingly, under VSC of 13EtOAc, the reaction rates are not modified. Kinetic action spectra measured as a function of cavity tuning (Figure 3 d) show no change in the rate for the 13EtOAc solvent system relative to the rate outside the cavity, in contrast to 12EtOAc. This is a confirmation that the VSC of 12EtOAc is acting cooperatively and perturbing the internal reaction coordinate of PNPA, affecting its bond‐dissociation energy, which is in agreement with recent theoretical predictions21 and will be discussed below.
To clarify how VSC is affecting the reaction barrier and whether it is truly catalyzing the reaction, the temperature dependence of the reaction rate was analyzed to extract the thermodynamic activation parameters. As immediately apparent from the slopes in Figure 4 a, the activation energy drops under VSC. The enthalpy of activation (ΔH ≠) calculated from the Eyring equation decreases substantially from 53.2 kJ mol−1 to 7.9 kJ mol−1 (Figure 4 a and the Supporting Information), indicating that the transition state (TS) is far lower in energy under VSC of 12EtOAc molecules. Another very interesting finding is that the entropy of activation (ΔS ≠) also decreases significantly from −70.9 J K mol−1 to −206.2 J K mol−1, partially offsetting the change in ΔH ≠. This large decrease in ΔS ≠ indicates that the TS is far more polar under VSC and thereby structuring the solvent cage which also decreases ΔH ≠. In other words, the solvent–solute interactions are much stronger under VSC, thereby favoring the BAc2 mechanism by stabilizing the intermediates.
Figure 4.
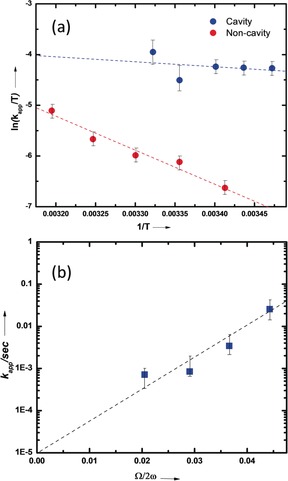
a) Eyring plot for reaction inside the cavity (blue circles) and non‐cavity (cell; red circles); b) Apparent rate constant as a function of Rabi splitting under VSC of 12EtOAc. Dotted lines are the corresponding linear fitting.
Next, we looked into the reaction‐rate variation with respect to the cooperative coupling strength of the 12EtOAc molecules. To do this, we fixed the concentration of PNPA (0.1 m) while varying the 12EtOAc concentration in a mixed anisole/12EtOAc solution. We chose anisole as a co‐solvent because the relative solvent‐polarity difference is only 0.03 compared to EtOAc and the rate is essentially the same in anisole and 12EtOAc in the absence of VSC (Figure S3; the 12EtOAc‐to‐reactant ratio is close to 1000, we therefore assume that the vibrational bath is still preserved at lower concentrations of 12EtOAc). An exponential rise in the reaction rate is observed upon increasing the concentration of 12EtOAc under strong coupling conditions, that is, with Rabi splitting (Figure 4 b). Such an exponential dependence has also been observed for both a decelerating desilylation reaction3 and for an accelerating hydrolysis reaction.7 This probably points to a more general feature by which the activation energy varies linearly with the Rabi splitting, whichever direction the barrier is moving under VSC.
In conclusion, the thermodynamic data unequivocally shows that the reaction is catalyzed under VSC of the solvent. The large decrease in the entropy of activation indicates that the electron‐density distribution is strongly modified in the TS under VSC, resulting in a more polar activated complex. The solvent‐concentration dependence and the kinetic isotope effect demonstrate that VSC can be transferred from the solvent to the solute as long as their vibrational resonances overlap. The spectral overlap itself is not enough to transfer VSC from the solvent to the reactant. Clearly, the solute–solvent interactions must be strong, for example, dipolar coupling, to induce the transfer, as has been recently shown in a theoretical study of ensemble‐induced strong coupling effects on single quantum objects.21 Other theoretical studies have considered chemical reactivity under VSC in the gas phase and confirm that the reactivity should be modified.13, 14, 15, 16 In view of the important role of solvation in the TS, further theoretical studies taking into account solvation effects under VSC would be very helpful. On the experimental side, many more classes of reactions need to be studied in order to extract general principles that govern reactions under VSC. The use of cooperative effects as demonstrated here should greatly facilitate this task, since it removes the requirement of a high concentration of reactants to achieve the strong‐coupling condition. Action spectra under VSC can be used as a spectroscopic tool to help elucidate which vibrations play a role in the reaction path. These findings should have a large impact on controlling and understanding chemical reactivity through VSC.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
J.G. would like to thank the Department of Chemical Sciences, IISER Mohali, for using the facilities; the DST‐SERB Core Research Grant, Govt. of India (EMR/2017/003455) is also acknowledged. J.L. and P.B. thank IISER Mohali for funding. T.W.E. acknowledges support of the International Center for Frontier Research in Chemistry (icFRC, Strasbourg), the ANR Equipex Union (ANR‐10‐EQPX‐52‐01), the Labex NIE projects (ANR‐11‐LABX‐0058 NIE), and CSC (ANR‐10‐LABX‐ 0026 CSC) within the Investissement d'Avenir program ANR‐10‐IDEX‐0002‐02 and the ERC (project no 788482 MOLUSC).
J. Lather, P. Bhatt, A. Thomas, T. W. Ebbesen, J. George, Angew. Chem. Int. Ed. 2019, 58, 10635.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.7531544.v1).
Contributor Information
Prof. Thomas W. Ebbesen, Email: ebbesen@unistra.fr.
Dr. Jino George, Email: jgeorge@iisermohali.ac.in.
References
- 1. Ebbesen T. W., Acc. Chem. Res. 2016, 49, 2403–2412. [DOI] [PubMed] [Google Scholar]
- 2. Hutchison J. A., Schwartz T., Genet C., Devaux E., Ebbesen T. W., Angew. Chem. Int. Ed. 2012, 51, 1592–1596; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1624–1628. [Google Scholar]
- 3. Thomas A., George J., Shalabney A., Dryzhakov M., Varma S. J., Moran J., Chervy T., Zhong X., Devaux E., Genet C., Hutchison J. A., Ebbesen T. W., Angew. Chem. Int. Ed. 2016, 55, 11462–11466; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11634–11638. [Google Scholar]
- 4. Munkhbat B., Wersäll M., Baranov D. G., Antosiewicz T. J., Shegai T., Sci. Adv. 2018, 4, eaas9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peters V. N., Faruk M. O., Asane J., Alexander R., Peters D. A., Prayakarao S., Rout S., Noginov M. A., Optica 2019, 6, 318–325. [Google Scholar]
- 6. Thomas A., Lethuillier-Karl L., Nagarajan K., Vergauwe R. M., George J., Chervy T., Shalabney A., Devaux E., Genet C., Moran J., Ebbesen T. W., Science 2019, 363, 615–619. [DOI] [PubMed] [Google Scholar]
- 7.H. Hiura, A. Shalabney, J. George, 10.26434/chemrxiv.7234721.v3, 2018. [DOI]
- 8. Shalabney A., George J., Hutchison J., Pupillo G., Genet C., Ebbesen T. W., Nat. Commun. 2015, 6, 5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Long J. P., Simpkins B. S., ACS Photonics 2015, 2, 130–136. [Google Scholar]
- 10. George J., Shalabney A., Hutchison J. A., Genet C., Ebbesen T. W., J. Phys. Chem. Lett. 2015, 6, 1027–1031. [DOI] [PubMed] [Google Scholar]
- 11. Flick J., Ruggenthaler M., Appel H., Rubio A., Proc. Natl. Acad. Sci. USA 2017, 114, 3026–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Ruggenthaler M., Tancogne-Dejean N., Flick J., Appel H., Rubio A., Nat. Rev. Chem. 2018, 2, 0118; [Google Scholar]
- 12b. Herrera F., Spano F. C., Phys. Rev. Lett. 2016, 116, 238301. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Du M., Ribeiro R. F., Yuen-Zhou J., Chem. 2019, 5, 1167–1181; [Google Scholar]
- 13b. Herrera F., Spano F. C., ACS Photonics 2018, 5, 65–79. [Google Scholar]
- 14.
- 14a.J. C. G. Angulo, R. F. Ribeiro, J. Yuen-Zhou, 2019, arXiv:1902.10264 [cond-mat, physics:physics, physics:quant-ph];
- 14b. Galego J., Garcia-Vidal F. J., Feist J., Nat. Commun. 2016, 7, 13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Climent C., Galego J., Garcia-Vidal F. J., Feist J., Angew. Chem. Int. Ed. 2019, 58, 8698–8702; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8790–8794. [Google Scholar]
- 16.J. Galego, C. Climent, F. J. Garcia-Vidal, J. Feist, 2018, arXiv:1807.10846 [physics, physics:quant-ph].
- 17. Park J. H., Meriwether B. P., Clodfelder P., Cunningham L. W., J. Biol. Chem. 1961, 236, 136–141. [PubMed] [Google Scholar]
- 18. Mitton C., Schowen R., Gresser M., Shapley J., J. Am. Chem. Soc. 1969, 91, 2036–2044. [Google Scholar]
- 19. Mitton C. G., Gresser M., Schowen R. L., J. Am. Chem. Soc. 1969, 91, 2045–2047. [Google Scholar]
- 20. Owrutsky J. C., Raftery A. D., Hochstrasser R. M., Annu. Rev. Phys. Chem. 1994, 45, 519–555. [DOI] [PubMed] [Google Scholar]
- 21.S. Schütz, J. Schachenmayer, D. Hagenmüller, V. Sandoghdar, T. W. Ebbesen, C. Genes, G. Pupillo, 2019, arXiv:1904.08888 [quant-ph]. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary