

Abstract
The selective cleavage of thermodynamically stable C(sp3)−C(sp3) single bonds is rare compared to their ubiquitous formation. Herein, we describe a general methodology for such transformations using homogeneous copper‐based catalysts in the presence of air. The utility of this novel methodology is demonstrated for Cα−Cβ bond scission in >70 amines with excellent functional group tolerance. This transformation establishes tertiary amines as a general synthon for amides and provides valuable possibilities for their scalable functionalization in, for example, natural products and bioactive molecules.
Keywords: air, amines, C−C cleavage, copper, morpholines
Carbon–carbon single bonds are arguably one of the least reactive “functional” groups in chemistry and biology.1 While in the past decades, the site‐selective activation of C−H bonds attracted widespread interest,2 related transformations of C−C bonds are rare in the context of synthetic chemistry.3 Obviously, nonpolar C−C σ‐bonds are both thermodynamically stable and lesser accessible,4 which makes their selective cleavage one of the most challenging transformationsin chemistry.5 In contrast, several types of such transformations catalyzed by metalloenzymes are known in biology.6 Interestingly, in the active site of most of these enzymes (dioxygenases) iron or copper metal ions are found,7 which productively use both dioxygen atoms in the metabolism of steroids (cholesterol)8 and amino acids (tryptophan)9 as well as xenobiotics.10 Notably, in synthetic chemistry the oxidative cleavage of C−C σ‐bonds is only achieved using (over)stoichiometric amounts of hazardous oxidants such as O3,11 NaIO4, H5IO6, Pb(OAc)4, and KMnO4, which result often in poor functional group tolerance. Very recently, it has been demonstrated that these latter limitations can be overcome by so‐called deconstructive functionalizations using AgNO3 and ammonium persulfate as final oxidants.12 From a green and practical perspective there is still substantial need for selective methodologies, which make use of more benign oxidants. In this respect, air is the ideal reagent in terms of price and waste generation.13
The oxidation of amines plays a vital role in nature from both transformative and mechanistic aspects.14 The main transformations currently known are summarized in Figure 1. Most prominent is the preparation of amine oxides using different oxidants such as hydrogen peroxide (Figure 1 a). In addition, N‐dealkylation processes are known to be catalyzed by oxidases to yield the corresponding secondary amines (Figure 1 b).15 Much less explored is the synthesis of enamines from amines in the presence of stoichiometric amounts of mercuric(II) acetate as shown in Figure 1 c.16 Finally, the preparation of amides by α‐C−H oxidation has been reported to some extent (Figure 1 d).17 Complementary to all of these known transformations, we herein communicate a general copper‐catalyzed selective cleavage of C(sp3)−C(sp3) single bonds within amines using simply air (Figure 1 e).
Figure 1.
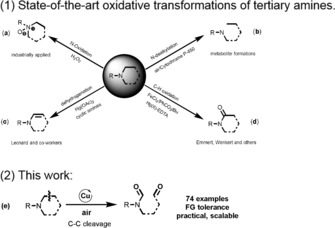
Oxidative transformation of tertiary amines. (a) N‐Oxidation to amine oxides. (b) N‐dealkylation (c) Oxidative dehydrogenation. (d) C−H oxidation. (e) Selective cleavage of C(sp3)−C(sp3) bonds in tertiary amines.
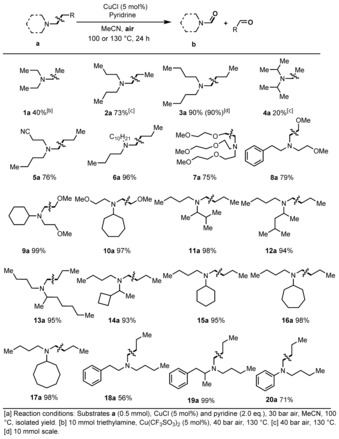
We commenced this work studying the C−H oxidation of industrially relevant tri‐n‐butylamine as a benchmark system using a broad range of metal salts, ligands, oxidants, and solvents. Surprisingly, employing copper salts under pressure of air resulted in the cleavage of C−C bonds leading to dibutylformamide 3 b.18 After optimization we obtained product 3 b in 90 % yield with very high selectivity by using 5 mol % CuCl and 2 equiv pyridine at 30 bar air and 100 °C in acetonitrile (Tables S1 and S2). It is worth noting that this reaction can be easily performed on a gram scale even in the presence of water.
Next, we were interested in evaluating the reactivity of other aliphatic amines. As shown in Table 1, 20 different amines underwent smooth and selective C−C bond cleavage to give the corresponding formamides 1 b–20 b in general in high yields. Using inexpensive triethylamine shows the general possibility to synthesize N,N‐diethylformamide, which is a less toxic alternative to the frequently used solvent N,N‐dimethylformamide. However, for practical applications the procedure still has to be improved. Given the importance of nonsymmetrical amines, we explored the regioselective cleavage of carbon–carbon bonds in substrates containing different alkyl substituents or more than one potential site for functionalization. Gratifyingly, this protocol exhibits both excellent chemoselectivity and regioselectivity (4 a–10 a). For example, several substituted butylamines containing C2–C6 branched alkyl groups and C4–C8 cyclic alkyl groups were converted into the desired products with ≥93 % yields (11 b–17 b). Notably, N,N‐dibutyl 2‐phenylethan‐1‐amine afforded the butyl‐cleaved product in 56 % yield along with trace amounts of benzaldehyde and N,N‐bis(2‐methoxyethyl)formamide, which is observed by GC–MS (18 b) (Scheme S2). The more sterically encumbered N,N‐dibutyl (1‐phenylpropan‐2‐yl) amine gave the desired product quantitatively (19 b). Finally, N,N‐dibutylaniline afforded the oxidized product (20 b) in 71 % yield.
Table 1.
C−C Bond cleavage reactions: tertiary amines.[a]
|
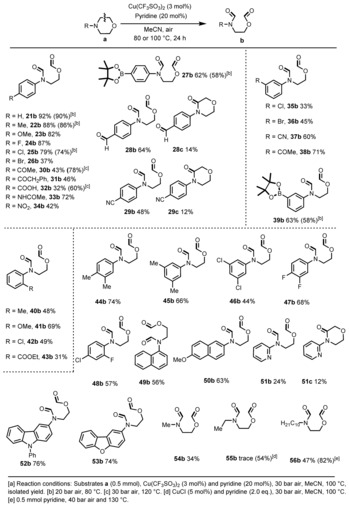
Nitrogen heterocycles such as morpholines and piperazines constitute privileged scaffolds in modern drugs.19 In fact, several out of the top 50 pharmaceuticals belong to this class of compounds. Thus, their preparation and derivatization continues to attract considerable attention. As shown in Table 2, 33 different N‐(hetero)aryl morpholines provided the desired products under a set of standard conditions (3 mol % Cu(CF3SO3)2, 20 mol % pyridine, MeCN, 100 °C, 24 hours) with often excellent selectivity (Tables S3 and S4; see the Supporting Information for further experimental details.) The transformations can be easily run on a gram scale as shown by conversion of N‐phenylmorpholine (21 a), which gave 68 % isolated yield of 21 b [Eq. (S2)]. More sensitive benzylic C−H bonds in 22 b, 40 b, 44 b and 45 b, as well as a methoxy substituent in 23 b are well tolerated giving the desired products in 48–87 % isolated yields. N‐Aryl morpholines bearing ‐F, ‐Cl, ‐Br, ‐CN, and ‐COOMe groups are shown to be compatible with this procedure, too (24 b, 25 b, 26 b, 29 b, 35 b, 36 b, 37 b, 42 b, 43 b, 46 b, 47 b, and 48 b). Although homogeneous aerobic copper catalysis is known to cleave α‐C−C bonds of ketones,20 the ketone groups were untouched with our [Cu]/air system (30 b, 31 b, and 38 b). Most surprisingly, even the formyl‐substituted starting material gave mainly the C−C bond‐cleavage product 28 b in 64 % yield and no expected acid was formed. To the best of our knowledge this is a rare example of oxidative C−C cleavage in the presence of an aldehyde. Interestingly, along with C−C cleavage also formation of the amide 28 c resulted. Similar side products were observed in the case of 29 c and 51 c. Substrates bearing one or more electron‐withdrawing functional groups such as ketones, nitro, carboxylic acid, and ester were also active (30 b, 31 b, 32 b, 34 b, 38 b, and 43 b). Boron‐containing compounds represent important building blocks for all kinds of life science molecules and allow for numerous further valorizations. Gratifyingly, 27 a and 39 a gave the preferred products in 62 % and 63 % yield. Moreover, a 72 % yield of the amide‐containing product 33 b was isolated under the standard conditions. In addition, 4‐(naphthalen‐1‐yl)morpholine (49 a) and 4‐(6‐methoxynaphthalen‐2‐yl)morpholine (50 a) provided 49 b and 50 b in 56 % and 63 % yield, respectively. Even nitrogen‐ and oxygen‐heteroaryl‐substituted morpholines such as 4‐(pyridin‐2‐yl)morpholine (51 a), 4‐(9‐phenyl‐9H‐carbazol‐3‐yl)morpholine (52 a), and 4‐(dibenzo[b,d]furan‐2‐yl)morpholine (53 a) underwent smooth oxidative cleavage in the presence of the catalytic system. We were pleased to find that the alkyl‐substituted derivatives undergo a similar transformation. Indeed, N‐methylmorpholine, N‐ethylmorpholine, and N‐decylmorpholine yielded the cleavage products 54 b–56 b in 34–82 % yield. For comparison,18a we used commercially available CuO and Cu2O as the catalysts for the cleavage of morpholines such as 4‐(p‐tolyl)morpholine (22 a), 4‐morpholinobenzaldehyde (28 a), and 1‐(4‐morpholinophenyl)ethan‐1‐one (30 a) (Table S5). All these results (yields <32 %) show that the heterogeneous systems CuO and Cu2O are not comparable to the Cu(CF3SO3)2/pyridine system and are less efficient, especially for functionalized substrates (Tables S2, S4, and S5).
Table 2.
C−C Bond cleavage reactions of morpholines.[a]
|
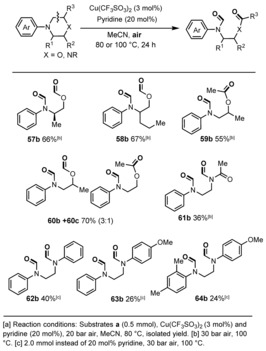
As shown in Table 3, ring‐substituted morpholines reacted in a similar manner and provided the corresponding products (57 b–60 b). Notably, a methyl group in C2 position of the morpholine ring allowed for selective cleavage of the C5−C6 vs. the C2−C3 bond (75:25), while alkyl substituents in the 3‐position led exclusively to activation of the nonsubstituted bond. Apart from morpholines, also the oxidative cleavage of piperazines was investigated. Although somewhat lower yields of the corresponding products (61 b–64 b) were observed, the products can be easily isolated due to their different physical properties. In addition, we have also tried N‐phenylpyrrolidine and N‐phenylpiperidine under the standard conditions, but no C−C cleavage products were observed.
Table 3.
C−C Bond cleavage reactions: morpholines and piperazines.[a]
|
To demonstrate the utility of this copper‐catalyzed oxidation reaction, we evaluated late‐stage C−C bond‐cleavage reactions of functionalized natural products and bioactive molecules including derivatives of isophorone, terpenoids such as ionone, citronellal, piperonylacetone, and 5‐cholesten‐3‐one. As shown in Table 4, the corresponding amides were obtained in high yield and selectivity (65 b, 66 b, 67 b, 68 b, 71 b, and 72 b). Notably, in the case of 72 b the cleavage of the Cα(sp3)−Cβ(sp3) single bond proceeded selectively in the presence of a C=C double bond! Furthermore, the amination product of pentoxifylline, a xanthine derivative used to treat muscle pain, was evaluated and the corresponding products were smoothly isolated (69 b and 70 b). Finally, to showcase late‐stage drug modifications as well as to prepare putative drug metabolites, we performed the reactions of linezolid (73 a), which is a morpholine‐containing antimicrobial used for the treatment of infections caused by Gram‐positive bacteria, and the nitrogen‐containing heterocycle emorfazone (74 a), a nonsteroidal anti‐inflammatory drug used to treat dental pain and inflammation. Under aerobic conditions in the presence of homogeneous copper, 73 b and 74 b were formed in good yield with excellent selectivity.
Table 4.
Late‐stage C−C bond cleavage of modified natural products and bioactive molecules.[a]
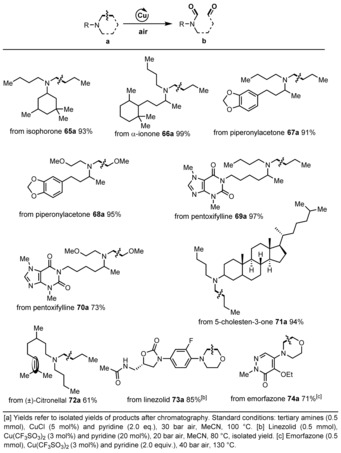
|
To understand the mechanism of this general oxidative cleavage reaction, several experiments and in situ spectroscopic investigations were performed using N‐phenylmorpholine (21 a). Firstly, to prove the stability of the cocatalyst/ligand, which significantly improves the conversion, deuterated [D5]pyridine was employed instead of pyridine. However, a standard catalytic experiment revealed only [D5]pyridine was detected by GC–MS after 24 h [Eq. (S3)]. To understand the formation of the oxidative cleavage products, possible intermediates 4‐phenylmorpholin‐3‐one (75 a) and 4‐phenylmorpholine‐2,3‐dione (76 a) were submitted to the regular reaction conditions [Eqs. (S4) and (S5)]. Both compounds proved to be stable and no formation of 21 b was observed. Thus, these compounds can be excluded as putative intermediates. On the other hand, trapping experiments demonstrate the importance of radical intermediates in this transformation. When 2 equivalents of TEMPO (2,2,6,6‐tetramethyl‐1‐piperidinyloxy) or BHT (2,6‐di‐tert‐butyl‐4‐methylphenol) were added, the reaction completely stopped [Eq. (S6)], along with the dehydrogenated product 4‐phenyl‐3,4‐dihydro‐2H‐1,4‐oxazine detected by GC‐MS analysis [Eq. (S7), Scheme S3]. A related intermediate was observed by GC–MS in the oxidation of tri‐n‐butylamine, too (Figure S1 and Scheme S1).
Notably, in the absence of air N‐phenylmorpholine (21 a) gave no desired product, even in the presence stoichiometric amounts of copper(II) pre‐catalyst [Eq. (S8)]. In order to gain more details on this copper/air catalysis, we performed kinetic studies utilizing 21 a under the standard conditions. As shown in Figure S2, no induction period is required for the in situ generation of the active catalytic species and 32 % yield of the product 21 b is formed in the first hour. However, no intermediates were detected by gas chromatography during the whole process. Finally, we added 18O‐labeled water to the model reaction under standard conditions; however, no 18O‐incorporated product was observed. This clearly demonstrates that only O2 from the air acts as the oxygen donor in this transformation [Eq. (S9)]. Finally, EPR and UV/Vis investigations were performed to further explore the reaction steps. The CuII precursor reacts with 21 a via single‐electron transfer (SET) to generate the free‐radical cation A and CuI [Eq. (1)]. This assumption is confirmed by EPR investigations, which showed that addition of N‐phenylmorphline to a CuII solution under inert atmosphere resulted in a fast disappearance of the CuII EPR signal and the formation of a temporary signal at g=2.004 with unresolved hyperfine structure typical for an organic radical A (Figure S3).
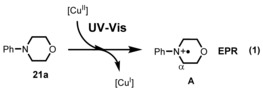
The reduction of CuII to CuI by 21 a is also evident from UV/Vis measurements, which showed the decay of the ligand‐to‐metal charge transfer (LMCT) and the weak d–d transition bands of CuII below 300 nm and around 750 nm, while new metal‐to‐ ligand charge transfer bands (MLCT) of CuI at 320 and 466 nm appeared (Figure S4). In addition, transient UV/Vis bands at 913 and 1034 nm were detected, which we assign tentatively to the formation of radical intermediate A [Eq. (1)]. No further detailed knowledge has been obtained from in situ EPR investigations, since the next, very complex step in the catalytic reaction might involve H or proton abstraction from amino radical as well as activation of O2 by CuI or intermediate A to form superoxide species.
In conclusion, we have developed a general protocol for the aerobic cleavage of Cα(sp3)−Cβ(sp3) single bonds in amines (>70 examples) using a practical and inexpensive copper catalyst. This system is effective for the conversion of industrial bulk amines as well as for the late‐stage functionalization of modified natural products and bioactive molecules. Complementary to other oxidation reactions of amines, excellent site‐selectivity and functional‐group tolerance are observed, for example, aldehyde and olefins remained untouched.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge the support from the Federal Ministry of Education and Research (BMBF) and the State of Mecklenburg‐Vorpommern. We thank Dr. Wolfgang Baumann, Susann Buchholz, and Dr. Christine Fischer for their excellent analytical support, and Dr. Haijun Jiao, Zhihong Wei, Bianca Wendt, and Dr. Basudev Sahoo (all at LIKAT) for valuable discussions.
W. Li, W. Liu, D. K. Leonard, J. Rabeah, K. Junge, A. Brückner, M. Beller, Angew. Chem. Int. Ed. 2019, 58, 10693.
References
- 1.
- 1a. Murakami M., Ishida N., J. Am. Chem. Soc. 2016, 138, 13759–13769; [DOI] [PubMed] [Google Scholar]
- 1b. Chen F., Wang T., Jiao N., Chem. Rev. 2014, 114, 8613–8661; [DOI] [PubMed] [Google Scholar]
- 1c. Murakami M., Ishida N., Nat. Chem. 2017, 9, 298–299. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Lyons T. W., Sanford M. S., Chem. Rev. 2010, 110, 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. He J., Wasa M., Chan K. S. L., Shao Q., Yu J. Q., Chem. Rev. 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Roque J. B., Kuroda Y., Gottemann L. T., Sarpong R., Science 2018, 361, 171–174; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Xia Y., Lu G., Liu P., Dong G., Nature 2016, 539, 546–550; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Liu J., Qiu X., Huang X., Luo X., Zhang C., Wei J., Pan J., Liang Y., Zhu Y., Qin Q., Song S., Jiao N., Nat. Chem. 2019, 11, 71–77; [DOI] [PubMed] [Google Scholar]
- 3d. Ota E., Wang H., Frye N. L., Knowles R. R., J. Am. Chem. Soc. 2019, 141, 1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Masarwa A., Didier D., Zabrodski T., Schinkel M., Ackermann L., Marek I., Nature 2014, 505, 199–203; [DOI] [PubMed] [Google Scholar]
- 4b. Gozin M., Weisman A., Ben-David Y., Milstein D., Nature 1993, 364, 699–701. [Google Scholar]
- 5. Allpress C. J., Berreau L. M., Coord. Chem. Rev. 2013, 257, 3005–3029. [Google Scholar]
- 6.
- 6a. Stahl S. S., Angew. Chem. Int. Ed. 2004, 43, 3400–3420; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3480–3501; [Google Scholar]
- 6b. Elwell C. E., Gagnon N. L., Neisen B. D., Dhar D., Spaeth A. D., Yee G. M., Tolman W. B., Chem. Rev. 2017, 117, 2059–2107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Esguerra K. V. N., Lumb J. P., Angew. Chem. Int. Ed. 2018, 57, 1514–1518; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1530–1534. [Google Scholar]
- 7.
- 7a. Osberger T. J., Rogness D. C., Kohrt J. T., Stepan A. F., White M. C., Nature 2016, 537, 214–219; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Wendlandt A. E., Suess A. M., Stahl S. S., Angew. Chem. Int. Ed. 2011, 50, 11062–11087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11256–11283. [Google Scholar]
- 8. al Kandari H., Katsumata N., Alexander S., Rasoul M. A., J. Clin. Endocrinol. Metab. 2006, 91, 2821–2826. [DOI] [PubMed] [Google Scholar]
- 9. Basran J., Efimov I., Chauhan N., Thackray S. J., Krupa J. L., Eaton G., Griffith G. A., Mowat C. G., Handa S., Raven E. L., J. Am. Chem. Soc. 2011, 133, 16251–16257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varfaj F., Zulkifli S. N., Park H. G., Challinor V. L., De Voss J. J., Ortiz de Montellano P. R., Drug Metab. Dispos. 2014, 42, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Saliu F., Orlandi M., Bruschi M., ISRN Org. Chem. 2012, 281642; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Suarez-Bertoa R., Saliu F., Bruschi M., Rindone B., Tetrahedron 2012, 68, 8267–8275. [Google Scholar]
- 12. Roque J. B., Kuroda Y., Gottemann L. T., Sarpong R., Nature 2018, 564, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Hruszkewycz D., McCann S., Stahl S., Liquid Phase Aerobic Oxidation Catalysis, Wiley-VCH, Weinheim, 2016, pp. 67–83; [Google Scholar]
- 13b. L. Que, Jr. , Tolman W. B., Nature 2008, 455, 333–340; [DOI] [PubMed] [Google Scholar]
- 13c. Schultz M. J., Sigman M. S., Tetrahedron 2006, 62, 8227–8241; [Google Scholar]
- 13d. Wu Q., Luo Y., Lei A., You J., J. Am. Chem. Soc. 2016, 138, 2885–2888; [DOI] [PubMed] [Google Scholar]
- 13e. Wu K., Huang Z., Ma Y., Lei A., RSC Adv. 2016, 6, 24349–24352. [Google Scholar]
- 14. Schümperli M. T., Hammond C., Hermans I., ACS Catal. 2012, 2, 1108–1117. [Google Scholar]
- 15. Rose J., Castagnoli N., Med. Res. Rev. 1983, 3, 73–88. [DOI] [PubMed] [Google Scholar]
- 16. Leonard N. J., Hay A. S., Fulmer R. W., Gash V. W., J. Am. Chem. Soc. 1955, 77, 439–444. [Google Scholar]
- 17. Legacy C. J., Wang A., O'Day B. J., Emmert M. H., Angew. Chem. Int. Ed. 2015, 54, 14907–14910; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15120–15123. [Google Scholar]
- 18.For a similar copper oxide catalyzed reaction of tri-n-butyl amine see:
- 18a. Wang M., Gu X.-K., Su H.-Y., Lu J.-M., Ma J.-P., Yu M., Zhang Z., Wang F., J. Catal. 2015, 330, 458–464; for photocatalytic cleavage reactions see: [Google Scholar]
- 18b. Zhao Y., Cai S., Li J., Wang D. Z., Tetrahedron 2013, 69, 8129–8131; [Google Scholar]
- 18c. Ji W., Li P., Yang S., Wang L., Chem. Commun. 2017, 53, 8482–8485; for metabolite studies using a Cu catalyst see: [DOI] [PubMed] [Google Scholar]
- 18d. Genovino J., Lutz S., Sames D., Toure B. B., J. Am. Chem. Soc. 2013, 135, 12346–12352. [DOI] [PubMed] [Google Scholar]
- 19. Al-Ghorbani M., Begum B. A., Zabiulla, Mamatha S. V., Khanum S. A., J. Chem. Pharm. Res. 2015, 7, 281–301. [Google Scholar]
- 20. Tsang A. S., Kapat A., Schoenebeck F., J. Am. Chem. Soc. 2016, 138, 518–526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary