

Abstract
Objective
While pulmonary arterial hypertension (PAH) is rare in infants and children, it results in substantial morbidity and mortality. In recent years, prognosis has improved, coinciding with the introduction of new PAH‐targeted therapies, although much of their use in children is off‐label. Evidence to guide the treatment of children with PAH is less extensive than for adults. The goal of this review is to discuss the treatment recommendations for children with PAH, as well as the evidence supporting the use of prostanoids, endothelin receptor antagonists (ERAs), and phosphodiesterase type 5 inhibitors (PDE5i) in this setting.
Data Sources
Nonsystematic PubMed literature search and authors’ expertise.
Study Selection
Articles were selected concentrating on the nitric oxide (NO)‐soluble guanylate cyclase (sGC)‐cyclic guanosine monophosphate (cGMP) pathway in PAH. The methodology of an ongoing study evaluating the sGC stimulator riociguat in children with PAH is also described.
Results
Despite recent medical advances, improved therapeutic strategies for pediatric PAH are needed. The efficacy and tolerability of riociguat in adults with PAH have been well trialed.
Conclusion
The pooling of data across trials, supplemented by registry data, will help to confirm the safety and tolerability of prostanoids, ERAs, and PDE5i in children. Ongoing studies will clarify the place of sGC stimulators in the treatment strategy for pediatric PAH.
Keywords: PAH, pediatrics, riociguat, sGC stimulators
Abbreviations
- 6MWD
6‐Minute walking distance
- AVT
Acute vasoreactivity testing
- BNP
Brain natriuretic peptide
- CCBs
Calcium channel blockers
- cGMP
Cyclic guanosine monophosphate
- CHD
Congenital heart disease
- EMA
European Medicines Agency
- ERAs
Endothelin receptor antagonists
- FDA
Food and Drug Administration
- iPAH
Idiopathic PAH
- mPAP
Mean pulmonary artery pressure
- NO
Nitric oxide
- NT‐proBNP
N‐terminal prohormone of BNP
- PAH
Pulmonary arterial hypertension
- PAWP
Pulmonary arterial wedge pressure
- PDE5i
Phosphodiesterase type 5 inhibitors
- PDEs
Phosphodiesterases
- PH
Pulmonary hypertension
- PVO2
Peak oxygen consumption
- PVR
Pulmonary vascular resistance
- REMS
Risk Evaluation and Mitigation Strategy
- RHC
Right heart catheterization
- SAEs
Serious adverse events
- sGC
Soluble guanylate cyclase
- tid
Three times daily
- WHO FC
World Health Organization functional class
- WSPH
World Symposium on Pulmonary Hypertension
1. INTRODUCTION
Pulmonary hypertension (PH) is a rare condition in infants and children that results in substantial morbidity and mortality.1 According to international guidelines,2 the definition of PH in children (aged ≥ 3 months), as in adults, is a mean pulmonary artery pressure (mPAP) ≥ 25 mm Hg. In the current international classification, five broad subgroups of PH are defined: pulmonary arterial hypertension (PAH) (Group 1), PH due to left heart disease (Group 2), PH due to lung diseases and/or hypoxia (Group 3), chronic thromboembolic pulmonary hypertension (Group 4), and PH with unclear and/or multifactorial mechanisms (Group 5).2, 3 PAH is specifically defined as PH with pulmonary arterial wedge pressure (PAWP) ≤15 mm Hg and pulmonary vascular resistance (PVR) ≥3 Wood units × m2 (≈240 dyn s cm–5), in the absence of lung disease and Group 4 or 5 PH.1, 2, 3 The 6th World Symposium on Pulmonary Hypertension (WSPH) (2018) proposed an mPAP cutoff of >20 mmHg and suggested a new definition for Group 4 to include chronic thromboembolic pulmonary hypertension (CTEPH) and other pulmonary artery obstructions.1, 3
1.1. Presentation of pediatric PH
1.1.1. Epidemiology
The two most common forms of PH in children are PAH (Group 13), which accounts for the majority of cases, and PH associated with lung disease (Group 3), of which the most common form is bronchopulmonary dysplasia.1, 4
The most frequent causes of PAH in children are idiopathic (iPAH), heritable gene defects, and congenital heart disease (CHD); however, connective tissue disease, human immunodeficiency virus, drugs, and portopulmonary hypertension are rare causes in pediatric populations.1, 4 The prevalence of iPAH has been determined as 2.1 to 4.4 cases per million children, with an incidence of 0.5 to 1‐2 cases per million children per year.5, 6, 7, 8, 9 PAH‐CHD was more common, comprising 75% of total PAH cases in an American study,8 with prevalence of 10.1 to 15.6 cases per million and incidence of 1.9 to 2.2 cases per million per year in European studies.5, 6, 7 Importantly, hospitalizations for pediatric PH are increasing, particularly for children with non‐CHD‐related PH.10, 11
1.1.2. Diagnosis and assessment
The diagnosis of PH in children is challenging as the initial symptoms are nonspecific. In infants, these include failure to thrive, tachypnea, and irritability, while in older children, initial symptoms can be exercise intolerance (dyspnea on exertion), with or without chest pain.12, 13 In more severe disease, cyanosis may also be observed. While various algorithms have been suggested for the diagnosis of PAH in children, none of them is universally accepted, and diagnostic practices vary between centers.1, 14 Infants and young children may be unable to undergo all necessary tests, thus making algorithms difficult to apply. Guidelines recommend pulmonary function testing as well as chest and heart imaging (including echocardiography and chest radiography) for the initial assessment of suspected PH.1, 15 Patients with PH should be referred to a specialist center for further evaluation, including right heart catheterization (RHC). RHC measures hemodynamic parameters, including cardiac output, mPAP, and PAWP, from which further parameters (such as PVR) can be calculated to confirm the diagnosis, provide information on prognosis, and guide the choice of therapy.1, 16, 17 Thorough clinical investigation is required to detect all potential causes of PH and diagnose iPAH, as this remains a diagnosis of exclusion.1, 14, 18, 19
Biomarkers, such as brain natriuretic peptide (BNP) and N‐terminal prohormone of BNP (NT‐proBNP), are correlated with hemodynamics and right ventricular function and can be used to assess disease severity and prognosis, and to define treatment goals.20, 21, 22, 23 The prognosis of pediatric PAH varies widely depending on etiology, with better survival in iPAH than in associated PAH.12, 24 For example, the UK Pulmonary Hypertension Service for Children reported 5‐year survival rates of 71.9% in iPAH and 56.9% in associated PAH.25 Patients with CHD whose PAH persists after surgical repair have very poor survival.24, 25 However, a review of 134 children with CHD found no survival difference between four groups of patients with PAH‐CHD (Eisenmenger syndrome; left‐to‐right shunts; coincidental CHD; postoperative PAH).26 Other predictors of survival include World Health Organization functional class (WHO FC), BNP/NT‐proBNP, mean right atrial pressure, PVR, cardiac index, and response to acute vasoreactivity testing (AVT) (see below).27
1.2. Prognosis
The reported prognosis of pediatric PAH has improved in recent years, coinciding with the widespread use of new PAH‐targeted therapies, although few are licensed for use in children. Evidence to guide treatment of children with PAH is less extensive than for adults, partly because of challenges in designing and conducting clinical trials of new therapies in this population. These include strict requirements from regulatory bodies; ethical restrictions; recruitment and retention of patients; creation of suitable formulations for use by children; and selection of suitable endpoints.28, 29 Innovative trial designs, novel endpoints, and incorporation of technology are likely to be required for the full evaluation of drugs in pediatric populations.30, 31
2. CURRENT MANAGEMENT OF PEDIATRIC PAH
2.1. Guideline recommendations
An algorithm for the management of PAH in children was developed by the Pediatric Task Force of the 5th World Symposium on Pulmonary Hypertension (WSPH) in 2013 and updated in 2018 following the 6th WSPH.1, 32 The update emphasizes serial reassessment to determine whether patients are reaching treatment goals (Figure 1).1 The algorithm recommends that supportive therapy, such as diuretics, oxygen, anticoagulation, or digoxin, be considered on an individual basis, although clinical experience suggests that there is limited benefit for children.
Figure 1.
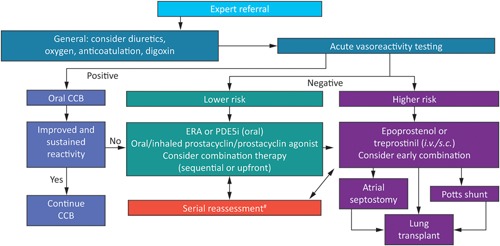
6th WSPH consensus treatment algorithm for the management of pediatric PAH. Reproduced with permission of the ERS 2019. European Respiratory Journal Jan 2019, 53 (1) 1801916; https://doi.org/10.1183/13993003.01916‐20181. CCB, calcium channel blocker; ERA, endothelin receptor antagonist; iv, intravenous; PAH, pulmonary arterial hypertension; PDE5i, phosphodiesterase type 5 inhibitor; sc, subcutaneous; #patients deteriorating or not meeting treatment goals. [Color figure can be viewed at wileyonlinelibrary.com]
In AVT, a vasodilator (eg, inhaled nitric oxide; NO) is administered. A decrease in mPAP by ≥10 mm Hg to a value of <40 mmHg with sustained cardiac output (the Sitbon criteria) identifies patients who are likely to have a good long‐term prognosis when treated with calcium channel blockers (CCBs).1 These children should receive long‐term oral CCBs. Registry data have shown, however, that only 23% of children, judged by their physician to have a positive acute response to AVT, actually received CCBs without additional PAH‐targeted therapy.33 Determination of response to AVT using the Sitbon criteria34 is a more reliable predictor of response to CCBs than physician judgment.
Initiation of PAH‐targeted therapy is based on the level of risk, with low‐risk patients receiving an oral endothelin receptor antagonist (ERA) or phosphodiesterase type 5 inhibitor (PDE5i), or an oral or inhaled prostacyclin analog (prostanoid), with a trend toward combining oral therapies in countries where it is possible. High‐risk patients should receive an injectable prostanoid alone or in combination with other drugs. Early combination therapy, including triple combination therapy, should be considered for high‐risk patients as has previously been reported in adults.35, 36 Those who do not respond to medical therapy may require atrial septostomy, a Potts shunt, or lung transplantation. The ultimate goals of treatment are to improve long‐term outcomes and quality of life. These endpoints are difficult to measure, however, and surrogate endpoints are often used in clinical trials. These include hemodynamic parameters, biomarkers, and exercise capacity.37, 38
This review concentrates on the role of the NO‐soluble guanylate cyclase (sGC)‐cyclic guanosine monophosphate (cGMP) pathway in PAH, therefore data on prostanoids and ERAs are discussed briefly.
2.2. Prostanoids
Endogenous prostacyclin (prostaglandin I2) activates prostacyclin receptors in pulmonary vascular smooth muscle cells and other tissues, increasing production of cyclic adenosine monophosphate, which induces pulmonary vasodilation as well as antithrombotic, antiproliferative, antimitogenic, and immunomodulatory effects.24, 39, 40 Prostacyclin levels are reduced in patients with PAH,41, 42 providing a rationale for treatment with prostacyclin analogs (prostanoids).43 In the form of epoprostenol, prostacyclin has been used since the 1980s for treatment of PAH in adults, and several newer prostanoids have since been developed, including treprostinil and iloprost, as well as the nonprostanoid prostacyclin receptor agonist selexipag.
In an early study in children with PAH, intravenous epoprostenol significantly improved survival in nonresponders to AVT, and in responders who did not improve with the CCB therapy.44 Subsequently, a retrospective review indicated that epoprostenol improved the survival of children with severe PAH.45 The dosing regulation and delivery of epoprostenol are complex, and therefore alternative agents may be preferable for children. A study in pediatric patients with PAH treated with epoprostenol or treprostinil, indicated that, while hemodynamic improvements are not sustained at 5 years, both drugs improved survival compared with historical controls.46 In children with severe PAH, subcutaneous treprostinil has been shown to improve WHO FC, exercise tolerance, and hemodynamic parameters.47 Numerous case studies and small trials suggest that inhaled iloprost may have a role in neonates and children with acute PH, although appropriate dosing is unclear.48 For example, a study in 22 children with iPAH or PAH‐CHD found that inhaled iloprost reduced mPAP, but also reduced forced expiratory volume in 1 second in some children.49
Several prostanoids or compounds acting on the prostanoid pathway are being evaluated in trials with wholly or partly pediatric cohorts with PAH. These include treprostinil (ClinicalTrials.gov: NCT01027949), iloprost (NCT02825160), and selexipag (NCT03492177), while ralinepag is being evaluated in adults with PAH (NCT02279745, NCT03626688).
2.3. ERAs
Endogenous endothelin‐1 (ET‐1) acts via endothelin A and B receptors to cause vasoconstriction and promote the proliferation of smooth muscle cells in the pulmonary vasculature.43 Endothelin receptors are upregulated in patients with PAH.50 ERAs, including bosentan, ambrisentan, and macitentan, act by antagonizing the activity of ET‐1. Ambrisentan is a selective ETA receptor antagonist: macitentan and bosentan are dual ETA and ETB receptor antagonists. In children with PAH, bosentan has been shown to improve exercise capacity and functional status.51, 52, 53 The efficacy and tolerability of bosentan are maintained during long‐term treatment.54, 55 In practice, bosentan is often combined with PDE5i, mainly sildenafil, although there is no clinical trial evidence to support this combination.
In the USA and several other countries, bosentan is licensed for use in children aged ≥3 years, while in Europe, bosentan is approved for use in children aged ≥1 year. In many countries, bosentan is available as a dispersible pediatric formulation, with similar tolerability to the adult formulation.53, 55 Bosentan may be better tolerated in children than in adults, particularly with regard to raised liver aminotransferases.56 A small study in children suggested that ambrisentan is well tolerated with some evidence of efficacy.57 Studies of children with PAH being treated with ambrisentan (NCT01342952 and NCT01406327) and another ERA, macitentan (NCT02932410 and NCT00667823) are ongoing.
2.4. Drugs acting on the NO‐sGC‐cGMP pathway
2.4.1. Role of the NO‐sGC‐cGMP pathway in PAH
NO signaling plays a critical role in vasodilation and vascular remodeling in the pulmonary circulation.58 NO stimulates production of sGC, which in turn catalyzes the conversion of guanosine triphosphate to cGMP, the primary mediator of NO signaling, leading to vasorelaxation. Disruption of the NO‐sGC‐cGMP pathway is central to the pathogenesis of PAH, with reduced NO levels present in the lungs of affected patients.58
2.4.2. PDE5 inhibitors
cGMP is deactivated by phosphodiesterases (PDEs), notably PDE5. PDE5i such as sildenafil and tadalafil prevent the breakdown of cGMP.58 The consequent increase in intracellular cGMP levels results in vasodilation and inhibition of vascular smooth muscle proliferation.59
Sildenafil, licensed for use in adults with PAH in Europe and the USA, has been extensively studied in children and adolescents with PAH (key studies are summarized in Table 1). The largest trial was STARTS‐1, in which 235 children and adolescents (age, 1–17 years) with PAH were randomized double‐blind to sildenafil at low (10 mg three times daily [tid]), medium (10–40 mg tid), or high (20–80 mg tid) dose, or placebo for 16 weeks.60 The primary endpoint was the percentage change in peak oxygen consumption (PVO2) for sildenafil vs placebo. The difference in PVO2 between sildenafil and placebo was not significant, with a mean (±SE) treatment difference of 7.7 ± 4.0% (P = .056). PVO2, WHO FC, and hemodynamic parameters did, however, improve in some dose groups vs placebo. Sildenafil was generally well tolerated, although two treatment‐related serious adverse events (SAEs) occurred in the high‐dose group. The risk‐benefit profile therefore favored medium dosing. Patients who completed STARTS‐1 were eligible to enter a long‐term open‐label extension (STARTS‐2), in which those who had previously received sildenafil continued their previous dose, while those who had received placebo were randomized to high‐, medium‐, or low‐dose sildenafil.61 The result showed an increase in mortality with high‐dose sildenafil compared with low and medium doses. Analyses adjusted for baseline characteristics, including PAH etiology, PVR index, and right atrial pressure, however, produced reduced hazard rations61 and cast doubt on the relationship between dose and survival. The European Medicines Agency (EMA) subsequently added a warning to the labeling for sildenafil (originally approved in 2011) to avoid use of the high dose. By contrast the US Food and Drug Administration (FDA) released a strong warning in August 2012 against the (chronic) use of sildenafil for pediatric patients with PAH (https://www.fda.gov/Drugs/DrugSafety/ucm317123.htm). In 2014, however, the FDA clarified this warning, stating that there may be situations in which the risk‐benefit profile may be acceptable in individual children, although it is still not recommended (https://www.fda.gov/drugs/drugsafety/ucm390876.htm). Additional pediatric trials of sildenafil in PAH have been performed but are not discussed here because of low patient numbers and/or lack of placebo control.62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75
Table 1.
Summary of key studies evaluating sildenafil for the treatment of children and adolescents with PAH
| Study | Design | Treatment arms | Key findings |
|---|---|---|---|
| Sabri et al76 | Randomized, open‐label study in 42 infants (age, 3‐24 mo) with a large septal defect and PAH | Sildenafil 1–3 mg/kg/day pre‐ and post surgery | • No significant difference in pulmonary artery‐to‐aortic pressure ratio and sPAP in the first 48 h after surgery |
| Tadalafil 1 mg/kg/day pre‐ and post surgery | • No significant differences in ICU stay, mechanical ventilation time, clinical findings of low cardiac output state, and echocardiographic data | ||
| • Both treatments were well tolerated | |||
| STARTS‐160 | Randomized, placebo‐controlled trial in 235 treatment‐naïve children (age, 1‐17 y) weighing ≥8 kg with idiopathic or hPAH, or PAH associated with connective tissue disease or CHD | Low‐dose sildenafil (10 mg tid) | • Estimated mean change in PVO2 (primary endpoint) for sildenafil (pooled) vs placebo was 7.7% (95% CI, −0.2‐15.6%; P = .056) |
| Medium‐dose sildenafil (10–40 mg tid) | • PVO2, FC, and hemodynamics improved with medium and high doses vs placebo; low‐dose sildenafil was ineffective | ||
| High‐dose sildenafil (20–80 mg tid) | • Most adverse events were mild to moderate in severity | ||
| Placebo | |||
| STARTS‐1 (sub‐analysis)77 | Post hoc analysis of 48 children (age, 1‐17 y) with PAH and Down syndrome | Low‐dose sildenafil (10 mg tid) | • Sildenafil had no effect on PVRI and mPAP |
| Medium‐dose sildenafil (10–40 mg tid) | • Sildenafil was well tolerated in children with Down syndrome | ||
| High‐dose sildenafil (20–80 mg tid) | |||
| Placebo | |||
| STARTS‐261 | Long‐term open‐label extension in 220 children who completed STARTS‐1 | Low‐dose sildenafil (10 mg tid) | • Deaths reported in 37 patients, of whom 28 had idiopathic or hPAH |
| Medium‐dose sildenafil (10–40 mg tid) | • Deaths more likely in patients in FC III/IV (38%) than the overall cohort (15%), and in patients with worse baseline hemodynamics | ||
| High‐dose sildenafil (20–80 mg tid) | • 3‐y survival rates: 94%, 93%, and 88% for low‐, medium‐, and high‐dose sildenafil, respectively (HR [95% CI] = 3.95 [1.46‐10.65] for high vs low and 1.92 [0.65‐5.65] for medium vs low) | ||
| Xia et al78 | Open‐label, randomized, controlled study in 50 children (age, 2 mo‐2 y) with high‐altitude heart disease and severe PAH | Sildenafil 1 mg/kg/day | • Sildenafil reduced mPAP and increased arterial pO2, cardiac output, cardiac index, and oxygenation index vs controls (all P < .05) |
| Conventional therapy (control) | • No significant changes in mean arterial pressure, routine blood parameters and blood biochemical parameters, and no major adverse event | ||
| Humpl et al79 | Open‐label pilot study in 14 children (age, 5.3‐18 y) with symptomatic PAH who could reliably perform a 6‐min walk test | Sildenafil 0.25–0.5 mg/kg qid (0.1 mg/kg qid in 1 patient) | • 6MWD (primary endpoint) increased from 278 m to 443 m at 6 mo (P = .02) and 432 m at 12 mo (P = .005) |
| • The difference between 6 and 12 mo was not significant | |||
| • Median mPAP decreased from 60 mm Hg to 50 mm Hg (P = .014) | |||
| • Median PVR decreased from 15 Wood units m2 to 12 Wood units m2 (P = .024) | |||
| Karatza et al80 | Case series including one child with idiopathic PAH (age, 13 y) and two children with PAH‐CHD (ages, 6 and 10 y) | 0.5–2.0 mg/kg 4‐hourly | • Increased exercise capacity and FC in all three patients |
| • 6MWD increased by 74%, 75%, and 25% | |||
| • Oxyhemoglobin saturations increased from 79%, 97%, and 80% to 93%, 100%, and 93%, respectively. | |||
| • There were no side effects and no fall in systemic blood pressure | |||
| Mourani et al81 | Retrospective chart review of 25 children (age, <2 y) with chronic lung disease and PH | 1.5–8.0 mg/kg/day | • Hemodynamic improvement in 88% of patients (median follow‐up: 40 d) (primary endpoint) a |
| • Eleven of 13 patients with interval estimates of systolic pulmonary artery pressure with echocardiogram showed clinically significant reductions in PH | |||
| • Five deaths (20%) | |||
| • Adverse events leading to cessation or interruption of therapy in two patients | |||
| Lunze et al82 | Open‐label pilot study in 11 patients (median age, 12.9 y; range, 5.5‐54.7 y) | Sildenafil (mean dose, 2.1 mg/kg) + bosentan (mean dose, 2.3 mg/kg) | • No major liver‐ or blood pressure‐related side effects |
| • FC generally improved by one NYHA class, with increased transcutaneous oxygen saturation (89.9–92.3%; P = .037), maximum oxygen uptake (18.1–22.8 mL/kg min; P = .043), and 6MWD (351–451 m; P = .039) | |||
| • mPAP decreased (62‐46 mm Hg; P = .041) |
Abbreviations: 6MWD, 6‐minute walking distance; CHD, congenital heart disease; CI, confidence interval; FC, functional class; hPAH, heritable pulmonary arterial hypertension; HR, hazard ratio; ICU, intensive care unit; mPAP, mean pulmonary artery pressure; NYHA, New York Heart Association; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; pO2, partial pressure of oxygen; PVO2, peak oxygen consumption; PVR, pulmonary vascular resistance; PVRI, pulmonary vascular resistance index; qid, four times daily; sPAP, systolic pulmonary artery pressure; tid, three times daily.
Defined as ≥20% decrease in the ratio of pulmonary to systemic systolic arterial pressure or improvement in the degree of septal flattening assessed by serial echocardiograms.
Tadalafil has also been evaluated in children with PAH.76, 83, 84, 85, 86, 87 These open‐label, uncontrolled trials, some of which included patients previously treated with sildenafil,83, 84, 85 suggested that tadalafil improves hemodynamics and exercise capacity, and is generally well tolerated (Table 2). While tadalafil is licensed for use in adults with PAH in Europe and the USA, it is not licensed for use in children in either region.
Table 2.
Summary of key published studies evaluating tadalafil for the treatment of children and adolescents with PAH
| Study | Design | Treatment arms | Key findings |
|---|---|---|---|
| Takatsuki et al84 | Retrospective chart review of 33 children (age, 4–18 y) with PAH a | Tadalafil (mean dose, 1.0 mg/kg/day) | • In 14 patients who underwent repeat catheterization, improvements after switching from sildenafil were observed in mPAP (53.2 vs 47.4 mm Hg; P < .05) and PVRI (12.2 vs 10.6; P < .05) |
| • In four treatment‐naïve patients, clinical improvement was noted | |||
| • Side effect profiles were similar in treatment‐naïve and switched patients | |||
| • Two patients discontinued tadalafil due to adverse events (migraine; allergic reaction) | |||
| Sabri et al85 | Prospective open‐label study of 18 children and young adults (age, 4–24 y) with PAH previously receiving sildenafil | Tadalafil 1 mg/kg/day (maximum: 40 mg/day) | • No change in FC after 6 wk, despite some symptom improvement |
| • Mean oxygen saturation improved compared with baseline before and after 6‐minute walk test (P = .005 and .036, respectively) | |||
| • Mean 6MWD improved (P = .005) | |||
| • No significant side effects of tadalafil | |||
| Shiva et al83 | Prospective open‐label study of 25 children (age, 2 mo‐5 y) with PAH | Tadalafil 1 mg/kg/day | • Significant improvements in mPAP at all four monthly visits (P < .01), including in six patients who had previously received sildenafil for >6 mo |
| • Tadalafil was generally safe and well tolerated; nausea was the most frequently reported adverse event (n = 3) | |||
| Sabri et al76 | Randomized, open‐label study in 42 Infants (age, 3–24 mo) with a large septal defect and PAH | Sildenafil | • No significant difference in pulmonary artery‐to‐aortic pressure ratio and sPAP in the first 48 h after surgery |
| 1–3 mg/kg/day pre‐ and post surgery | • No significant differences in ICU stay, mechanical ventilation time, clinical findings of low cardiac output state, and echocardiographic data | ||
| Tadalafil 1 mg/kg/day pre‐ and post surgery | • Both treatments were well tolerated |
Abbreviations: 6MWD, 6‐minute walking distance; FC, functional class; ICU, intensive care unit; mPAP, mean pulmonary artery pressure; PAH, pulmonary arterial hypertension; PVRI, pulmonary vascular resistance index; sPAP, systolic pulmonary artery pressure.
29 patients switched from sildenafil (mean, 3.4 mg/kg/day) to tadalafil.
Ongoing trials of sildenafil in children with PAH include trials in neonates (ClinicalTrials.gov: NCT01720524), children with β‐thalassemia associated with PH (NCT03402191), and Japanese children (NCT03364244, NCT01642407). Pharmacokinetic (NCT01484431) and safety and efficacy (NCT01824290) studies of tadalafil are also under way.
Combination therapy that targets multiple pathways in the pathogenesis of PAH is an attractive option to improve the efficacy of treatment.1, 2, 88 It may be prescribed upfront, or sequentially with the addition of one or two drugs to initial monotherapy, but there are no definitive trials in children. Several studies35, 89, 90, 91, 92 have suggested that combination therapy may improve hemodynamic parameters and exercise capacity and reduce clinical worsening in adults with PAH. For example, patients in the placebo‐controlled AMBITION study were randomized to initial therapy with ambrisentan, tadalafil, or both.92 Upfront combination therapy significantly reduced the risk of clinical failure events compared with either agent as monotherapy. It also significantly improved exercise capacity, NT‐proBNP, and clinical response compared with the pooled monotherapy arms. In a meta‐analysis of 17 studies (n = 4095), combination therapy was associated with a significant reduction in the risk of clinical worsening compared with monotherapy.93 A retrospective case review of data from three major referral centers for pediatric PAH found that combination therapy was independently associated with improved survival compared with monotherapy.94
In the 6th WSPH treatment algorithm, PDE5i or ERA are considered first‐line treatments in patients with lower‐risk PAH, with the option of upfront or sequential combination of both classes, especially in those who deteriorate on ERA or PDE5i alone.1 Combination therapy (parenteral prostanoid plus ERA/PDE5i) should be considered early in higher‐risk patients with PAH.1 At present, there is no clear evidence as to which combination or strategy is optimal.93 Data from the Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) registry showed that treatment was initiated with 1, 2, or 3 PAH‐targeted drugs in 51.6%, 18.0%, and 2.3% of patients, respectively, but there are no robust data on outcomes of upfront vs sequential combination therapy in children.95
2.4.3. Riociguat
Riociguat, the first member of the drug class of sGC stimulators, acts by directly stimulating sGC, independent of NO, and by increasing the sensitivity of sGC to NO, increasing cGMP levels.96, 97, 98 The ability of riociguat to stimulate sGC independently of NO means that it may retain its efficacy under conditions of NO depletion, such as PAH, in which intracellular concentrations of cGMP are low and the effectiveness of PDE5i may be reduced.99, 100, 101
The efficacy and tolerability of riociguat in patients with PAH were demonstrated in the double‐blind, placebo‐controlled Phase 3 PATENT‐1 study.102 Patients (n = 443) were randomized to placebo; riociguat individually dose‐adjusted up to 2.5 mg tid; or an exploratory arm in which riociguat was capped at 1.5 mg tid. At week 12, significant improvements in 6‐minute walking distance (6MWD [the primary endpoint]) and several secondary endpoints, were seen with riociguat 2.5 mg tid‐maximum compared with placebo, and riociguat was generally well tolerated. On the basis of PATENT‐1, riociguat is indicated for the treatment of adults with PAH. Patients who completed PATENT‐1 were eligible for a long‐term, open‐label extension (PATENT‐2) in which all patients (n = 396) received riociguat 2.5 mg tid‐maximum.103 The improvements in 6MWD and WHO FC observed in PATENT‐1 were maintained after 2 years of riociguat treatment, with survival rates at 1 and 2 years of 91% and 83%, respectively. The RESPITE study reported that switching to riociguat was effective and generally well tolerated in patients with PAH who had experienced an inadequate clinical response with PDE5i treatment.104 A second study of switching from PDE5i to riociguat (REPLACE) is currently recruiting105 (ClinicalTrials.gov: NCT02891850). Riociguat is contraindicated in pregnant women based on reproductive toxicity and placental transfer in animal studies.106, 107 In the USA, riociguat is available to female patients only through a restricted program called the Adempas Risk Evaluation and Mitigation Strategy (REMS).107, 108, 109
Riociguat has not yet been evaluated in children. The PATENT‐CHILD study (Clinical Trials.gov: NCT02562235) (Bayer AG, data on file), an open‐label, single‐arm, dose‐adjustment study, is in progress to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of riociguat in children and adolescents with PAH. At least 20 patients aged 6–17 years with WHO FC I‐III PAH and receiving treatment with ERAs and/or prostanoids for ≥12 weeks before the baseline visit, will be enrolled. Concomitant use of riociguat and PDE5i is contraindicated106, 107 and is therefore not permitted. Pretreatment with sildenafil or tadalafil is, however, allowed up to 1 day and 3 days, respectively, before starting riociguat. During the washout period, patients should be in a stable clinical condition and receiving ERAs and/or prostanoids. PATENT‐CHILD consists of a 24‐week main phase, followed by an optional long‐term extension phase. Following safety review of an initial group of patients aged ≥ 12 years by an independent Data Monitoring Committee, recruitment has been opened to the 6–11 years of age group. Patients will receive riociguat as film‐coated tablets, adjusted to doses of 1.0–2.5 mg tid, or an oral suspension, providing equivalent exposure to adult doses of 0.5 mg–2.5 mg tid.
Growth and development in children entail constant hormonal and metabolic changes, necessitating thorough assessments throughout clinical trials in PAH.1 In preclinical studies, riociguat induced alterations to bone morphology in juvenile and adolescent rats (Bayer AG, data on file). These findings were seen at 3–4 times greater exposure than in clinical use, were reversible, and were not considered prohibitive for the initiation of PATENT‐CHILD (Bayer AG, data on file). To assess bone safety during the study, a bone X‐ray of the left hand will be performed at baseline, at week 24, and every 12 months until growth plates are closed, and bone age and morphology will be determined centrally by a specialist. Safety (the primary endpoint) will also be assessed in terms of adverse events and SAEs, vital signs, and laboratory parameters. Selection of efficacy endpoints for children with PAH is difficult and is a matter of debate among experts.1, 110 Importantly, the demonstration of efficacy in terms of 6MWD in adults cannot be directly translated to the pediatric population. Pediatric‐specific biomarkers, such as functional classification and NT‐proBNP, have been evaluated,1 and are exploratory efficacy endpoints (in addition to 6MWD) in PATENT‐CHILD.
3. CONCLUSIONS
Diagnosis and management of pediatric PAH are challenging and should be conducted only in expert centers. The introduction of prostanoids, ERAs, and PDE5i has coincided with an improvement in the reported prognosis of pediatric PAH, and guidelines recommend use of these agents, alone or in combination, for patients who do not respond to acute vasoreactivity testing. Despite these improvements, the prognosis in children with nonvasoreactive PAH remains poor. There is therefore a need for new therapies and regimens. Data from clinical trials, both for monotherapy and for agents added to standard of care, are now becoming available. Pooling of data across trials, supplemented by registry data, will help to confirm the safety and tolerability of these agents in children. Older strategies based on therapy escalation at the time of clinical worsening, should be challenged by investigating switching and combination therapy in clinical trials and registries. There are limitations to using RHC in children. Innovative efficacy endpoints, such as those that combine measures of improvement and worsening, should be explored. Registry data will be important in the validation of composite disease progression endpoints.
The PATENT‐CHILD trial is in progress to assess the pharmacokinetics, safety, and exploratory efficacy of riociguat in children and adolescents with PAH. Until results from PATENT‐CHILD are available, we strongly recommend that riociguat should be given to children only when participating in a clinical trial, and that clinicians should not use riociguat empirically in pediatric patients.
CONFLICTS OF INTEREST
MB is a Consultant for Actelion Pharmaceuticals, Arena, Bayer AG, GlaxoSmithKline, Eli Lilly, and Pfizer; and has received grant support from Actelion and Bayer AG, and personal fees from Actelion Pharmaceuticals, Arena, GlaxoSmithKline, Eli Lilly, Pfizer, and Bayer Healthcare.
The University of Colorado receives fees for DDI to be a consultant for and perform research studies for Actelion Pharmaceuticals, Bayer AG, Eli Lilly, and United Therapeutics. DDI receives grant funding from the National Institutes of Health and the FDA.
DB is a consultant or steering committee member for Actelion Pharmaceuticals, Bayer AG, Novartis, and Eli Lilly.
MG serves on an advisory board for Bayer AG and receives lecture fees: from Pfizer, MSD, and Actelion Pharmaceuticals.
SM serves on advisory boards or steering committees for Bayer AG and Actelion Pharmaceuticals.
ACKNOWLEDGMENTS
Funding for the PATENT‐CHILD study was provided by Bayer AG, Berlin, Germany and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Medical writing support was provided by Adelphi Communications Ltd (Bollington, UK), which was in accordance with Good Publication Practice (GPP3) guidelines and funded by Bayer AG (Muellerstrasse 178, 13342 Berlin, Germany).
Beghetti M, Gorenflo M, Ivy DD, Moledina S, Bonnet D. Treatment of pediatric pulmonary arterial hypertension: a focus on the NO‐sGC‐cGMP pathway. Pediatric Pulmonology. 2019;54:1516‐1526. 10.1002/ppul.24442
References
REFERENCES
- 1. Rosenzweig EB, Abman SH, Adatia I, et al. Paediatric pulmonary arterial hypertension: pdates on definition, classification, diagnostics and management. Eur Respir J. 2019;53:1801916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46:903‐975. [DOI] [PubMed] [Google Scholar]
- 3. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53:1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berger RM, Beghetti M, Humpl T, et al. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet. 2012;379:537‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moledina S, Hislop AA, Foster H, Schulze‐Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart. 2010;96:1401‐1406. [DOI] [PubMed] [Google Scholar]
- 6. van Loon RL, Roofthooft MT, Hillege HL, et al. Pediatric pulmonary hypertension in The Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation. 2011;124:1755‐1764. [DOI] [PubMed] [Google Scholar]
- 7. del Cerro Marin MJ, Sabate Rotes A, Rodriguez Ogando A, et al. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med. 2014;190:1421‐1429. [DOI] [PubMed] [Google Scholar]
- 8. Li L, Jick S, Breitenstein S, Hernandez G, Michel A, Vizcaya D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm Circ. 2017;7:126‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saji T. Update on pediatric pulmonary arterial hypertension. Differences and similarities to adult disease. Circ J. 2013;77:2639‐2650. [DOI] [PubMed] [Google Scholar]
- 10. Frank DB, Crystal MA, Morales DL, et al. Trends in pediatric pulmonary hypertension‐related hospitalizations in the United States from 2000‐2009. Pulm Circ. 2015;5:339‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maxwell BG, Nies MK, Ajuba‐Iwuji CC, Coulson JD, Romer LH. Trends in hospitalization for pediatric pulmonary hypertension. Pediatrics. 2015;136:241‐250. [DOI] [PubMed] [Google Scholar]
- 12. Vorhies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Paediatr Drugs. 2014;16:43‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53:1801904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beghetti M, Berger RM, Schulze‐Neick I, et al. Diagnostic evaluation of paediatric pulmonary hypertension in current clinical practice. Eur Respir J. 2013;42:689‐700. [DOI] [PubMed] [Google Scholar]
- 15. Latus H, Kuehne T, Beerbaum P, et al. Cardiac MR and CT imaging in children with suspected or confirmed pulmonary hypertension/pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart. 2016;102(Suppl 2):ii30‐ii35. [DOI] [PubMed] [Google Scholar]
- 16. Rosenkranz S, Preston IR. Right heart catheterisation: Best practice and pitfalls in pulmonary hypertension. Eur Respir Rev. 2015;24:642‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beghetti M, Schulze‐Neick I, Berger RM, et al. Haemodynamic characterisation and heart catheterisation complications in children with pulmonary hypertension: Insights from the global TOPP registry (tracking outcomes and practice in paediatric pulmonary hypertension). Int J Cardiol. 2016;203:325‐330. [DOI] [PubMed] [Google Scholar]
- 18. Pfarr N, Fischer C, Ehlken N, et al. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Respir Res. 2013;14:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozlik‐Feldmann R, Hansmann G, Bonnet D, Schranz D, Apitz C, Michel‐Behnke I. Pulmonary hypertension in children with congenital heart disease (PAH‐CHD, PPHVD‐CHD). Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart. 2016;102(Suppl 2):ii42‐ii48. [DOI] [PubMed] [Google Scholar]
- 20. Pattathu J, Gorenflo M, Hilgendorff A, et al. Genetic testing and blood biomarkers in paediatric pulmonary hypertension. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart. 2016;102(Suppl 2):ii36‐ii41. [DOI] [PubMed] [Google Scholar]
- 21. Kheyfets VO, Dunning J, Truong U, Ivy DD, Hunter KA, Shandas R. Assessment of N‐terminal prohormone B‐type natriuretic peptide as a measure of vascular and ventricular function in pediatric pulmonary arterial hypertension. Pulm Circ. 2015;5:658‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ploegstra MJ, Douwes JM, Roofthooft MT, Zijlstra WM, Hillege HL, Berger RM. Identification of treatment goals in paediatric pulmonary arterial hypertension. Eur Respir J. 2014;44:1616‐1626. [DOI] [PubMed] [Google Scholar]
- 23. Takatsuki S, Wagner BD, Ivy DD. B‐type natriuretic peptide and amino‐terminal pro‐B‐type natriuretic peptide in pediatric patients with pulmonary arterial hypertension. Congenit Heart Dis. 2012;7:259‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frank BS, Ivy DD. Diagnosis evaluation and treatment of pulmonary arterial hypertension in children. Children (Basel). 2018;5:E44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK pulmonary hypertension service for children 2001‐2006. Heart. 2009;95:312‐317. [DOI] [PubMed] [Google Scholar]
- 26. Zijlstra WM, Douwes JM, Ploegstra MJ, et al. Clinical classification in pediatric pulmonary arterial hypertension associated with congenital heart disease. Pulm Circ. 2016;6:302‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ploegstra MJ, Zijlstra WM, Douwes JM, Hillege HL, Berger RM. Prognostic factors in pediatric pulmonary arterial hypertension: A systematic review and meta‐analysis. Int J Cardiol. 2015;184:198‐207. [DOI] [PubMed] [Google Scholar]
- 28. Beghetti M, Berger RM. The challenges in paediatric pulmonary arterial hypertension. Eur Respir Rev. 2014;23:498‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joseph PD, Craig JC, Caldwell PH. Clinical trials in children. Br J Clin Pharmacol. 2015;79:357‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sable CA, Ivy DD, Beekman RH 3rd, et al. 2017 ACC/AAP/AHA health policy statement on opportunities and challenges in pediatric drug development: Learning from sildenafil. J Am Coll Cardiol. 2017;70:495‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torok RD, Li JS, Kannankeril PJ, et al. Recommendations to enhance pediatric cardiovascular drug development: report of a multi‐stakeholder think tank. J Am Heart Assoc. 2018;7:e007283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol. 2013;62:D117‐D126. [DOI] [PubMed] [Google Scholar]
- 33. Douwes JM, Humpl T, Bonnet D, et al. Acute vasodilator response in pediatric pulmonary arterial hypertension: Current clinical practice from the TOPP registry. J Am Coll Cardiol. 2016;67:1312‐1323. [DOI] [PubMed] [Google Scholar]
- 34. Sitbon O, Humbert M, Jais X, et al. Long‐term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105‐3111. [DOI] [PubMed] [Google Scholar]
- 35. Sitbon O, Jais X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43:1691‐1697. [DOI] [PubMed] [Google Scholar]
- 36. Sitbon O, Sattler C, Bertoletti L, et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J. 2016;47:1727‐1736. [DOI] [PubMed] [Google Scholar]
- 37. McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D73‐D81. [DOI] [PubMed] [Google Scholar]
- 38. Galie N, McLaughlin VV, Rubin LJ, Simonneau G. Improving patient outcomes in pulmonary hypertension. Eur Respir Rev. 2015;24:550‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ivy D. Pulmonary hypertension in children. Cardiol Clin. 2016;34:451‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2018;53:1801889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70‐75. [DOI] [PubMed] [Google Scholar]
- 42. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925‐1932. [DOI] [PubMed] [Google Scholar]
- 43. Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71:73‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation. 1999;99:1197‐1208. [DOI] [PubMed] [Google Scholar]
- 45. Yung D, Widlitz AC, Rosenzweig EB, Kerstein D, Maislin G, Barst RJ. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation. 2004;110:660‐665. [DOI] [PubMed] [Google Scholar]
- 46. Siehr SL, Ivy DD, Miller‐Reed K, Ogawa M, Rosenthal DN, Feinstein JA. Children with pulmonary arterial hypertension and prostanoid therapy: Long‐term hemodynamics. J Heart Lung Transplant. 2013;32:546‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levy M, Del Cerro MJ, Nadaud S, et al. Safety, efficacy and management of subcutaneous treprostinil infusions in the treatment of severe pediatric pulmonary hypertension. Int J Cardiol. 2018;264:153‐157. [DOI] [PubMed] [Google Scholar]
- 48. Mulligan C, Beghetti M. Inhaled iloprost for the control of acute pulmonary hypertension in children: a systematic review. Pediatr Crit Care Med. 2012;13:472‐480. [DOI] [PubMed] [Google Scholar]
- 49. Ivy DD, Doran AK, Smith KJ, et al. Short‐ and long‐term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davie N, Haleen SJ, Upton PD, et al. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. 2002;165:398‐405. [DOI] [PubMed] [Google Scholar]
- 51. Barst RJ, Ivy D, Dingemanse J, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372‐382. [DOI] [PubMed] [Google Scholar]
- 52. Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart. 2006;92:664‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Beghetti M, Haworth SG, Bonnet D, et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE‐1 study. Br J Clin Pharmacol. 2009;68:948‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hislop AA, Moledina S, Foster H, Schulze‐Neick I, Haworth SG. Long‐term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur Respir J. 2011;38:70‐77. [DOI] [PubMed] [Google Scholar]
- 55. Berger RM, Haworth SG, Bonnet D, et al. FUTURE‐2: Results from an open‐label, long‐term safety and tolerability extension study using the pediatric formulation of bosentan in pulmonary arterial hypertension. Int J Cardiol. 2016;202:52‐58. [DOI] [PubMed] [Google Scholar]
- 56. Beghetti M, Hoeper MM, Kiely DG, et al. Safety experience with bosentan in 146 children 2‐11 years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance program. Pediatr Res. 2008;64:200‐204. [DOI] [PubMed] [Google Scholar]
- 57. Takatsuki S, Rosenzweig EB, Zuckerman W, Brady D, Calderbank M, Ivy DD. Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr Pulmonol. 2013;48:27‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tonelli AR, Haserodt S, Aytekin M, Dweik RA. Nitric oxide deficiency in pulmonary hypertension: Pathobiology and implications for therapy. Pulm Circ. 2013;3:20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5:689‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barst RJ, Ivy DD, Gaitan G, et al. A randomized, double‐blind, placebo‐controlled, dose‐ranging study of oral sildenafil citrate in treatment‐naive children with pulmonary arterial hypertension. Circulation. 2012;125:324‐334. [DOI] [PubMed] [Google Scholar]
- 61. Barst RJ, Beghetti M, Pulido T, et al. STARTS‐2: long‐term survival with oral sildenafil monotherapy in treatment‐naive pediatric pulmonary arterial hypertension. Circulation. 2014;129:1914‐1923. [DOI] [PubMed] [Google Scholar]
- 62. Apitz C, Reyes JT, Holtby H, Humpl T, Redington AN. Pharmacokinetic and hemodynamic responses to oral sildenafil during invasive testing in children with pulmonary hypertension. J Am Coll Cardiol. 2010;55:1456‐1462. [DOI] [PubMed] [Google Scholar]
- 63. Bigdelian H, Sedighi M. The role of preoperative sildenafil therapy in controlling of postoperative pulmonary hypertension in children with ventricular septal defects. J Cardiovasc Thorac Res. 2017;9:179‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De A, Shah P, Szmuszkovicz J, Bhombal S, Azen S, Kato RM. A retrospective review of infants receiving sildenafil. J Pediatr Pharmacol Ther. 2018;23:100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Douwes JM, Roofthooft MT, Van Loon RL, et al. Sildenafil add‐on therapy in paediatric pulmonary arterial hypertension, experiences of a national referral centre. Heart. 2014;100:224‐230. [DOI] [PubMed] [Google Scholar]
- 66. Fender RA, Hasselman TE, Wang Y, Harthan AA. Evaluation of the tolerability of intermittent intravenous sildenafil in pediatric patients with pulmonary hypertension. J Pediatr Pharmacol Ther. 2016;21:419‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Juarez Olguin H, Camacho Reyes L, Roldan Arce A, Calderon Guzman D. Treatment of pulmonary arterial hypertension using extemporaneous formulation of sildenafil in Mexican children. Pediatr Cardiol. 2015;36:1019‐1023. [DOI] [PubMed] [Google Scholar]
- 68. Olguin HJ, Martinez HO, Perez CF, et al. Pharmacokinetics of sildenafil in children with pulmonary arterial hypertension. World J Pediatr. 2017;13:588‐592. [DOI] [PubMed] [Google Scholar]
- 69. Oliveira EC, Amaral CF. Sildenafil in the management of idiopathic pulmonary arterial hypertension in children and adolescents. J Pediatr (Rio J). 2005;81:390‐394. [DOI] [PubMed] [Google Scholar]
- 70. Palma G, Giordano R, Russolillo V, et al. Sildenafil therapy for pulmonary hypertension before and after pediatric congenital heart surgery. Tex Heart Inst J. 2011;38:238‐242. [PMC free article] [PubMed] [Google Scholar]
- 71. Raposo‐Sonnenfeld I, Otero‐Gonzalez I, Blanco‐Aparicio M, Ferrer‐Barba A, Medrano‐Lopez C. Treatment with sildenafil, bosentan, or both in children and young people with idiopathic pulmonary arterial hypertension and eisenmenger's syndrome. Rev Esp Cardiol. 2007;60:366‐372. [PubMed] [Google Scholar]
- 72. Tan K, Krishnamurthy MB, O'Heney JL, Paul E, Sehgal A. Sildenafil therapy in bronchopulmonary dysplasia‐associated pulmonary hypertension: a retrospective study of efficacy and safety. Eur J Pediatr. 2015;174:1109‐1115. [DOI] [PubMed] [Google Scholar]
- 73. Trottier‐Boucher MN, Lapointe A, Malo J, et al. Sildenafil for the treatment of pulmonary arterial hypertension in infants with bronchopulmonary dysplasia. Pediatr Cardiol. 2015;36:1255‐1260. [DOI] [PubMed] [Google Scholar]
- 74. Zeng WJ, Lu XL, Xiong CM, et al. The efficacy and safety of sildenafil in patients with pulmonary arterial hypertension associated with the different types of congenital heart disease. Clin Cardiol. 2011;34:513‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang ZN, Jiang X, Zhang R, et al. Oral sildenafil treatment for eisenmenger syndrome: a prospective, open‐label, multicentre study. Heart. 2011;97:1876‐1881. [DOI] [PubMed] [Google Scholar]
- 76. Sabri MR, Bigdelian H, Hosseinzadeh M, Ahmadi A, Ghaderian M, Shoja M. Comparison of the therapeutic effects and side effects of tadalafil and sildenafil after surgery in young infants with pulmonary arterial hypertension due to systemic‐to‐pulmonary shunts. Cardiol Young. 2017;27:1686‐1693. [DOI] [PubMed] [Google Scholar]
- 77. Beghetti M, Rudzinski A, Zhang M. Efficacy and safety of oral sildenafil in children with Down syndrome and pulmonary hypertension. BMC Cardiovasc Disord. 2017;17:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xia YL, Yan WX, Chen H. [Efficacy and safety of sildenafil in the treatment of high altitude heart disease associated with severe pulmonary arterial hypertension in children: A preliminary evaluation]. Zhongguo Dang Dai Er Ke Za Zhi. 2014;16:745‐748. [PubMed] [Google Scholar]
- 79. Humpl T, Reyes JT, H, Stephens D, Adatia I. Beneficial effect of oral sildenafil therapy on childhood pulmonary arterial hypertension: twelve‐month clinical trial of a single‐drug, open‐label, pilot study. Circulation. 2005;111:3274‐3280. [DOI] [PubMed] [Google Scholar]
- 80. Karatza AA, Bush A, Magee AG. Safety and efficacy of sildenafil therapy in children with pulmonary hypertension. Int J Cardiol. 2005;100:267‐273. [DOI] [PubMed] [Google Scholar]
- 81. Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long‐term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr. 2009;154:379‐384. 384 e371‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lunze K, Gilbert N, Mebus S, et al. First experience with an oral combination therapy using bosentan and sildenafil for pulmonary arterial hypertension. Eur J Clin Invest. 2006;36(Suppl 3):32‐38. [DOI] [PubMed] [Google Scholar]
- 83. Shiva A, Shiran M, Rafati M, et al. Oral tadalafil in children with pulmonary arterial hypertension. Drug Res (Stuttg). 2016;66:7‐10. [DOI] [PubMed] [Google Scholar]
- 84. Takatsuki S, Calderbank M, Ivy DD. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol. 2012;33:683‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sabri MR, Beheshtian E. Comparison of the therapeutic and side effects of tadalafil and sildenafil in children and adolescents with pulmonary arterial hypertension. Pediatr Cardiol. 2014;35:699‐704. [DOI] [PubMed] [Google Scholar]
- 86. Kohno H, Ichida F, Hirono K, et al. Plasma concentrations of tadalafil in children with pulmonary arterial hypertension. Ther Drug Monit. 2014;36:576‐583. [DOI] [PubMed] [Google Scholar]
- 87. Yamazaki H, Kobayashi N, Taketsuna M, Tajima K, Suzuki N, Murakami M. Safety and effectiveness of tadalafil in pediatric patients with pulmonary arterial hypertension: A sub‐group analysis based on japan post‐marketing surveillance. Curr Med Res Opin. 2017;33:2241‐2249. [DOI] [PubMed] [Google Scholar]
- 88. Sitbon O, Morrell N. Pathways in pulmonary arterial hypertension: the future is here. Eur Respir Rev. 2012;21:321‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kemp K, Savale L, O'Callaghan DS, et al. Usefulness of first‐line combination therapy with epoprostenol and bosentan in pulmonary arterial hypertension: an observational study. J Heart Lung Transplant. 2012;31:150‐158. [DOI] [PubMed] [Google Scholar]
- 90. Hoeper MM, McLaughlin VV, Barbera JA, et al. Initial combination therapy with ambrisentan and tadalafil and mortality in patients with pulmonary arterial hypertension: a secondary analysis of the results from the randomised, controlled AMBITION study. Lancet Respir Med. 2016;4:894‐901. [DOI] [PubMed] [Google Scholar]
- 91. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809‐818. [DOI] [PubMed] [Google Scholar]
- 92. Galiè N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834‐844. [DOI] [PubMed] [Google Scholar]
- 93. Lajoie AC, Lauziere G, Lega JC, et al. Combination therapy versus monotherapy for pulmonary arterial hypertension: a meta‐analysis. Lancet Respir Med. 2016;4:291‐305. [DOI] [PubMed] [Google Scholar]
- 94. Zijlstra WMH, Douwes JM, Rosenzweig EB, et al. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63:2159‐2169. [DOI] [PubMed] [Google Scholar]
- 95. Humpl T, Berger RMF, Austin ED, et al. Treatment initiation in paediatric pulmonary hypertension: insights from a multinational registry. Cardiol Young. 2017;27:1123‐1132. [DOI] [PubMed] [Google Scholar]
- 96. Stasch JP, Evgenov OV. Soluble guanylate cyclase stimulators in pulmonary hypertension. Handb Exp Pharmacol. 2013;218:279‐313. [DOI] [PubMed] [Google Scholar]
- 97. Grimminger F, Weimann G, Frey R, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33:785‐792. [DOI] [PubMed] [Google Scholar]
- 98. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kielstein JT, Bode‐Boger SM, Hesse G, et al. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2005;25:1414‐1418. [DOI] [PubMed] [Google Scholar]
- 100. Migneault A, Sauvageau S, Villeneuve L, et al. Chronically elevated endothelin levels reduce pulmonary vascular reactivity to nitric oxide. Am J Respir Crit Care Med. 2005;171:506‐513. [DOI] [PubMed] [Google Scholar]
- 101. Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330‐340. [DOI] [PubMed] [Google Scholar]
- 103. Ghofrani HA, Grimminger F, Grunig E, et al. Predictors of long‐term outcomes in patients treated with riociguat for pulmonary arterial hypertension: data from the PATENT‐2 open‐label, randomised, long‐term extension trial. Lancet Respir Med. 2016;4:361‐371. [DOI] [PubMed] [Google Scholar]
- 104. Hoeper MMS G, Corris PA, Ghofrani H‐A, et al. RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase55 inhibitors. Eur Respir J. 2017;(3):50 10.1183/13993003.02425-2016 pii: 1602425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hoeper MM, Ghofrani H‐A, Benza RL, et al. Rationale and design of the REPLACE: Riociguat rEplacing Phosphodiesterase 5 Inhibitor (PDE5i) Therapy evaLuated Against Continued PDE5i thErapy in patients with pulmonary arterial hypertension (PAH). Am J Respir Crit Care Med. 2017;195:A2296. [Google Scholar]
- 106. Adempas EU summary of product characteristics . [last accessed 8 April 2019]. https://www.ema.europa.eu/en/documents/product‐information/adempas‐epar‐product‐information_en.pdf
- 107. Adempas (riociguat) tablets, for oral use. Highlights of prescribing information . [last accessed 8 April 2019]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/204819s011lbl.pdf
- 108. Approved risk evaluation and mitigation strategies (REMS) . [last accessed 5 July 2019]. https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=IndvRemsDetails.page&REMS=149
- 109. Adempas REMS program . [last accessed 7 July 2019]. https://www.adempasrems.com/#MainContent
- 110. Adatia I, Haworth SG, Wegner M, et al. Clinical trials in neonates and children: Report of the pulmonary hypertension academic research consortium pediatric advisory committee. Pulm Circ. 2013;3:252‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]