

Abstract
Tumor‐associated lymphangiogenesis and lymphatic invasion of tumor cells correlate with poor outcome in many tumor types, including breast cancer. Various explanations for this correlation have been suggested in the past, including the promotion of lymphatic metastasis and an immune‐inhibitory function of lymphatic endothelial cells (LECs). However, the molecular features of tumor‐associated lymphatic vessels and their implications for tumor progression have been poorly characterized. Here, we report the first transcriptional analysis of tumor‐associated LECs directly isolated from the primary tumor in an orthotopic mouse model of triple negative breast cancer (4T1). Gene expression analysis showed a strong upregulation of inflammation‐associated genes, including endothelial adhesion molecules such as VCAM‐1, in comparison to LECs derived from control tissue. In vitro experiments demonstrated that VCAM‐1 is not involved in the adhesion of tumor cells to LECs but unexpectedly promoted lymphatic permeability by weakening of lymphatic junctions, most likely through a mechanism triggered by interactions with integrin α4 which was also induced in tumor‐associated LECs. In line with this, in vivo blockade of VCAM‐1 reduced lymphatic invasion of 4T1 cells. Taken together, our findings suggest that disruption of lymphatic junctions and increased permeability via tumor‐induced lymphatic VCAM‐1 expression may represent a new target to block lymphatic invasion and metastasis.
Keywords: lymphangiogenesis, breast cancer, lymphatic invasion, metastasis
Short abstract
What's new?
Tumor‐associated lymphatic vessels serve important roles in tumor progression and metastasis. Nonetheless, little is known about the molecular changes in these vessels that give rise to a tumor‐promoting phenotype. In this study, transcriptional analysis was performed on lymphatic endothelial cells (LECs) isolated from a mouse model of triple‐negative breast cancer. Endothelial adhesion molecules, including tumor‐induced VCAM‐1, were strongly upregulated in tumor‐associated LECs. Additional experiments showed that VCAM‐1 upregulation influences lymphatic permeability and that its inhibition attenuates lymphatic breast cancer cell invasion. The findings identify VCAM‐1 as a potential target for the blockade of lymphatic invasion of tumor cells.
Introduction
Tumor‐associated lymphangiogenesis and lymphatic expansion are common phenomena in many tumor types, and frequently correlate with metastasis and poor outcome.1, 2 Typically, they affect the primary tumor tissue as well as draining collecting vessels and lymph nodes (LNs) which can markedly increase in size during tumor progression.3 In addition, lymphangiogenesis may also occur at distant metastatic sites, where it correlates with poor outcome in patients with melanoma metastasis to the lung.4 With regard to breast cancer, clinical studies have revealed that the extent of lymphangiogenesis is very heterogenous, as is the correlation with the clinical outcome,1, 2 probably reflecting the wide spectrum of breast cancer subtypes which differ extensively in their cellular, molecular and pathological characteristics and prognosis. Nonetheless, a recent meta‐analysis including a total of 19 clinical studies involving various breast cancer subtypes found an overall positive correlation between lymphatic vessel density, lymphatic invasion and reduced disease‐free and overall survival.5
There are several potential explanations for this correlation.6 First, tumor‐ (or metastasis‐) associated lymphatic vessels are directly involved in LN metastasis, which can give rise to distant metastasis both in tumor models and in cancer patients.7, 8, 9, 10, 11 A critical step toward lymphatic metastasis is the invasion of tumor cells into tumor‐associated lymphatic vessels. Consequently, the presence of lymphatic invasion also correlates with poor outcome.1, 2 In addition, lymphatic vessels have been proposed to serve as a niche for stem‐like cancer cells that may promote recurrence after phases of dormancy.12 Finally, lymphatic endothelial cells (LECs), especially those lining the lymphatic sinuses of LNs, have been implicated in the inhibition of adaptive T‐cell immunity through various mechanisms including antigen presentation and expression of coinhibitory molecules, such as PD‐L1 (reviewed in Ref. 13).
Despite the multiple roles of the lymphatic system in cancer progression, surprisingly little is known about the molecular changes occurring in tumor‐associated lymphatic vessels that may explain their tumor‐promoting function and could reveal new targets for cancer therapy. So far, only few studies have investigated gene expression signatures of tumor‐associated lymphatic endothelium, including tumor‐derived, in vitro expanded LECs,14 tumor‐associated lymphatic collectors15 and LECs isolated from tumor‐draining LNs.16 Correspondingly, the mechanisms regulating lymphatic invasion of tumor cells are still not fully understood. It has been suggested that chemokines and adhesion molecules expressed by tumor‐associated LECs are involved in this process,14, 17, 18, 19 indicating that at least in some cases, cancer cells may employ similar pathways as leukocytes to enter lymphatic vessels. On the other hand, lymphatic invasion may also be mediated by high levels of lymphangiogenic signaling leading to poor lymphatic vascular integrity,20 by destruction of lymphatic endothelium by tumor cells21 or by bystander cells such as fibroblasts,22 innate lymphoid cells23 or macrophages.24
Here, using an orthotopic, syngeneic mouse model of triple‐negative breast cancer (4T1), we report the first gene expression signature of LECs directly isolated from primary tumor tissue. The upregulated genes were highly enriched for inflammation‐associated genes, such as adhesion molecules and chemokines. Interestingly, both VCAM‐1 and its receptor integrin α4 were induced on tumor‐associated LECs. We found that blockade of VCAM‐1 in vitro and in vivo reduces tumor‐induced lymphatic permeability and lymphatic invasion, suggesting that integrin α4—VCAM‐1 interactions among tumor‐associated LECs could be a potential new target to impede lymphatic metastasis.
Materials and Methods
Cell lines
4T1‐luc2 cells (RRID:CVCL_L899, Caliper, Newton, MA) were maintained in DMEM with L‐glutamine and 10% FBS (all Thermo). B16‐F10‐luc2 cells (RRID:CVCL_5J17, Caliper) expressing human VEGF‐C25 were maintained in DMEM with GlutaMax, pyruvate, 10% FBS and 1.5 mg/ml G418 (Roche, Basel, Switzerland). Immortalized mouse LECs26 were maintained on collagen type‐1 (Advanced Biomatrix)/fibronectin (Millipore, Burlington, MA) coated dishes (10 μg/ml each) in DMEM/F12 with 20% FBS, 56 μg/ml heparin (Sigma, St. Louis, MO), and 10 μg/ml endothelial cell growth supplement (BioRad, San Diego, CA). LECs were cultured at 33°C in the presence of 1 U/ml IFN‐γ (Peprotech, Rocky Hill, NJ) for maintenance, and at 37°C without IFN‐γ for experiments.
The CrispR‐Cas9n double nickase approach was used to generate VCAM‐1 knockout clones of the 4T1‐luc2 cell line as described.27 In brief, a pair of guide‐RNAs was designed for target sequences in exon 3 of the mouse VCAM‐1 gene (Fig. 3a) using the online tool at http://crispr.mit.edu, and were cloned into pSpCas9n(BB)‐2A‐GFP (Addgene #48140). Vectors were transfected into 4T1‐luc2 cells using polyethylenimine. Forty‐eight hours later, single GFP+ clones were sorted using an ARIAII sorter (BD), expanded and screened for successful VCAM‐1 knockout by FACS and genomic PCR followed by sequencing. One clone with complete loss of VCAM‐1 protein expression and a confirmed deletion in exon 3 (1F8) was selected for all further experiments. A clone transfected with pSpCas9n(BB)‐2A‐GFP without guide‐RNAs (XF9) was used as control.
All cell lines were routinely tested for mycoplasma contamination using the Mycoscope kit (Genlantis, San Diego, CA).
Conditioned media
Conditioned media (CM) were collected from 4T1‐1F8 cells cultured for 72 hr in DMEM + 1% FBS. DMEM + 1% FBS served as control. CM were centrifuged, filtered (0.2 μm) and stored at −80°C until use. In some cases, CM were depleted of tumor‐cell secreted SPARC using goat anti‐SPARC (R&D AF942) coupled to protein‐G dynabeads (Thermo) by O/N incubation at 4°C under constant rotation.
VCAM‐1 blocking antibody
The rat antimouse VCAM‐1 blocking antibody clone 6C7.1 (rat IgG128) was generated in house by protein‐G (GE Healthcare) purification from supernatant of hybridoma cells (a kind gift of Prof. Dietmar Vestweber, MPI Münster) cultured in Hybridoma SFM medium (Thermo) using the Äkta Start system (GE Healthcare, Chicago, IL). Purified antibody was concentrated and rebuffered to PBS, depleted of endotoxin using Detoxi‐Gel columns (Thermo), and sterile filtered before use.
Mice and tumor models
All animal procedures were approved by the Cantonal Veterinary Office Zurich (license number ZH012/15). Balb/c and C57Bl6/N mice (Janvier) were housed in an SPF facility. Parental 4T1‐luc2 or 4T1‐1F8 cells (1 × 105) were implanted into the 4th mammary fat pad, and B16F10 cells (1 × 105) were injected intradermally into the flank. Tumors were allowed to grow for up to 3 weeks (4T1) or 2 weeks (B16F10), and the volume was monitored by caliper measurements and calculated as vol = (length × width2 × π/6). For ex vivo bioluminescence imaging, mice were i.p. injected with 3 mg luciferine/20 g body weight. Ten minutes later, mice were euthanized and tumors, LNs, lung and liver were imaged separately in an IVIS Spectrum instrument (Perkin Elmer, Waltham, MA). For VCAM‐1 blockade, 4T1‐1F8 and B16F10‐VEGFC tumor‐bearing mice were treated every other day with 100 μg of VCAM‐1 blocking antibody (clone 6C7.1) or control rat IgG1 (BE0080, BioXCell, West Lebanon, NH) by i.p. injection.
Sorting of tumor‐associated LECs and RNA sequencing
Mice were euthanized on Day 21 after tumor implantation and tumors together with the overlying skin were collected, minced and digested in 10 mg/ml collagenase IV (Sigma), 20 μg/ml DNAse I and 2.25 μM CaCl2 for 30 min at 37°C. Abdominal skin from naïve mice served as control. The tissue was passed through a 70 μm strainer, depleted of erythrocytes using PharmLyse (BD), washed and filtered through a 40 μm cell strainer.
Cell suspensions were stained with CD45.2‐FITC (BD 553772), CD31‐APC (BD 551262) and hamster antipodoplanin (clone 8.1.1, DSHB Iowa) followed by goat antihamster‐PE (Thermo HA6104) and labeling with 7‐AAD (Biolegend, San Jose, CA) for life/dead discrimination. LECs (CD45− CD31+ podoplanin+) were sorted directly into RLT buffer (Qiagen, Hilden, Germany) using an ARIA II sorter (BD), and stored immediately at −80°C until RNA extraction using the RNeasy micro kit (Qiagen). RNA quality check, library preparation (Smart‐seq ultra‐low RNA input kit v2, Clontech, Mountain View, MA) and RNA sequencing (2 × 50 bp paired‐end reads on a HiSeq 2500, Illumina) were performed at the New York Genome Center (NYGC, nygenome.org) according to the manufacturers’ protocols. Basic analysis (mapping, counting) was also performed at the NYGC. Differential gene expression analysis by DESeq2 v1.22.129 was done in R v3.5.1 using the corresponding packages from Bioconductor v3.8. Genes with a false discovery rate (FDR) < 0.01 and a log2FC > 2 between control skin and tumor‐associated LECs were considered as differentially expressed and are listed in Supporting Information Table S1. Gene ontology BP (biological process) annotation was done using DAVID 6.8 (david.ncifcrf.gov,30, 31).
FACS analysis of tissues and cultured cells
Cell suspensions of control skin, 4T1 tumors and tumor‐draining LNs were prepared as described.32 For staining, the following antibodies were used: CD45‐APC/Cy7 or CD45‐PacificBlue (Biolegend 103116, 103126), CD31‐APC (BD 551262), podoplanin‐PE (eBioscience, San Diego, CA, 12‐5381‐82), CD3‐PE/Cy7 (Biolegend 100220), CD11b‐BV605 (Biolegend 101257), Ly6C‐PE (Biolegend 128008), Ly6G‐FITC (Biolegend 127606), F4/80‐Alexa647 (BioRad MCA497G), VE‐cadherin‐APC (Biolegend 138012) and goat antimouse VCAM‐1 (R&D AF643), followed by washing and labeling with a secondary donkey antigoat‐Alexa488 antibody. For staining of cultured cells, cells were harvested using trypsin and subsequently stained with Itga4‐FITC (Biolegend 103606), Itgb1‐FITC (Biolegend 102206), goat antimouse Itga9 (R&D, Minneapolis, MN, AF3827) or goat antimouse VCAM‐1 (R&D AF643), followed by a donkey antigoat‐Alexa488 antibody. 7‐AAD or Zombi‐Aqua (both Biolegend) was used for life/dead discrimination. Samples were analyzed on an ARIA II or a Canto (both BD) or a Cytoflex S (Beckman Coulter, Brea, CA), and analyzed using FlowJo v10.4.2 (FlowJo LLC, Ashland, OR).
Quantitative PCR
Monolayers of mouse LECs were starved O/N in DMEM/F12 + 1% FBS and were subsequently treated with mouse VEGF‐A (20 ng/ml, Cell Sciences, Newburyport, MA), VEGF‐C (200 ng/ml, R&D), TNF‐α (40 ng/ml, Peprotech), IFN‐γ (100 ng/ml, Peprotech) or 50% 4T1‐1F8 CM or the corresponding control medium. RNA was extracted at the indicated timepoints using the NucleoSpin RNA kit (Macherey‐Nagel, Düren, Germany), and reverse transcribed using the high capacity cDNA synthesis kit (Applied Biosystems, Foster City, CA). Quantitative real‐time PCR was performed on a 7900HT FAST instrument (Applied Biosystems) using SYBRGreen Master Mix (Roche), with mouse RPLP0 serving as reference gene. The following primer sequences were used: mRPLP0 fwd: AGATTCGGGATATGCTGTTGG, rev: TCGGGTCCTAGACCAGTGTTC; mVCAM1 fwd: TGGGAAGCTGGAACGAAGTA, rev: CTCTGGATCCTTGGGGAAA.
Adhesion assay
Monolayers of mouse LECs were starved O/N in 1% FBS in DMEM/F12 and treated with TNF‐α (40 ng/ml) for 24 hr. Subsequently, cells were treated with a VCAM‐1 blocking antibody (clone 6C7.1) or control rat IgG1 (both 25 μg/ml) for 1 hr. Tumor cells were labeled using the PKH26 dye (Sigma) according to the manufacturer's instructions and were added to the treated LEC monolayers for 15 min. Then, monolayers were washed with PBS to remove nonadherent cells and fixed in 4% PFA. Four microscopic images around the center of each well were taken with an inverted Axioskop 2 (Zeiss) with a 5× objective and the number of adherent labeled tumor cells was counted in Fiji.33 Three wells/condition were analyzed within each individual experiment.
In vitro permeability assay
Mouse LECs were seeded on fibronectin/collagen‐coated transwell inserts (0.4 μm pore size, Costar, Washington, DC) and starved O/N in DMEM/F12 + 1% FBS with or without TNF‐α (40 ng/ml, Peprotech). The monolayers were subsequently treated with VCAM‐1 blocking antibodies (clone 6C7.1) or control rat IgG1 for 1 hr before the addition of 4T1‐1F8 CM or control medium for another 6 hr. The final concentrations of antibody and CM were 25 μg/ml and 50%, respectively. Next, transwell inserts were transferred to wells with fresh medium and 70 kDa FITC‐dextran (Sigma) was added to the inner well to a final concentration of 1 mg/ml. Samples from the outer well were taken at the indicated timepoints and the amount of fluorescence was measured using a Gemini plate reader (Molecular Devices, San Jose, CA). Three wells/condition were analyzed within each individual experiment.
Immunofluorescence and immunohistochemistry
Immunofluorescence staining of 7 μm cryosections of skin and tumors was performed essentially as before.32 The following antibodies were used: goat antimouse VCAM‐1 (R&D AF643), rabbit antimouse LYVE‐1 (AngioBio, San Diego, CA, 11‐034), goat antimouse LYVE‐1 (R&D AF2125), rat antimouse MECA32 (BD 553849), rabbit antipan‐keratin (Dako Z0622), rabbit anti‐gp100 (abcam ab137078), goat antimouse VE‐cadherin (R&D AF1002), rat antimouse Itga4 (EMD Millipore CBL1304), goat antimouse Itga9 (R&D AF3827), rat antimouse Itgb1 (BD 553715) and corresponding secondary antibodies donkey antirabbit, antigoat and antirat conjugated to Alexa488 or Alexa594 (all Thermo, Waltham, MA). Images were taken with an Axioskop 2, an LSM780 or an LSM880 confocal microscope (all Zeiss). For fluorescence staining of cultured cells, LEC monolayers were treated as described above, washed and fixed in 4% PFA. Cells were stained with goat antimouse VE‐cadherin (R&D AF1002) in presence of 0.3% Triton X100 followed by a secondary donkey antigoat Alexa594 antibody. Quantification of the VE‐cadherin positive area was measured by manual thresholding in Fiji and normalized to the number of nuclei/image (nuclei on edges were counted as half). Immunohistochemistry was performed on sections of formalin‐fixed, paraffin‐embedded 4T1‐1F8 tumors and tumor‐draining LNs. Sections were deparaffinized, followed by epitope retrieval (10 mM Na‐citrate pH 6.0) and stained with a pan‐keratin antibody (Dako Z0622) followed by a goat antirabbit biotin secondary antibody and detection with the ABC kit and AEC substrate (all Vector Laboratories, Burlingame, CA).
Statistical analysis
All data analyses apart from the RNA sequencing data were done using Prism v8 (GraphPad Software Inc., San Diego, CA). Bars/horizontal lines indicate mean values ± standard deviations. Student's t‐test (to compare two groups), one‐way ANOVA (to compare more than two groups) or two‐way ANOVA (to compare groups defined by two variables) were used to test for statistical significance. A p‐value < 0.05 was considered significant.
Results
Inflammation‐related genes are upregulated in tumor‐associated lymphatic vessels
To investigate molecular changes in tumor‐associated lymphatic vessels, we employed the orthotopic 4T1 triple‐negative breast cancer model which spontaneously metastasizes to the lung and displays robust peri‐ and intratumoral lymphangiogenesis (Fig. 1 a). Tumor cells were implanted into the fourth mammary fat pad of wild‐type Balb/c mice. Three weeks later, we collected the tumors (including the thin overlying skin layer) and isolated CD45− CD31+ podoplanin+ LECs by FACS sorting (Fig. 1 b). Abdominal skin from naïve Balb/c mice was used as control. On average, we could isolate around 500 LECs/mouse, which were then subjected to RNA sequencing. Due to the extremely low RNA input and some unavoidable RNA degradation which led to a 3′ bias in the mapped reads, only the 3′ ends of the detected transcripts (500 bp) were used for all further analyses, including a total of four control and five 4T1 tumor‐associated LEC samples. Of note, expression of pan‐endothelial markers such as CD31 (Pecam1), VE‐cadherin (Cdh5) and VEGFR‐2 (Kdr) as well as lymphatic markers such as VEGFR‐3 (Flt4), podoplanin (Pdpn), Prox1 and CCL21 was robust in all samples and not significantly altered between the groups, whereas CD45 (Ptprc) was hardly detectable, indicating that the isolation of LECs had been effective (data not shown). Differential gene expression analysis resulted in the identification of 831 genes that were significantly upregulated, and 92 genes that were significantly downregulated in tumor‐associated LECs (FDR < 0.01, log2FC > 2; Fig. 1 c and Supporting Information Table S1). Gene ontology (GO) analysis suggested that the upregulated genes were highly enriched in transcripts associated with inflammation, chemotaxis and immune‐related processes (Fig. 1 d), including various chemokines as well as cell adhesion molecules such as VCAM‐1, GLYCAM and ALCAM (Supporting Information Table S1).
Figure 1.
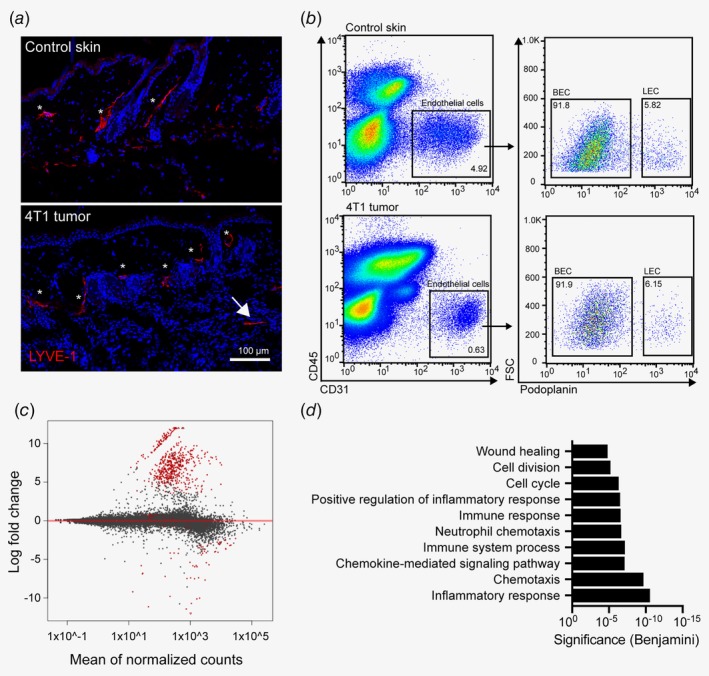
Transcriptional characterization of tumor‐associated lymphatic vessels in the 4T1 breast cancer model. (a) Representative images of abdominal skin of a naïve Balb/c mouse (top) and of a 4T1 tumor‐bearing mouse (bottom), showing lymphatic vessels stained for LYVE‐1 (red). Dermal/peritumoral lymphatic vessels are marked with asterisks, intratumoral lymphatic vessels with an arrow. (b) Representative FACS plots of control abdominal skin (top) and 4T1 tumor (bottom) single‐cell suspensions (pregated for living singlets) to illustrate the strategy for sorting of LECs. (c) Representation of RNA sequencing results of four control skin and five 4T1 tumor samples. Red dots indicate significantly (FDR < 0.01, log2FC > 2) differentially expressed genes in tumor‐associated LECs compared to control LECs. (d) Gene Ontology analysis (biological process) of significantly up‐regulated genes in 4T1‐associated LECs compared to control skin LECs. [Color figure can be viewed at wileyonlinelibrary.com]
VCAM‐1 and integrin α4 are upregulated by tumor‐associated LECs
We were particularly intrigued by the generally high expression level and strong upregulation (log2FC > 10) of VCAM‐1, which is typically considered as a leukocyte‐to‐endothelial adhesion molecule. However, in line with a previous study,34 our RNA sequencing data demonstrated that tumor‐associated LECs also upregulated integrin α4 (Itga4), and constitutively expressed high levels of integrin α9 and β1 (Itgb1) (Supporting Information Table S1). As integrin α4/β1 and α9/β1 are canonical VCAM‐1 receptors, this suggested that lymphatic VCAM‐1 may also have an LEC autonomous function, mediating interactions between neighboring LECs that could influence lymphatic function. To investigate this hypothesis further, we first investigated expression of VCAM‐1 and its receptors on tumor‐associated LECs on the protein level. Immunofluorescence staining of 4T1 tissue sections revealed a strong VCAM‐1 expression by 4T1 tumor cells themselves as previously published35 (Supporting Information Fig. S1a), which made it difficult to assess the expression of VCAM‐1 in LECs using this approach. On the other hand, α4, α9 and β1 integrins could be readily detected in 4T1‐associated LECs, and integrin α9 was also expressed in LECs in control abdominal skin (Supporting Information Fig. S1b). In order to confirm the expression of VCAM‐1 in tumor LECs, we therefore used flow cytometry and found that on Day 21 after tumor inoculation, VCAM‐1 was strongly induced in tumor LECs compared to control abdominal skin LECs (Fig. 2 a). No difference in VCAM‐1 expression in draining LN LECs could be detected by FACS (Supporting Information Fig. S1c). We made very similar observations in a second, unrelated tumor model. In B16F10 tumors engineered to overexpress VEGF‐C in order to increase the amount of tumor‐associated LECs,25 we found increased VCAM‐1 expression in LECs within the primary tumor, but not in the draining LN (Supporting Information Figs. S1d and S1e).
Figure 2.

VCAM‐1 is upregulated on tumor‐associated LECs in vivo and by inflamed LECs in vitro. (a) Left panel: Representative FACS histogram of VCAM‐1 expression on 4T1‐associated LECs (black curve) compared to control skin LECs (gray curve) on day 21 after tumor implantation. The gray dotted line indicates staining with control goat IgG. Right panel: Quantification of VCAM‐1 expression by FACS in control skin and 4T1‐associated LECs (n = 4 mice/group; one out of two independent experiments with equivalent results is shown). (b) Immortalized mouse LECs were treated with the indicated growth factors or cytokines for 6, 24 or 48 hr, and the expression of VCAM‐1 mRNA was determined by qPCR (n = 3 technical replicates; one representative of three independent experiments is shown). (c) Upregulation of surface VCAM‐1 on TNF‐α and IFN‐γ treated mouse LECs was validated by FACS after 24 hr (n = 3). (d) Cultured mouse LECs were treated with control medium or 4T1 tumor cell‐conditioned medium (CM, 50%) for the indicated time periods before qPCR analysis of VCAM‐1 mRNA expression (n = 3 technical replicates; one representative of three independent experiments is shown). (e) Upregulation of surface VCAM‐1 on 4T1 CM treated mouse LECs was validated by FACS after 24 hr (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001).
VCAM‐1 in LECs is regulated by inflammatory cytokines
To characterize how VCAM‐1 is regulated in LECs, we used an in vitro system of immortalized mouse LECs,26 which we treated with a panel of tumor‐relevant (lymph‐) angiogenic growth factors and inflammatory cytokines, namely VEGF‐C, VEGF‐A, TNF‐α and IFN‐γ. Using qPCR, we found that only TNF‐α and, to a lesser extent, IFN‐γ induced VCAM‐1 in cultured LECs (Fig. 2 b). This was confirmed by FACS (Fig. 2 c). VCAM‐1 expression was also induced when cultured LECs were exposed to conditioned medium (CM) of 4T1 cells (Figs. 2 d and 2e), which is known to contain several inflammatory cytokines.36
Generation of a VCAM‐1 knockout 4T1 clone
Targeting lymphatic VCAM‐1 to assess its in vivo function is difficult in the context of a primary tumor that itself expresses high levels of VCAM‐1. We therefore genetically deleted VCAM‐1 in parental 4T1 cells by targeting two adjacent PAM motifs in exon 3 with the help of specific guide RNAs and the double nickase (Cas9n) approach to minimize off‐target mutations (Fig. 3 a). After transfection, we selected tumor cell clones with reduced VCAM‐1 expression and analyzed them by genomic PCR, sequencing and flow cytometry. One of these clones, designated as 1F8, displayed a deletion in exon 3 starting at the targeted genomic site (data not shown) and had lost VCAM‐1 protein expression entirely (Fig. 3 b). Primary tumors formed after orthotopic implantation of this clone grew at a comparable rate to parental 4T1 tumors (Supporting Information Fig. S1f) and still induced VCAM‐1 expression in tumor‐associated LECs (Fig. 3 c). Therefore, this clone was chosen for functional experiments in vivo.
Figure 3.
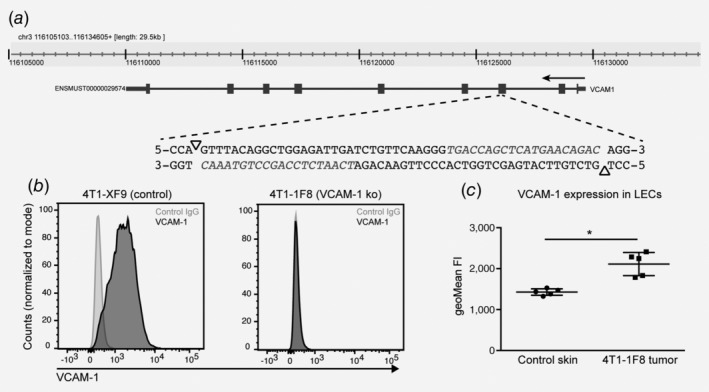
VCAM‐1 is up‐regulated in tumor‐associated LECs in 4T1 tumors deficient of VCAM‐1. (a) Schematic representation of the strategy to delete VCAM‐1 in 4T1 tumor cells. Two neighboring PAM motifs on the top and the bottom strand were identified within exon 3 of the vcam1 gene (triangles). Guide‐RNAs for adjacent target sequences (in italics) together with the Cas9n enzyme were transfected into 4T1 tumor cells and single knockout clones were isolated by FACS sorting. (b) Representative FACS histogram of VCAM‐1 staining using a polyclonal antibody in a control clone (XF9) transfected with Cas9n only and a complete VCAM‐1 knockout clone (1F8). (c) 4T1‐1F8 cells were implanted into Balb/c mice and the expression of VCAM‐1 on tumor‐associated LECs was determined by FACS on Day 21 after implantation (n = 5 mice/group; *p < 0.05).
VCAM‐1 blockade in vivo reduces lymphatic invasion of tumor cells
To investigate the function of lymphatic VCAM‐1 during tumor progression, we inoculated syngeneic Balb/c mice with VCAM‐1 deficient 4T1‐1F8 tumor cells and treated them every other day with a VCAM‐1‐blocking antibody for 21 days. Previously, it has been suggested that VCAM‐1 is involved in tumor angiogenesis in breast cancer.37 However, in our model, there was no measurable effect on tumor angiogenesis, lymphangiogenesis, or blood vessel permeability at the endpoint (Day 21) (Supporting Information Fig. S2a–S2c). Additionally, VCAM‐1 blockade had no major effect on leukocyte infiltration into the tumor (Supporting Information Fig. S2d). In line with this, we did not observe a significant impact on primary tumor growth, although 3 out of 10 anti‐VCAM‐1 treated mice developed only very small tumors (Figs. 4 a and 4b). We also assessed the weight of tumor‐draining inguinal and axillary LNs. Although statistically not significant, there was a clear tendency toward smaller LNs in anti‐VCAM‐1 treated mice compared to control rat IgG1 treated mice (Fig. 4 c). A reduction in draining LN weight could be due to various reasons, including a reduction of lymphatic metastasis, lymphatic drainage or immune cell recruitment via high endothelial venules. Therefore, we next assessed the invasion of tumor cells into the tumor‐associated lymphatic vasculature within the primary tumor. Importantly, the frequency of tumor‐cell containing lymphatic vessels was significantly reduced on Day 21 after anti‐VCAM‐1 antibody treatment (Fig. 4 d). A similar trend was also observed at an earlier timepoint (Day 8), although it did not yet reach statistical significance due to an overall lower rate of lymphatic invasion at this timepoint (Supporting Information Fig. S2e). Additionally, we also investigated lymphatic invasion in mice bearing B16F10‐VEGFC tumors. However, lymphatic invasion was largely absent in this model and was not affected by VCAM‐1 blockade (Supporting Information Fig. S2f).
Figure 4.
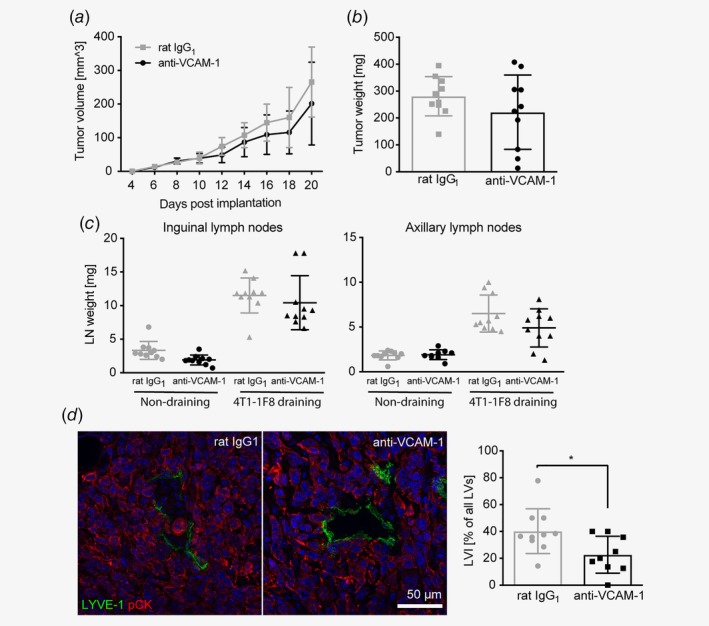
VCAM‐1 blockade in vivo does not affect 4T1 tumor growth but inhibits lymphatic invasion. Tumor volumes (a) and endpoint (Day 21) tumor weights (b) of VCAM‐1 deficient 4T1‐1F8 tumors in mice treated with a VCAM‐1 blocking antibody or a corresponding IgG control every other day (n = 10 mice/group). (c) Weight of inguinal (left) and axillary (right) lymph nodes at the endpoint (Day 21; n = 10 mice/group). (d) Left panel: Representative microscopic images showing frequent invasion of tumor cells (stained for pan‐cytokeratin, red) into lymphatic vessels (LYVE‐1, green) in control‐treated tumors but not in tumors treated with the VCAM‐1 blocking antibody. Right panel: Quantification of the frequency of tumor cell‐invaded lymphatic vessels in control and anti‐VCAM‐1 treated tumors (n = 10 control/9 treated tumors; *p < 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
4T1 cells have been reported to colonize tumor‐draining LNs very inefficiently.16, 38, 39, 40, 41, 42 In line with this, with the exception of one mouse, we were not able to detect LN metastases in our model, neither histologically nor by bioluminescence (Supporting Information Figs. S2g and S2h), preventing us from further studying the role of lymphatic VCAM‐1 in lymphogenous metastasis. Distant metastasis to lungs and liver was also low and not significantly affected by anti‐VCAM‐1 treatment (Supporting Information Fig. S2i), most likely due to a critical function of 4T1‐intrinsic VCAM‐1 in this process as reported previously.35
VCAM‐1 regulates permeability of lymphatic vessels through junction remodeling
We first hypothesized that VCAM‐1 expressed by LECs serves as an adhesion molecule for tumor cells, facilitating their entry into lymphatic vessels. However, using flow cytometry, we found that whereas 4T1‐1F8 cells express high levels of integrin β1 (Itgb1), they completely lacked the two α‐chains α4 and α9 that are needed to form the classic VCAM‐1 receptors α4/β1 and α9/β1 (Fig. 5 a). This was in contrast to cultured mouse LECs which expressed all three integrin chains in a largely TNF‐independent manner, with the exception of β1 which was slightly induced by TNF‐α treatment (Fig. 5 b). In line with this, 4T1‐1F8 cells adhered poorly to mouse LEC monolayers in vitro, and neither TNF‐α treatment of the LECs (to induce VCAM‐1 expression) nor VCAM‐1 blockade had any consistent effect on this adhesion (Fig. 5 c). Interestingly, the case was quite different for B16F10‐VEGFC cells. In agreement with a previous report,43 we found that these cells express high levels of integrin α4, α9 and β1 (Supporting Information Fig. S3a), and adhere to LEC monolayers in a TNF‐α and VCAM‐1 dependent manner (Supporting Information Fig. S3b).
Figure 5.

VCAM‐1 blockade reduces lymphatic permeability. (a, b) Representative FACS histograms (left panels) and staining quantification (right panels) of the classic VCAM‐1 receptors integrin α4 (Itga4), integrin α9 (Itga9) and integrin β1 (Itgb1) in 4T1‐1F8 cells (a) and cultured mouse LECs with or without TNF‐α treatment for 24 hr (b; n = 3). (c) Adhesion of fluorescently labeled 4T1‐1F8 cells to monolayers of cultured mouse LECs pretreated with TNF‐α or not and in the presence of control IgG or VCAM‐1 blocking antibodies (n = 3 wells/condition; 1 representative of 6 individual experiments is shown). (d) In vitro permeability of mouse LEC monolayers grown on transwell inserts (0.4 μm pore size) toward 70 kD FITC‐dextran. LEC monolayers were pretreated with TNF‐α overnight and subsequently incubated with control or 4T1 CM in presence of control IgG or anti‐VCAM‐1 antibodies (n = 3 wells/condition; one representative of three individual experiments is shown). Statistical test refers to 4T1‐1F8 CM—rat IgG1 compared to 4T1‐1F8 CM—anti‐VCAM‐1. (e) Representative images (left panels) and quantification (right panel) of VE‐cadherin staining in mouse LEC monolayers that were pretreated with TNF‐α overnight and subsequently incubated with control or 4T1 CM in presence of control IgG or anti‐VCAM‐1 antibodies (n = 10 random images/condition; *p < 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
We next hypothesized that VCAM‐1 on LECs might be involved in the organization of cell‐to‐cell junctions and in the regulation of permeability, as has been reported to be the case in blood vessels.44, 45, 46 Indeed, using an in vitro permeability assay of mouse LEC monolayers and a 70kD FITC‐dextran tracer, we observed that treatment with 4T1‐1F8 CM increased permeability, which could be entirely blocked by preincubation of the LECs with a VCAM‐1 blocking antibody (Fig. 5 d). This effect required pretreatment of the LECs with TNF‐α, most likely to induce sufficiently high levels of VCAM‐1 (Supporting Information Fig. S3c).
Next, we aimed to elucidate the mechanisms of VCAM‐1 dependent permeability regulation in LECs. Recently, it was reported that the tumor‐cell secreted protein SPARC can bind VCAM‐1 on blood vascular endothelial cells, activating an intracellular signaling cascade leading to barrier disruption and increased permeability.45 As 4T1 cells express SPARC (data not shown), we hypothesized that a similar mechanism might operate in LECs. However, antibody‐mediated SPARC depletion from the conditioned medium of 4T1‐1F8 cells did not reduce the permeability‐reducing effect of VCAM‐1 blockade, suggesting that SPARC did not play a major role in this case (Supporting Information Fig. S3d).
Leukocyte binding to VCAM‐1 on blood vessel endothelial cells via integrin α4/β1 has also been reported to stimulate junctional weakening by displacing ZO‐146 and by reducing VE‐cadherin mediated cell adhesion via VE‐PTP displacement from VE‐cadherin.44 Cultured mouse LECs expressed α4, α9 and β1 integrins (Fig. 5 b) and α4, together with VCAM‐1, was induced in 4T1‐associated LECs (Supporting Information Table S1). We therefore considered the possibility that LEC‐LEC interactions through α4/β1 and VCAM‐1 could similarly induce a weakening of lymphatic junctions, leading to increased permeability. To investigate this further, we stained cultured LECs treated with TNF‐α and 4T1‐1F8 CM in the presence of a VCAM‐1 blocking or control rat IgG1 for VE‐cadherin and quantified brightly stained junctional areas. Doing so, we found that VCAM‐1 blockade increased junctional VE‐cadherin staining (Fig. 5 e), whereas the total VE‐cadherin level was not affected (Supporting Information Fig. S3e). Interestingly, we noted a trend towards increased VE‐cadherin staining intensity in LYVE‐1+ lymphatic vessels in 4T1‐1F8 tumors treated with anti‐VCAM‐1, and towards a higher frequency of VE‐cadherin+ LECs determined by FACS (Supporting Information Fig. S3f). We also analyzed ZO‐1 expression in cultured LECs treated with 4T1‐1F8 and TNF‐α but found no differences in the total protein level or in its Ser‐phosphorylation (Supporting Information Fig. S3g). Taken together, these data indicate that tumor‐derived factors in combination with inflammatory signals induce VCAM‐1 and integrin α4 on tumor‐associated LECs, leading to a junctional displacement of VE‐cadherin that increases permeability and facilitates lymphatic invasion of tumor cells (Fig. 6).
Figure 6.
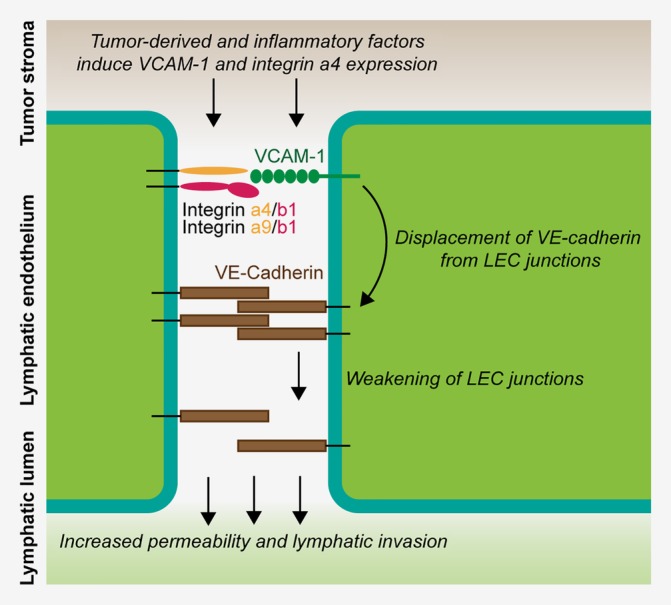
Model of VCAM‐1/integrin mediated permeability regulation in tumor‐associated lymphatic vessels. Schematic of the proposed mechanism of how permeability and lymphatic invasion are regulated through VCAM‐1. Tumor‐derived factors, in combination with inflammatory signals from the tumor microenvironment, induce expression of VCAM‐1 and integrin α4 in associated LECs. VCAM‐1 engagement by integrin α4/β1 or α9/β1 complexes on adjacent LECs then acts on VE‐cadherin complexes in LEC junctions and induces their destabilization, possibly involving VE‐PTP detachment from VE‐cadherin. This leads to the displacement of VE‐cadherin from the junctions, increasing paracellular permeability for tissue fluid but also facilitating tumor cell invasion into the lymphatic lumen. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Despite their implication in tumor progression and correlation with poor prognosis in many cancer types, the molecular features of primary tumor‐associated lymphatic vessels have remained largely unknown, most likely due to the technical challenge to isolate sufficient numbers of LECs from tumor tissue. The only previously reported gene expression signatures of tumor‐associated LECs were thus obtained from a VEGF‐C‐overexpressing tumor model and involved in vitro expansion of LECs (which likely affected their phenotype),14 concentrated on tumor‐draining lymphatic collectors,15 or on tumor‐draining LNs.16 Using FACS sorting of LECs in combination with an RNAseq protocol for ultra‐low input, we provide here the first complete and direct transcriptional characterization of primary tumor‐associated lymphatic vessels in a model of triple‐negative breast cancer (4T1). Even with this approach, obtaining sufficient high‐quality RNA was challenging, and some RNA degradation could not be avoided due to the long and relatively harsh digestion procedure required for both the tumor and the control tissue. This was most likely the reason for a certain 3′ bias we observed among the mappable reads, which we mitigated by including only the 3′ ends (500 bp) of the transcriptome into the differential gene expression analysis. Whereas this approach led to the loss of information about the 5′ regions, differential splicing, etc., our dataset remains very valuable to study tumor‐induced gene expression changes in LECs.
Our results are striking in terms of the strong inflammatory gene expression signature of 4T1‐associated LECs, which included induction of many chemokines and adhesion molecules. We also found a trend (log2FC = 1.96, FDR = 0.68) toward increased expression of the T cell inhibitory protein PD‐L1, which is in line with our own previous observations.32 Clearly, this proinflammatory gene expression signature is likely to have a plethora of effects on tumor progression, metastasis and host immunity. Here, we focused on the role of lymphatic VCAM‐1. VCAM‐1 is best known as adhesion molecule mediating leukocyte adhesion and transmigration through blood vessel endothelium under inflammatory conditions.47 Additionally, VCAM‐1 expression has also been found on lymphatic vessels during skin inflammation and has been reported to facilitate LN migration of DCs48 and neutrophils.49
Besides its classic role in leukocyte‐to‐endothelial adhesion in inflammatory situations, VCAM‐1 expression by cancer cells has been found to transmit survival signals for metastatic cells in the lung (including 4T1 cells),35 and to facilitate LN colonization.50 In line with this, we found a rather low rate of lung metastasis in mice bearing VCAM‐1 deficient 4T1 tumors. VCAM‐1 has furthermore been implicated in breast cancer angiogenesis by mediating adhesion between endothelial and perivascular cells37 as well as in the regulation of blood vessel permeability in tumors and in association with leukocyte diapedesis. Several mechanisms have been proposed for this. Tichet et al.45 reported that melanoma cell‐derived SPARC binds to VCAM‐1 on blood vessel endothelial cells, triggering a signaling cascade that leads to actin remodeling and vascular barrier disruption. Furthermore, leukocyte binding to VCAM‐1 via integrin α4/β1 was reported to destabilize endothelial junctions via dissociation of the endothelial phosphatase VE‐PTP from VE‐cadherin complexes44 and phosphorylation of ZO‐1,46 leading to loosening of endothelial junctions. Our data suggest that although 4T1 tumor cells express SPARC (data not shown), SPARC is not involved in the regulation of lymphatic permeability, at least in vitro. On the other hand, in agreement with a previous report demonstrating that breast cancer‐associated lymphatic vessels upregulate integrin α4, possibly in response to lymphangiogenic growth factors,34 we found that 4T1‐associated LECs upregulate integrin α4 and robustly expressed integrin α9, indicating that LEC—LEC interactions via α4/β1 or α9/β1—VCAM‐1 binding might contribute to the regulation of lymphatic vessel permeability. Finally, it is also possible that lymphatic VCAM‐1 regulates vessel permeability by recruiting immune cells releasing permeability‐inducing factors. Our data however indicate that VCAM‐1 can control LEC permeability independently of immune cells, at least in vitro.
Our in vivo and in vitro results further suggest that VCAM‐1 mediated permeability regulation involves reduction of junctional VE‐cadherin, possibly through a mechanism involving VE‐PTP, which according to our sequencing data is robustly expressed by control and tumor‐associated LECs (data not shown). It is tempting to speculate that this pathway represents a mechanism how lymphatic vessels are activated to take up more tissue fluid in, for example, inflammatory conditions, where increased liquid filtration from blood vessels requires increased lymphatic drainage to keep tissue swelling at bay. In a tumor context, however, increased lymphatic permeability and fluid uptake likely come at the cost of facilitated lymphatic invasion and dissemination of tumor cells. In this regard, it is interesting to note that the mechanism of VCAM‐1 induced destabilization of VE‐cadherin complexes in endothelial junctions is highly related to the mechanism of VEGF‐A induced vessel permeability.44 Furthermore, VEGF‐C that activates similar intracellular signaling cascades as VEGF‐A was recently shown to destabilize VE‐cadherin complexes in lymphatic junctions in colorectal cancer, leading to increased lymphatic invasion and metastasis.20 Thus, it appears that both inflammatory signals (via induction of VCAM‐1 and integrin α4 in LECs) and VEGF signaling converge to increase permeability in lymphatic vessels via a reduction of VE‐cadherin in endothelial junctions. In line with this, lymphatic VCAM‐1 blockade in the 4T1 model inhibited tumor cell lymphatic invasion and appeared to reduce LN swelling. In agreement with our data, blocking of α4/β1 integrin has previously been shown to reduce LN metastasis in several mouse tumor models.34, 50 Although this was at least in part ascribed to an antilymphangiogenic effect of α4/β1 blockade or to capture of VCAM‐1+ tumor cells by LN LECs, an inhibition of VCAM‐1 mediated lymphatic permeability might have contributed to those findings as well.
Supporting information
Appendix S1: Supporting Information
Table S1
Acknowledgements
The authors would like to acknowledge technical and experimental support by Yuliang He, Dr. Qiaoli Ma, Dr. Marco D'Addio, and Noelle Leary (all ETH Zurich). This work was supported by Swiss National Science Foundation grants 310030_166490 and 310030B_185392, European Research Council grant LYVICAM and Oncosuisse (all to MD).
Conflict of interest: The authors declare no potential conflict of interest.
Contributor Information
Lothar C. Dieterich, Email: lothar.dieterich@pharma.ethz.ch.
Michael Detmar, Email: michael.detmar@pharma.ethz.ch.
Data availability
The entire sequencing data are available at the European Nucleotide Archive (ENA) under accession number PRJEB30889. Other data will be made available upon reasonable request.
References
- 1. Dieterich LC, Detmar M. Tumor lymphangiogenesis and new drug development. Adv Drug Deliv Rev 2016;99:148–60. [DOI] [PubMed] [Google Scholar]
- 2. Stacker SA, Williams SP, Karnezis T, et al. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer 2014;14:159–72. [DOI] [PubMed] [Google Scholar]
- 3. Hirakawa S, Kodama S, Kunstfeld R, et al. VEGF‐A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med 2005;201:1089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma Q, Dieterich LC, Ikenberg K, et al. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci Adv 2018;4:eaat4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang S, Zhang D, Gong M, et al. High lymphatic vessel density and presence of lymphovascular invasion both predict poor prognosis in breast cancer. BMC Cancer 2017;17:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma Q, Dieterich LC, Detmar M. Multiple roles of lymphatic vessels in tumor progression. Curr Opin Immunol 2018;53:7–12. [DOI] [PubMed] [Google Scholar]
- 7. Naxerova K, Reiter JG, Brachtel E, et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017;357:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong MK, Macintyre G, Wedge DC, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun 2015;6:6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown M, Assen FP, Leithner A, et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018;359:1408–11. [DOI] [PubMed] [Google Scholar]
- 11. Pereira ER, Kedrin D, Seano G, et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018;359:1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim M, Koh YJ, Kim KE, et al. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res 2010;70:10411–21. [DOI] [PubMed] [Google Scholar]
- 13. Maisel K, Sasso MS, Potin L, et al. Exploiting lymphatic vessels for immunomodulation: rationale, opportunities, and challenges. Adv Drug Deliv Rev 2017;114:43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clasper S, Royston D, Baban D, et al. A novel gene expression profile in lymphatics associated with tumor growth and nodal metastasis. Cancer Res 2008;68:7293–303. [DOI] [PubMed] [Google Scholar]
- 15. Karnezis T, Shayan R, Caesar C, et al. VEGF‐D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012;21:181–95. [DOI] [PubMed] [Google Scholar]
- 16. Commerford CD, Dieterich LC, He Y, et al. Mechanisms of tumor‐induced lymphovascular niche formation in draining lymph nodes. Cell Rep 2018;25:3554–63.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dua RS, Gui GP, Isacke CM. Endothelial adhesion molecules in breast cancer invasion into the vascular and lymphatic systems. Eur J Surg Oncol 2005;31:824–32. [DOI] [PubMed] [Google Scholar]
- 18. Wang Z, Wang Z, Li G, et al. CXCL1 from tumor‐associated lymphatic endothelial cells drives gastric cancer cell into lymphatic system via activating integrin beta1/FAK/AKT signaling. Cancer Lett 2017;385:28–38. [DOI] [PubMed] [Google Scholar]
- 19. Zhong G, Chen L, Yin R, et al. Chemokine (CC motif) ligand 21/CC chemokine receptor type 7 triggers migration and invasion of human lung cancer cells by epithelial mesenchymal transition via the extracellular signal regulated kinase signaling pathway. Mol Med Rep 2017;15:4100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tacconi C, Correale C, Gandelli A, et al. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 2015;148:1438–51 e8. [DOI] [PubMed] [Google Scholar]
- 21. Kerjaschki D, Bago‐Horvath Z, Rudas M, et al. Lipoxygenase mediates invasion of intrametastatic lymphatic vessels and propagates lymph node metastasis of human mammary carcinoma xenografts in mouse. J Clin Invest 2011;121:2000–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tatti O, Gucciardo E, Pekkonen P, et al. MMP16 mediates a proteolytic switch to promote cell‐cell adhesion, collagen alignment, and lymphatic invasion in melanoma. Cancer Res 2015;75:2083–94. [DOI] [PubMed] [Google Scholar]
- 23. Irshad S, Flores‐Borja F, Lawler K, et al. RORgammat(+) innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res 2017;77:1083–96. [DOI] [PubMed] [Google Scholar]
- 24. Evans R, Flores‐Borja F, Nassiri S, et al. Integrin‐mediated macrophage adhesion promotes lymphovascular dissemination in breast cancer. Cell Rep 2019;27:1967–78.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Proulx ST, Luciani P, Derzsi S, et al. Quantitative imaging of lymphatic function with liposomal indocyanine green. Cancer Res 2010;70:7053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vigl B, Aebischer D, Nitschke M, et al. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus‐dependent manner. Blood 2011;118:205–15. [DOI] [PubMed] [Google Scholar]
- 27. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Engelhardt B, Laschinger M, Schulz M, et al. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4‐integrin but not alpha4beta7‐integrin. J Clin Invest 1998;102:2096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang d W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 32. Dieterich LC, Ikenberg K, Cetintas T, et al. Tumor‐associated lymphatic vessels upregulate PDL1 to inhibit T‐cell activation. Front Immunol 2017;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garmy‐Susini B, Avraamides CJ, Schmid MC, et al. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res 2010;70:3042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM‐1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011;20:538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kano A. Tumor cell secretion of soluble factor(s) for specific immunosuppression. Sci Rep 2015;5:8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garmy‐Susini B, Jin H, Zhu Y, et al. Integrin alpha4beta1‐VCAM‐1‐mediated adhesion between endothelial and mural cells is required for blood vessel maturation. J Clin Invest 2005;115:1542–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adiseshaiah PP, Patel NL, Ileva LV, et al. Longitudinal imaging of cancer cell metastases in two preclinical models: a correlation of noninvasive imaging to histopathology. Int J Mol Imaging 2014;2014:102702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992;52:1399–405. [PubMed] [Google Scholar]
- 40. Kuonen F, Laurent J, Secondini C, et al. Inhibition of the kit ligand/c‐kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin Cancer Res 2012;18:4365–74. [DOI] [PubMed] [Google Scholar]
- 41. Tao K, Fang M, Alroy J, et al. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer 2008;8:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thiele W, Rothley M, Dimmler A, et al. Platelet deficiency in Tpo(−/−) mice can both promote and suppress the metastasis of experimental breast tumors in an organ‐specific manner. Clin Exp Metastasis 2018;35:679–89. [DOI] [PubMed] [Google Scholar]
- 43. Rebhun RB, Cheng H, Gershenwald JE, et al. Constitutive expression of the alpha4 integrin correlates with tumorigenicity and lymph node metastasis of the B16 murine melanoma. Neoplasia 2010;12:173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE‐PTP from VE‐cadherin. Blood 2013;122:2512–22. [DOI] [PubMed] [Google Scholar]
- 45. Tichet M, Prod'Homme V, Fenouille N, et al. Tumour‐derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun 2015;6:6993. [DOI] [PubMed] [Google Scholar]
- 46. Abdala‐Valencia H, Kountz TS, Marchese ME, et al. VCAM‐1 induces signals that stimulate ZO‐1 serine phosphorylation and reduces ZO‐1 localization at lung endothelial cell junctions. J Leukoc Biol 2018;104:215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7:678–89. [DOI] [PubMed] [Google Scholar]
- 48. Johnson LA, Clasper S, Holt AP, et al. An inflammation‐induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med 2006;203:2763–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rigby DA, Ferguson DJ, Johnson LA, et al. Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin‐dependent proteolysis and lipoxin‐induced junctional retraction. J Leukoc Biol 2015;98:897–912. [DOI] [PubMed] [Google Scholar]
- 50. Garmy‐Susini B, Avraamides CJ, Desgrosellier JS, et al. PI3Kalpha activates integrin alpha4beta1 to establish a metastatic niche in lymph nodes. Proc Natl Acad Sci USA 2013;110:9042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Table S1
Data Availability Statement
The entire sequencing data are available at the European Nucleotide Archive (ENA) under accession number PRJEB30889. Other data will be made available upon reasonable request.