

Abstract
Saccharides are ubiquitous biomolecules, but little is known about their interaction with, and assembly at, surfaces. By combining preparative mass spectrometry with scanning tunneling microscopy, we have been able to address the conformation and self‐assembly of the disaccharide sucrose on a Cu(100) surface with subunit‐level imaging. By employing a multistage modeling approach in combination with the experimental data, we can rationalize the conformation on the surface as well as the interactions between the sucrose molecules, thereby yielding models of the observed self‐assembled patterns on the surface.
Keywords: disaccharides, electrospray ion beam deposition, multistage modeling, scanning tunneling microscopy, self-assembly
Carbohydrates (also called saccharides) serve as the key structural component of plants, making up the majority of the biomass, and are a source of chemical energy for living organisms.1 Although these molecules are highly flexible in solution, they can form stable crystals as a result of the many hydrogen bonds formed between the hydroxy groups at their periphery. Their 3D crystal structures can be determined by diffraction,2 exploiting the symmetry of the crystal, which enables averaging over many molecules. The conformations as well as assembly motifs of saccharides in environments of reduced symmetry, for example, on surfaces and in contact with few/other molecules, are essentially unknown. The former is of paramount importance in biological signaling,3 and the interactions with solid substrates are of relevance, for example, in the technological use of saccharides in cement hydration4 or for medical applications such as biomineralization.5 In both domains, however, the conformation and interactions matter on the level of the individual molecule. Gaining this knowledge at a molecular level presents an enormous analytical challenge chemically and structurally, because monosaccharides are generally structural isomers, whose specific function lies in the subtle arrangement of functional groups and, furthermore, on the details of their conformations, which are constrained by the way the monosaccharides are connected.
Scanning probe microscopy is a suitable technique to gain information about adsorbed molecules because it allows the direct imaging of individual molecules with submolecular resolution.6 Here we present the study of sucrose molecules arranged in a molecular network formed on a Cu(100) surface after deposition. The structural complexity of saccharides is reflected in the behavior of sucrose, as this consists of two monosaccharides, linked by a glycosidic bond, that can exhibit different adsorption conformations. By employing a new approach that combines direct imaging of the monosaccharide building blocks of sucrose by scanning tunneling microscopy (STM) and multistage modeling, we gain insight into the conformation of individual sucrose molecules on a surface and can infer intra‐ and intermolecular interactions from the patterns observed for the assemblies.
Sucrose (α‐d‐glucopyranosyl‐(1→2)‐β‐d‐fructofuranoside), chosen for the study presented herein, is an abundant disaccharide that crystallizes in anhydrous form in a monoclinic 3D structure.2 Sucrose is nonreducing, and thus remains stable in aqueous solution because no isomerism through ring‐opening reactions is expected. As a consequence of its nonvolatility, we employed soft‐landing electrospray ion beam deposition (ES‐IBD)7 to deposit the deprotonated molecule as a negatively charged ion on Cu(100). This enabled us to employ mass spectrometry and mass filtering as well as to decelerate the ion beam for soft‐landing on the surface (see also the Supporting Information, SI‐A). This ensures the intact and highly pure deposition of the selected species and that imaging is performed on the intact, for example, nonfragmented or degraded molecular species (dehydrogenated sucrose).8
After deposition, the deprotonated sucrose molecules most likely lose their charge to yield dehydrogenated sucrose molecules. These are mobile on the Cu(100) surface at room temperature, as we do not observe stable aggregates by STM. After cooling the sample to 40 K, we observe that nearly all the molecules are assembled in 2D islands arranged in a periodic porous network that exhibits frayed ends (Figure 1; for an overview image, see Figure S2 in the Supporting Information). The apparent building blocks of this network are elongated double lobes, four of which come together to form a node. We assign each (double) lobe to a single molecule because the dimensions of the lobes of (1.0±0.2) nm in length and 0.5 nm in width fit the expected size of a sucrose molecule.
Figure 1.
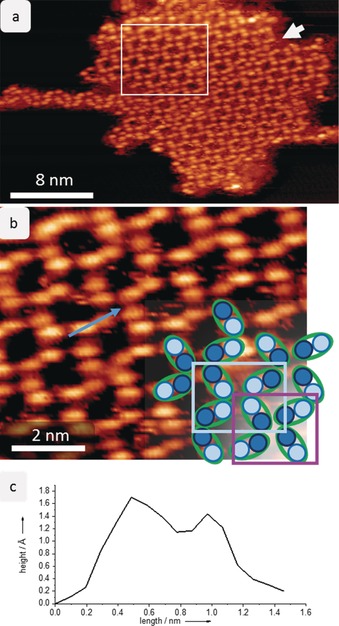
Self‐assembly of sucrose on Cu(100) at 40 K. a) STM image of one island of sucrose on Cu(100) showing the periodic network. The molecules are seen as oval lobes; a white arrow indicates a domain boundary that divides the entire island. An additional structure is found on the left side of the island. The white rectangle marks the area enlarged in (b). b) Magnification of the periodic network. The sucrose molecule is imaged as double lobes with two features of different intensity. In the lower right corner, a schematic representation is overlaid to illustrate the assembly. Two motifs are formed with either bright features (light blue rectangle) or lower‐intensity features (violet rectangle) together in a node. c) The line profile on a sucrose molecule shows the height of the two different features.
At high magnification (Figure 1 b), the entity representing the molecule (i.e. each double lobe) is resolved as two distinct, round features that differ by 0.3 Å in apparent height. Note that apparent height in an STM measurement is a convolution of actual topographic height and electron density. We assign these features to the two different monosaccharide building blocks of sucrose. On the right side of the STM image in Figure 1 b, the molecule is sketched as a green oval with two circles of different color, which correspond to the different intensities observed by STM. In the 2D network, the molecules assemble in such a way that each node is associated exclusively with one kind of feature, high or low, intermittently: either all the bright features come together to form a node (light blue rectangle) or all the dark features form a node (violet rectangle). Moreover, these nodes also differ in compactness: the node associated with the dark features measures (0.9±0.2) nm along the diagonal, whereas the bright feature node measures (1.1±0.2) nm diagonally across the node. Thus, there are two different nodes, which are formed exclusively either by the glucose or the fructose building blocks. However, an unambiguous assignment of the glucose or fructose unit to the bright or dark features is not possible based on the subunit‐resolved STM data alone.
To shed light on the molecular conformation and interactions in the observed assemblies, we modeled the structures at the atomic level. In contrast to the experiment, we considered neutral molecules without dehydrogenation in the calculations. We applied this approximation because we have no information regarding the position of the deprotonation within the molecule on the surface. Nevertheless, we assumed that the overall conformation of the sucrose molecule on the copper surface is only slightly affected by the dehydrogenation. This point is further discussed in the Supporting Information. We have to take into account the inherent flexibility of the molecule, which enables different adsorption configurations and intermolecular interactions (van der Waals and hydrogen bonds). High‐accuracy calculations are complicated and not feasible because of the large surface area that the assembly occupies. Thus, we employed a multistage procedure to accommodate all these needs (see the Supporting Information, SI‐B), as is often done in such a case.9
In a first step, we applied an iterative global exploration and local optimization (IGLOO) scheme, using an empirical potential, to explore the energy landscape of a single sucrose molecule on the surface with the aim of finding suitable candidate configurations (for details see the Supporting Information, SI‐B). Subsequently, we further optimized these candidate conformations using dispersion‐corrected DFT. By relaxing the constraints on bond lengths and bond angles imposed for the global exploration, the optimized conformations have improved molecule–surface interactions as well as intramolecular interactions through stronger hydrogen bonds. In a last step, we employed the single‐molecule conformations to obtain models of the assemblies according to constraints defined by the STM images, such as dimensions and symmetry, including chirality. It is important to note that only constrained translations of the molecule by the Cu unit‐cell parameter as well as rotations of 90°, 180°, and 270° are possible to keep the molecule on the surface in its optimized state. With these restrictions, only a very limited number of patterns can be constructed. For more details on the simulation procedure, see the Supporting Information (SI‐B).
Figure 2 shows representative conformations of a single sucrose molecule on Cu(100) in the three lowest energy basins found by the global exploration using the IGLOO method, referred to as s‐minA IGLOO, s‐minB IGLOO, and s‐minC IGLOO (see Figure 2 left). The subsequent DFT optimization of the single sucrose molecules produced only minor deformations (see Figure 2 right), the most significant being a modification of the ring puckering of the fructose moiety. The planar conformation in the model considered for the global search transforms to a C3′‐endo conformation after DFT minimization. These rather small structural changes upon switching from empirical potential to ab initio energies lend confidence to the expectation that we have identified the most relevant molecule–surface conformations during the global searches.
Figure 2.
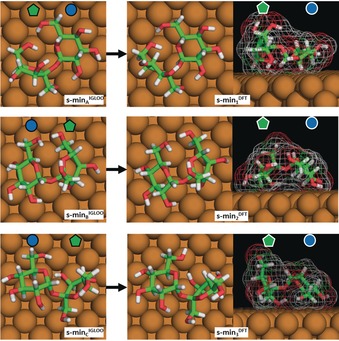
The three lowest energy conformations of sucrose on Cu(100) obtained by the IGLOO algorithm (left images) and after DFT minimization (right images). The glycan notation is used to indicate the glucose and fructose monosaccharide units.
These three low‐energy conformations are quite different from each other with respect to their position and orientation on the surface as well as in their overall molecular conformation, as seen in the side view (right panel). In s‐min1 DFT the fructose ring stands upright, while the glucose ring is almost flat on the surface, thereby leading to a global shape in which one side is higher than the other one by 2.3 Å. In s‐min2 DFT, the molecule is bent and both rings point towards the surface. This bowl‐like structure has a maximum height in its center part. Finally, s‐min3 DFT is characterized by a tilted glucose unit, in which the C4–C6 atoms point towards the surface. Consequently, the bridging oxygen atom also points towards the surface, while the fructose unit stands nearly up‐right so that the ring oxygen and the C6 atoms point away from the surface, thereby yielding a structure that exhibits a height difference of 1.4 Å between the two subunits. Although the conformations of these three low‐energy structures differ strongly, the IGLOO and DFT energy rankings are similar, with the total energy difference with respect to s‐min1 DFT being 12.6 kJ mol−1 for s‐min2 DFT and 22.2 kJ mol−1 for s‐min3 DFT, which is in the order of a hydrogen bond.10
As a consequence of these small differences in energy, we cannot unambiguously determine which of the conformations forms the assemblies solely on the basis of these single‐molecule models and their energy ranking. Thus, we tested all three molecular conformations as possible building blocks for the formation of the assembly pattern, as described above. The possible assembly patterns are discussed in detail in the Supporting Information (SI‐C). The assembly pattern that best reflects the dimensions and symmetry of the observed 2D network is obtained from s‐min3 DFT. We note that the conformation that best fits the assembly is not the lowest energy conformation for the isolated individual molecule on the surface. However, as intermolecular interactions might slightly change the conformation and thus the energies, it is reasonable that s‐min3 DFT is the most favorable conformation when part of the network, that is, a slightly higher energy at the individual molecule level can be compensated by a stronger interaction between molecules. It is also possible that the deprotonation of the molecule during the ESI workflow and the subsequent dehydrogenation on the surface leads to small energy changes, which might reverse the energetic ranking.
The model of the periodic 2D network exhibits two different types of nodes, in which only glucose or fructose units bind. In the glucose node, the molecules are rather flat, bending downwards at the end of the molecule, and can thus interact with each other and form three hydrogen bonds to adjacent molecules. This node is formed on a hollow site of the Cu(100) surface. The distance between two glucose units diagonally across the node is 1.1 nm, whereas the distance across the node of the four fructose building blocks is approximately 0.9 nm. However, as a consequence of the upright conformation of the fructose ring, no hydrogen bonds are formed between the molecules at this site. The absence of hydrogen bonds at the junction of fructose units is a general trend for all the conformations and assemblies tested. As the O4 atom points towards the surface and the O6 atom is oriented towards the molecule itself, there are no possibilities for a hydrogen bond to form.
The simulated STM image of this model (Figure 3) agrees very well with the experimental data, thus validating the model. The intensity distribution on the single molecule is very characteristic: the two monosaccharide building blocks are clearly distinguishable by a depression border between them, and the brightest feature is found on the fructose building block due to the upright‐pointing hydroxymethyl chain. Therefore, we can assign the bright feature in the STM image to the fructose building block, and the low‐intensity feature to the glucose unit.
Figure 3.
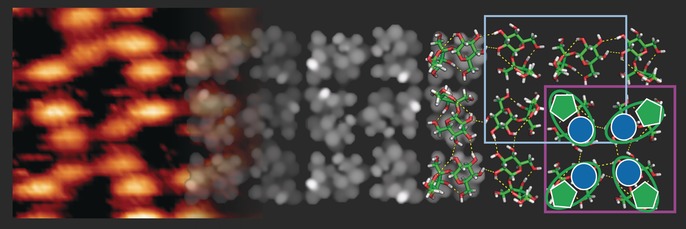
Model of the sucrose assembly pattern. Comparison of the measured STM image (left side) with the simulated STM image (center) demonstrating good qualitative agreement of the observed features, such as a porous structure and different intensities between the glucose (darker) and fructose (brighter) building blocks. On the right side, the molecular structures are presented, with hydrogen bonds indicated as yellow dotted lines.
In this context, we note that these 2D periodic networks do not appear to be related (e.g. as a cut‐out or cross‐section) to the three‐dimensional arrangements of sucrose molecules in the known molecular crystal modification of sucrose.
When considering the (energetic) driving force behind the formation of this periodic structure, four main contributions can be identified: molecule–surface interactions, intramolecular forces, intermolecular hydrogen bonds, and dispersion forces. Although the variation in the molecule–surface interaction and the internal energy of the molecule upon formation of the assembly need to be taken into account to judge the stability of the assemblies, the large number of intermolecular hydrogen bonds that are present in the proposed pattern suggests that they are central to the formation of the molecular assembly. Nevertheless, the molecules remain mobile at room temperature, thus indicating low barriers against lateral movement. This is likely promoted by the molecule's flexibility in conjunction with a large number of available binding sites on the metallic surface, as suggested by the observation of many similar, energetically closely spaced conformations found in the potential landscape searches. Thus, the specific type of periodic network arrangement is defined by the distribution and orientation of the hydrogen bonds, while the intermolecular dispersion (van der Waals) interactions produce an overall tendency to pack tightly.
In summary, we have imaged for the first time the disaccharide sucrose at submolecular resolution by STM, which has allowed the intuitive recognition of its subunits. This high‐resolution imaging was enabled by electrospray ion beam deposition, which can be used as a universal method to generate intact molecular adsorbates from nonvolatile species. Despite the large degree of flexibility of the sucrose molecule, we find a stable periodic arrangement at low temperatures. By combining simulation and imaging, we can identify the conformation of the disaccharide on the surface. The specific knowledge gained about the adsorption geometry of sucrose can help to understand the influence of sucrose on, for example, hydration properties. Moreover, at this level of resolution, STM—in combination with state‐of‐the‐art multistage simulation methods—is a very promising approach to characterize the structure of complex carbohydrates, for example, by identifying branching points. Thus, by employing a new approach for single‐molecule characterization, the present study lays the groundwork for understanding polysaccharide conformations at the submolecular level.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was granted access to the HPC resources of CALMIP supercomputing center under the allocation p16048. We thank Rico Gutzler for helpful discussions.
S. Abb, N. Tarrat, J. Cortés, B. Andriyevsky, L. Harnau, J. C. Schön, S. Rauschenbach, K. Kern, Angew. Chem. Int. Ed. 2019, 58, 8336.
References
- 1. Klemm D., Heublein B., Fink H.-P., Bohn A., Angew. Chem. Int. Ed. 2005, 44, 3358–3393; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3422–3458. [Google Scholar]
- 2.
- 2a. Beevers C. A., McDonald T. R. R., Robertson J. H., Stern F., Acta Crystallogr. 1952, 5, 689–690; [Google Scholar]
- 2b. Brown G. M., Levy H. A., Acta Crystallogr. Sect. B 1973, 29, 790–797. [Google Scholar]
- 3. Varki A., Glycobiology 2017, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Smith B. J., Rawal A., Funkhouser G. P., Roberts L. R., Gupta V., Israelachvili J. N., Chmelka B. F., Proc. Natl. Acad. Sci. USA 2011, 108, 8949–8954; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Smith B. J., Roberts L. R., Funkhouser G. P., Gupta V., Chmelka B. F., Langmuir 2012, 28, 14202–14217. [DOI] [PubMed] [Google Scholar]
- 5. Duchstein P., Kniep R., Zahn D., Cryst. Growth Des. 2013, 13, 4885–4889. [Google Scholar]
- 6.
- 6a. Baker A. A., Helbert W., Sugiyama J., Miles M. J., Biophys. J. 2000, 79, 1139–1145; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Abb S., Harnau L., Gutzler R., Rauschenbach S., Kern K., Nat. Commun. 2016, 7, 10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rauschenbach S., Stadler F. L., Lunedei E., Malinowski N., Koltsov S., Costantini G., Kern K., Small 2006, 2, 540–547. [DOI] [PubMed] [Google Scholar]
- 8. Rauschenbach S., Ternes M., Harnau L., Kern K., Annu. Rev. Anal. Chem. 2016, 9, 473–498. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Schön J. C., Oligschleger C., Cortes J., Z. Naturforsch. B 2016, 71, 351–374; [Google Scholar]
- 9b. Copie G., Makoudi Y., Krzeminski C., Chérioux F., Palmino F., Lamare S., Grandidier B., Cleri F., J. Phys. Chem. C 2014, 118, 12817–12825; [Google Scholar]
- 9c. Kalashnyk N., Nielsen J. T., Nielsen E. H., Skrydstrup T., Otzen D. E., Lægsgaard E., Wang C., Besenbacher F., Nielsen N. C., Linderoth T. R., ACS Nano 2012, 6, 6882–6889. [DOI] [PubMed] [Google Scholar]
- 10. Jabłoński M., Kaczmarek A., Sadlej A. J., J. Phys. Chem. A 2006, 110, 10890–10898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary