

Abstract
Some proteins are expressed as a result of a ribosome frameshifting event that is facilitated by a slippery site and downstream secondary structure elements in the mRNA. This review summarizes recent progress in understanding mechanisms of –1 frameshifting in several viral genes, including IBV 1a/1b, HIV‐1 gag‐pol, and SFV 6K, and in Escherichia coli dnaX. The exact frameshifting route depends on the availability of aminoacyl‐tRNAs: the ribosome normally slips into the –1‐frame during tRNA translocation, but can also frameshift during decoding at condition when aminoacyl‐tRNA is in limited supply. Different frameshifting routes and additional slippery sites allow viruses to maintain a constant production of their key proteins. The emerging idea that tRNA pools are important for frameshifting provides new direction for developing antiviral therapies.
Keywords: frameshifting, protein synthesis, recoding, regulation, ribosome, RNA, translation, tRNA
Abbreviation
IBV, infectious bronchitis virus
LSU, large ribosomal subunit
PRF, programmed ribosome frameshifting
SFV, Semliki Forest virus
smFRET, single‐molecule FRET
SSU, small ribosomal subunit
TRIT, tRNA Inhibition Therapy
Translation is a tightly regulated step of gene expression which ensures the synthesis of proteins according to the sequence of an mRNA produced during transcription. The ribosome, a macromolecular machine that synthesizes proteins in all cells, controls accurate decoding of mRNA triplets by the respective aminoacyl‐tRNAs (aa‐tRNAs) 1, 2, 3, 4, 5, 6. Stringent tRNA selection mechanisms ensure a very low overall error rate of mRNA decoding in the range of 10−7 to 10−5 7. Each time an amino acid is incorporated into the growing peptide chain, the ribosome moves by one codon along the mRNA to read the next codon. Keeping the codon‐wise step of translation is even more essential than preventing missense errors, because slippage by one or two nucleotides results in a completely altered sequence of the synthesized peptide. In fact, spontaneous frameshifting errors are rather infrequent, <10−5 per codon 8. However, in some cases, this rigorous reading frame control is abrogated allowing the ribosome to recode genetic information in response to specific stimulatory signals embedded in the mRNA sequence or structure 9, 10. Programmed ribosome frameshifting (PRF) is a recoding event that allows to produce multiple proteins from the same mRNA by shifting the reading frame in the forward (+PRF) or backward (–PRF) direction 9, 10, 11, 12. Slippage occurs typically by 1 nucleotide, although 2‐, 4‐, 5‐, and 6‐nucleotide shifts were also reported 13, 14, 15, 16, 17. –1PRF is found in all kingdoms of life including higher eukaryotes, but is particularly prevalent in viruses and mobile genetic elements 9, 10, 18. The biological significance of PRF is to increase the genome‐coding capacity, to control the stoichiometric ratio between proteins and to regulate gene expression by influencing mRNA stability 9, 10. Many human pathogenic viruses require frameshifting for their viability, because it ensures production of certain viral enzymes that are not encoded in the 0‐frame and modulates the ratio between viral structural proteins necessary for virion assembly 18, 19, 20, 21. Among the viral proteins produced upon –1PRF, many contribute to viral infectivity either directly by evading the host antiviral response 22 or indirectly by hindering viral particle formation and release, thereby decreasing viral titer 20, 23.
In this review, we summarize the recent progress in understanding the mechanisms of –1PRF, including the different routes of –1PRF and the mechanisms leading to alternative reading frames, for example, +1 and –2 frameshifting, which we call alternative slippages. In addition to two well‐studied cases of –1PRF in avian infectious bronchitis virus (IBV) and bacterial dnaX, we focus on examples of –1PRF from two phylogenetically distant human viruses, human immunodeficiency virus type 1 (HIV‐1), and Semliki Forest virus (SFV). We show how tRNA abundance can modulate the routes and efficiency of –PRF and discuss how different routes and additional slippery sites result in robust –1PRF which is needed for virus life cycle.
Frameshifting elements in the mRNA
Typically, –1PRF is governed by two cis‐acting elements—a slippery site (SS) and a downstream mRNA secondary structure (Fig. 1). The SS is a repetitive heptanucleotide sequence of the type X1 XXY4 YYZ7, which allows the two tRNAs that read the 0‐frame codons XXY and YYZ to re‐pair with their XXX and YYY codons after the slippage into the –1‐frame 24. The mRNA secondary structure—a stem‐loop (SL) or a pseudoknot (PK) or a kissing loop 25, 26, 27, 28—acts as a roadblock to hinder translocation and thereby promote frameshifting 29, 30. In addition, Shine–Dalgarno‐like (SD‐like) sequences in bacteria 31, trans‐acting proteins in viruses 32, 33, 34, 35, G‐quadruplexes 36, 37 and miRNAs in mammalian cells 32 can modulate the –1PRF efficiency.
Figure 1.
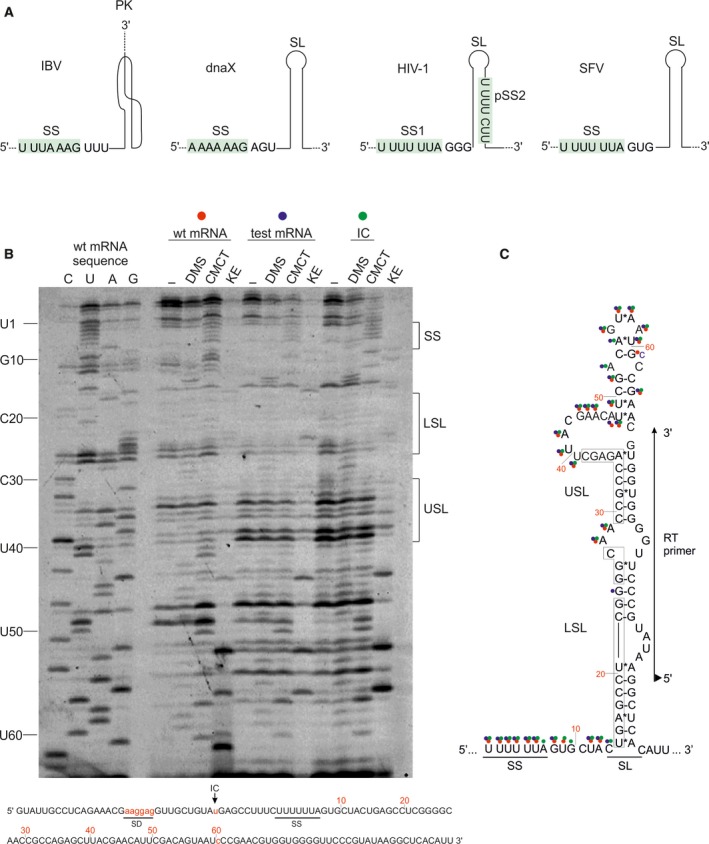
Sequence and structure of mRNA frameshifting motifs. (A) From left to right, schematics of frameshifting motifs of IBV 1a/1b, dnaX of E. coli, gag‐pol of HIV‐1, and 6K of SFV. Slippery sites (SS) are indicated and highlighted in green. The regulatory downstream mRNA element is a pseudoknot (PK) or a stem‐loop (SL), as indicated. pSS2 in HIV‐1 stands for the second putative SS. (B) Chemical probing of the mRNA secondary structure element in the SFV 6K mRNA. In vitro transcribed mRNA was treated with dimethyl sulfate (DMS; A‐ and C‐specific), 1‐cyclohexyl‐3‐(2‐morpholinoethyl) carbodiimide metho‐p‐toluenesulfonate (CMCT; U‐specific and low reactivity toward G) and β‐ethoxy‐α‐ketobutyraldehyde (kethoxal KE; G‐specific, and analyzed base modifications by primer extensions 109. (–) indicates untreated mRNA. Positions of reverse transcription (RT) stops due to modification were visualized on a sequencing gel using fluorescence primer complementary to positions 109–129 nucleotides of the mRNA (60–80 nucleotides downstream the SS). wt mRNA has the native sequence; test mRNA has been optimized for translation in E. coli (see lower panel; SD is Shine–Dalgarno sequence, AUG is the start codon, G61 is mutated to C to remove a potential initiation codon); IC is the initiation complex of test mRNA with 70S ribosomes. C, U, A, G are sequencing lanes. Numbered nucleotides to the left refer to the nucleotides in the SFV mRNA starting from the slippery site as indicated in c. LSL is lower stem‐loop, USL is upper stem‐loop. (C) Secondary structure of SFV 6K mRNA based on bioinformatics prediction 27 and probing results. Modified nucleotides are marked with circles: red for the wt mRNA, blue for the test mRNA and green for the test mRNA in the IC. Sequences in boxes indicate nucleotides forming lower (LSL) and upper (USL) stems. Primer‐binding site for RT is marked with an arrow; triangle on the 5′ of the primer indicates its fluorescence label Atto647N.
In most cases, the structure of the downstream mRNA secondary structure is known from bioinformatics, chemical probing, mutagenesis, or structural studies. For example, the PK at the frameshifting site of IBV was discovered based on mutational analysis and its structure predicted by bioinformatics (Fig. 1A), whereas the atomic structure of this PK is still lacking 28, 38. The structure of the SL in bacterial dnaX was initially suggested based on mutational analysis and structural probing 39; recent cryo‐EM studies determined the structure of this SL bound to the bacterial ribosome (Fig. 1A) 40. The SL structure of HIV‐1 is one of the best‐studied examples of mRNA secondary structures that modulate –1PRF. Its structure was solved using mutagenesis and enzymatic probing 41, thermodynamic and NMR analysis 28, 42, 43, as well as toeprinting and chemical probing in the presence of the bacterial ribosome (Fig. 1A) 44.
The secondary structure element of SFV was predicted to be an extended SL by bioinformatics and mutational analysis 27. We validated the structure by chemical probing, which can distinguish single‐ and double‐stranded RNA regions by their accessibility to chemical modification (Fig. 1B). The chemicals were chosen such as to modify the Watson–Crick positions of the nucleotide base; double‐stranded regions are protected from chemical modifications due to base pairing to the complementary strand. Modification causes a stop in the progression of the reverse transcriptase (RT) resulting in the production of short cDNA fragments, which can be then visualized by sequencing (Fig. 1B). In agreement with the bioinformatics predictions 27, the SL element in SFV contains a long lower stem encompassing nucleotides 15–26 after the SS (counting from nucleotide 1 of the SS), as seen from the lack of chemical modifications of this region (Fig. 1B,C). Nucleotides 28–29 form a small unstructured loop between the lower and upper stems, consistent with their accessibility to modifications. According to the bioinformatics analysis, C27 also belongs to this loop; however, its modification status is unclear. The upper stem was predicted to span nucleotides 30–35; however, our results suggest that also the adjacent nucleotides 36–39 are protected from modifications and thus might belong to the upper stem, although the accessibility of the complementary strand nucleotides (nucleotides 64–68) is unclear (Fig. 1C). The upper stem is closed by a large bulge spanning nucleotides 40–48, as predicted by the bioinformatics analysis and supported by the chemical probing data. Nucleotides 49–52 are predicted to form a small stem, but appear to be in a single‐stranded region according to chemical probing. According to the bioinformatics analysis, C54 is base paired to G61, suggesting that both should be inaccessible to chemical modification. This is, however, not the case: C54 is indeed inaccessible, but G61 is modified. In addition, when G61 is mutated to C61, it becomes protected, suggesting that the interaction pattern is more complex than predicted. Finally, we confirm the presence of the predicted AGUAAU loop closing the upper stem (Fig. 1C). Hence, chemical probing largely supports the structure of the SFV SL predicted by the bioinformatics analysis 27. The major differences concern the length of the upper stem, which appears to include nucleotides 36–39 based on chemical probing, and the absence of the predicted small stem spanning nucleotides 49–52. As expected, the SS sequence is single‐stranded. Nucleotides between the SS and the SL (nucleotides 8–14) show a very high degree of modification indicating that they do not belong to the SL but represent a single‐stranded spacer between the two frameshifting elements, as predicted 27. Binding of the ribosome to the start codon (initiation complex, IC) does not affect the mRNA structure, most likely because the ribosome binds to the mRNA region distal from the SL (Fig. 1B,C). Nucleotides C14, A53, A55 show different reactivity in the IC than in the free wt mRNA (Fig. 1B,C), however, this effect is most likely caused by the mutations in the test mRNA sequence introduced to ensure translation initiation in E. coli, rather than by the presence of the ribosome.
Mechanisms of –1PRF on IBV 1a/1b and E. coli dnaX mRNAs
–1PRF takes place during the elongation phase of translation, but due to a lack of kinetic data, the exact timing of –1PRF was until recently unknown. This led to the proposal of a number of different models that differ in the timing of the –1PRF during the elongation cycle, for example, some models predicted that –1PRF occurs during aa‐tRNA accommodation in the A site of the ribosome (integrated & 9 Å models), before peptidyl transfer (simultaneous slippage model), during hybrid state formation in translocation (dynamic model), in the post‐translocation state of the ribosome (mechanical model) or during the next round of elongation requiring three‐tRNA slippage (three‐tRNA model) (summarized in 12). The lack of a unifying model has prompted several groups to investigate –1PRF by ensemble kinetics or single molecule methods 14, 15, 29, 30, 45, 46. Compared to results obtained previously with cellular lysates (e.g., rabbit reticulocyte lysate), the in vitro assays used in these experiments have the advantage of providing a fully controlled environment where every elemental step of translation elongation can be dissected. Results for two unrelated frameshifting examples, gene 1a/1b of IBV 29 and dnaX of E. coli 14, 15, 30 suggested that –1PRF occurred predominantly during the tRNA translocation step and provided no support for any of the other models listed above. In the following we will discuss the current kinetic models of frameshifting.
The frameshifting motif of IBV gene 1a/1b consists of a SS motif U1 UUA4 AAG7 encoding Leu (UUA) and Lys (AAG) in 0‐frame, and a PK positioned 6 nucleotides downstream of the SS (Fig. 1A) 38. Comparison of the ensemble kinetics 29 and single molecule Fluorescence Resonance Energy Transfer (smFRET) 47, 48 results with the structural intermediates of translocation 49, 50, 51 suggests that frameshifting occurs when the 3′ ends of the P‐ and A‐site tRNAs have moved to the E and P site, respectively, on the large ribosomal subunit (LSU), but the tRNA anticodon domains are not yet fully translocated on the small ribosomal subunit (SSU) 29, 30, 46, 52, that is, frameshifting occurs when two tRNAs are bound to the ribosome. Because the ribosome adopts a conformation in which the SSU head domain swivels relatively to the body domain, the tRNA anticodons are placed in an intermediate position with respect to the SSU head and body domains 49, 52, 53. In this so‐called chimeric (with respect to SSU) hybrid (to LSU) state, the codon–anticodon interaction is destabilized, which favors frameshifting 54. The PK structure in the mRNA downstream of the SS impedes the closing movement of the 30S SSU head domain, which, in turn, hinders the release of the deacylated tRNA from the E site and the completion of translocation 29, 45. The presence of the mRNA secondary structure element leads to translational pausing and opens a kinetic window in which tRNAs can slip into a different reading frame. Notably, –1PRF appears favorable for translation because the ribosomes that shifted into the –1‐frame complete translocation and release EF‐G faster than those remaining in 0‐frame 29. Hence, –1PRF could be considered as a rescue mechanism to resolve a persistent translational block caused by a secondary structure and resume translation at its normal rate.
Another well‐studied example of –1PRF is on the dnaX mRNA of E. coli. The slippery site contains the SS motif A1 AAA4 AAG7 encoding two Lys (AAA and AAG) in 0‐frame, the downstream SL and an SD‐like sequence upstream of the SS (Fig. 1A) 39, 55, 56. Here –1PRF proceeds via two alternative routes, one of which is identical to that described for IBV 1a/1b, whereas the other is activated by aa‐tRNA limitation 15. –1PRF was studied on an dnaX model mRNA in detail using smFRET, mass spectrometry and rapid ensemble kinetics 14, 15, 30, 46. smFRET and ensemble kinetics show that the presence of the downstream SL hinders translocation, whereas the rates of A‐site tRNA delivery and peptidyl‐transfer are unchanged 15, 30, 45. The presence of the downstream SL in the dnaX mRNA also hinders the E‐site tRNA release 30, 45, as was shown with the PK of IBV 29. While pausing at the SL, ribosomes undergo multiple conformational transitions between classical and hybrid states in the presence of EF‐G 30, 46. EF‐G may take multiple attempts to complete translocation while the ribosome tries to resolve the secondary structure to continue canonical decoding in 0‐frame 14, 46, 57. This prevalent translocation‐dependent route, which corresponds to the two‐tRNA slippage mechanism suggested earlier 57, 58, 59, 60, 61, 62, is similar in IBV 1a/1b and E. coli dnaX.
Another pathway for frameshifting on dnaX was found to be operational when the A site remains vacant due to the absence of the cognate aa‐tRNA. In this case, delayed decoding eventually allows slippage of the single P‐site tRNA. Once the ribosome encounters a codon for which an aa‐tRNA is available, normal translation resumes. Compared to translocation‐dependent –1PRF, this so‐called ‘hungry’ or one‐tRNA frameshifting is very slow and does not require the presence of the downstream mRNA secondary structure 14, 15. Also, smFRET experiments suggest the presence of two branch points determining the reading frame: one during translocation, as described 15, 30 and one during tRNALys sampling in the A site 46. Mass spectrometry and optical tweezers data suggest that during translocation the ribosome attempts multiple trajectories or excursions along the mRNA and that the slippage into the new frame might occur from different 0‐frame codons 14. Many frameshift attempts fail due to codon–anticodon base pair mismatches in the alternative frame leading to the accumulation of incomplete peptides 14. This scenario explains many cases of alternative frameshifting events at conditions of in vitro translation, for example, −4 and +2 frameshifting and decoding‐related frameshifting observed on dnaX 14, 46 and may explain the heterogeneity of frameshifting products observed in previous in vivo studies 63, 64. The ‘hungry’ frameshifting also relates to a common mechanism for how frameshift suppressor tRNAs can rescue mRNA frameshift mutants by a –1 frameshift 65. For example, a translation defect caused by an G insertion in the mRNA resulting in a sequence ‐GGG‐GAA‐AGA‐ can be rescued by loss‐of‐function mutations in tRNAArg reading the AGA codon. The delay in decoding allows tRNAGlu bound at the P‐site GAA codon to slip into the –1‐frame, which restores translation in 0‐frame 66.
Given that –1PRF in IBV 1a/1b and E. coli dnaX proceeds via the same translocation‐dependent two‐tRNA slippage, the question arises whether the same mechanism is utilized in other cases as well or if any of the earlier proposed mechanisms can be identified. One of the most widely known cases of –1PRF is the gag‐pol overlap in HIV‐1, which prompted us to study the mechanism of –1PRF in this system. In addition, we studied –1PRF in SFV, a virus phylogenetically distant from HIV‐1, which has the same slippery site sequence as HIV‐1 with a remarkable degree of conservation in different virus subtypes 20. In the following section we will describe the mechanism of –1PRF on these two mRNAs.
Mechanism of –1PRF on slippery sites of gag‐pol in HIV and 6K in SFV
–1PRF in HIV‐1 takes place at the gag‐pol gene overlap and defines the ratio between the structural proteins of the capsid (Gag, 0‐frame) and the viral enzymes (Gag‐Pol, –1‐frame). Changes in the frameshift efficiency impede viral particle formation and are detrimental for viral viability and infectivity 21, 67. Notably, –1PRF on the gag‐pol mRNA results in two different –1‐frame products in addition to the 0‐frame peptide. Although numerous models were proposed to explain –1PRF in HIV‐1 21, 59, 61, 62, 68, the origin and biological significance of the two frameshifting products remained unknown, which prompted us to dissect the frameshifting event on the gag‐pol mRNA using our in vitro reconstituted translation system with either mammalian or bacterial components 69. The reported efficiency of gag‐pol –1PRF is about 10%, as measured with dual‐luciferase reporters in vivo in human cell culture 70, 71. We note that this frameshifting efficiency has been recapitulated in bacteria, yeast, and mammalian cells in vivo and in the respective in vitro translation systems, suggesting that –1PRF relies on the highly conserved components of the translational apparatus 21, 68, 72. The frameshifting motif of HIV‐1 consists of a main SS, SS1, with a sequence of U1 UUU4 UUA7 encoding Phe (UUU) and Leu (UUA) in 0‐frame with a downstream SL element (Figs 1A and 2) 21, 43. Of the two distinct frameshifting products, one contains the 0‐frame peptide Phe‐Leu followed by the –1‐frame amino acid sequence (FLR) and the other one a Phe incorporated instead of Leu, that is, Phe‐Phe followed by the –1‐frame sequence (FFR) (Fig. 2). The ratio of the two –1PRF products is about 70% to 30% 21, 61, 62, 64.
Figure 2.
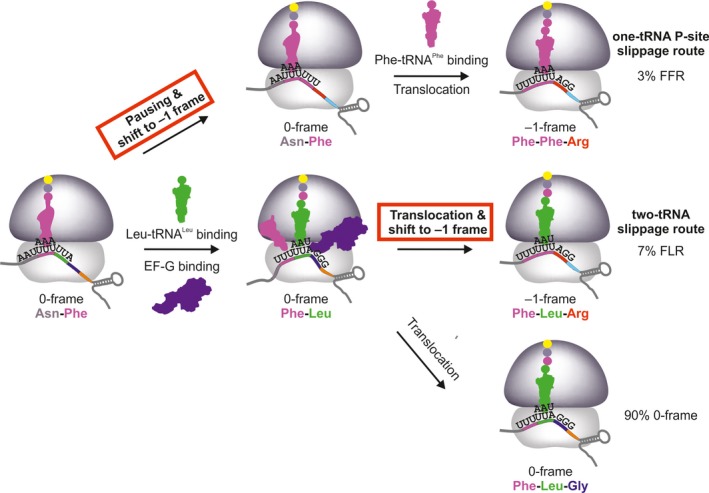
Kinetic mechanisms of FFR (upper) and FLR (lower) –1PRF pathways on the gag‐pol mRNA of HIV‐1. FFR results from one‐tRNA slippage with peptidyl‐tRNAP he in the P site (in magenta) when the A site is vacant due to low availability of Leu‐tRNAL eu( UAA ). FLR arises upon frameshifting during translocation of tRNAP he and peptidyl‐Phe‐Leu‐tRNAL eu( UAA ) (in green) and is prevalent at excess of Leu‐tRNAL eu( UAA ). After reading the slippery site, translation can continue in the –1‐frame by incorporating Arg at the AGG codon (red) or in 0‐frame by decoding Gly at the GGG codon (blue). The –1‐frame commitment steps on the FFR and FLR routes is marked in red. –1PRF on SFV 6K, IBV 1a/1b, and E. coli dnaX can, in principle, follow the same two routes. The existence of the two‐tRNA route is well‐documented 15, 29, 30, 45, 46, 57. The prevalence of the one‐tRNA route for SFV 6K depends on the concentration of tRNAL eu( UAA ) in the infected neuronal cells, which is not known (see text below). The one‐tRNA slippage on IBV 1a/1b could occur before decoding of the first slippery seqence codon UUA by the respective rare tRNAL eu( UAA ), however, the existence of the respective –1‐frame peptide product containing Phe‐Lys, rather than Leu‐Lys, has not been tested and the abundance of tRNAL eu( UAA ) in avian host cells is unknown. For dnaX, tRNAL ys that reads the slippery site codons is abundant and the one‐tRNA frameshifting pathway is only elicited by starvation.
Kinetic analysis shows that –1PRF in HIV‐1 proceeds via the same two routes as previously described for IBV 1a/1b and E. coli dnaX mRNAs (Fig. 2) 69. Intriguingly, we found that the two routes depend on the availability of Leu‐tRNALeu(UAA) to read the UUA codon of the SS1 (Fig. 2) 69. At limited supply of Leu‐tRNALeu(UAA) decoding is slow. While the ribosome waits for the tRNA delivery, the P‐site peptidyl‐tRNAPhe bound to the UUU codon slips backwards and re‐pairs with another UUU codon in –1 frame, resulting in the change of the identity of the A‐site codon from UUA to UUU and the incorporation of the second Phe into the peptide, which gives rise to the FFR –1‐frame product (consistent with earlier suggestions 59, 61, 64, 68) (Fig. 2). At low Leu‐tRNALeu(UAA) concentrations, the rate of Leu incorporation is lower than that of P‐site tRNAPhe slippage; hence, the one‐tRNA slippage and the respective FFR route prevail. With increasing Leu‐tRNALeu(UAA) concentration, its incorporation rate increases, which abolishes the FFR route and favors the two‐tRNA slippage resulting in FLR pathway of frameshifting (Fig. 2). Along with kinetic data 69, one of the main arguments supporting this finding is the lack of competition between tRNAGly (0‐frame) and tRNAArg (–1‐frame) reading the codon GGG (or AGG in –1‐frame) following the SS1 69. This observation indicates that the commitment to –1‐frame happens upon Leu incorporation but prior to the next codon decoding.
Another example of viral frameshifting is found in gene 6K of the alphavirus SFV. Here –1PRF defines the ratio between two structural proteins, 6K (0‐frame) and TransFrame (TF, –1‐frame), which play a role in the envelope protein processing, membrane permeabilization, virion assembly, virus budding, and contribute to infectivity 20. The efficiency of –1PRF in SFV measured with dual‐luciferase reporters in human cells is about 15% 27. Having probed the mRNA secondary structure element on the 6K mRNA (Fig. 1B,C), we studied the mechanism of –1PRF as described for IBV, dnaX, and HIV‐1. Because SFV and HIV‐1 have the same SS sequence U1 UUU4 UUA7, we hypothesized that –1PRF in SFV also results in two peptides, in this case FFS and FLS, depending on the presence of the Leu‐tRNALeu(UAA) isoacceptor (Fig. 3A). In the absence of Leu‐tRNALeu(UAA), the FFS product is formed and its yield depends on the concentration of Phe‐tRNAPhe (Fig. 3B), suggesting that the slippage occurs prior to and independent of tRNALeu(UAA) incorporation, similarly to ‘hungry’ slippage in HIV‐1. With the increase in Leu‐tRNALeu(UAA) concentration, the –1PRF efficiency decreases dramatically from about 70% in the absence of tRNALeu(UAA) to 18% at tRNALeu(UAA) saturation (Fig. 3C). Thus, the FFS route in SFV is operational when tRNALeu(UAA) is absent or in limited supply, whereas under saturating translation conditions the FLS route becomes prevalent. To better understand the FLS regime, we tested the competition between 0‐frame Val‐tRNAVal and –1‐frame Ser‐tRNASer for binding at the codon following the SS, GUG. Titration of these tRNAs in the presence of equimolar amounts of tRNALeu(UAA) (1:1 molar ratio to 70S) does not change the frameshifting efficiency appreciably (Fig. 3D), which suggests that, similarly to HIV‐1, FLS products result from the dual slippage of the SS tRNAs tRNAPhe and tRNALeu(UAA) in the late stage of translocation before the GUG codon is presented in the A site.
Figure 3.
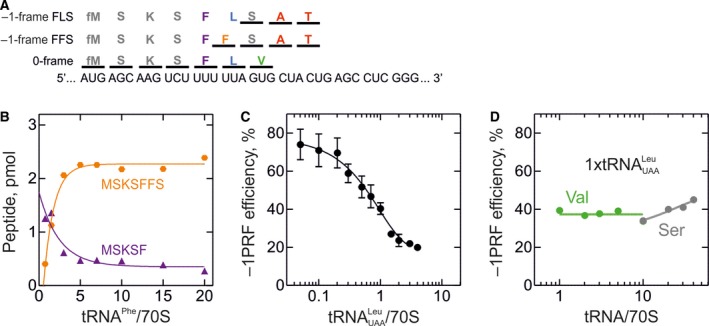
Mechanism of –1PRF on the SFV 6K mRNA. Translation was carried out in HiFi buffer at 37 °C as described in 69 for the gag‐pol mRNA; concentrations were Ser‐tRNAS er and Phe‐tRNAP he (with 0.8 μm each) and Lys‐tRNAL ys, Val‐tRNAV al, Ala‐tRNAA la and Thr‐tRNAT hr (0.25 μm each) and IC (0.08 μm) programmed with the 6K mRNA. Translation products were separated by reversed phase high‐performance liquid chromatography 69. 0‐frame products were identified based on the incorporation of [14C]Val, –1‐frame peptides using [14C]Ala and [14C]Thr. The –1PRF efficiency was calculated as a ratio between –1‐frame peptides and the sum of –1‐frame and all 0‐frame products, multiplied by 100%. (A) Schematic of the frameshifting site. The model SFV mRNA containing native SS and SL is optimized for translation in E. coli by introducing a SD sequence and a start codon AUG followed by AAG (Lys) to improve translation efficiency. (B) Effect of Phe‐tRNAP he on FFS peptide formation in the absence of Leu‐tRNAL eu( UAA ). Translation was carried out using tRNAs aminoacylated with M, S, K, F. (C) Dependence of –1PRF on Leu‐tRNAL eu( UAA ) concentration. Translation was carried out with M, S, K, F, L, V, A, and T aa‐tRNAs. (D) Effect of Val‐tRNAV al (green circles) and Ser‐tRNAS er (gray circles) concentrations on –1PRF efficiency. Translation was carried out using an equimolar concentrations of Leu‐tRNAL eu( UAA ) and aa‐tRNAs as in C. The large excess of Ser‐tRNA is required to ensure efficient translation of the SFV mRNA, which contains three Ser codons read by different tRNAS er isoacceptors in the total tRNAS er used in these experiments.
Effect of the tRNA pool on frameshifting and the implications for potential antiviral therapies
As described above, the Leu‐tRNALeu isoacceptor reading the UUA codon of the SS in HIV‐1 and SFV acts as the main modulator of the –1PRF efficiency and defines the frameshifting route in these two viruses. Interestingly, the UUA codon along with other A‐ending codons is rare in humans, but accounts for more than 45% of all Leu‐coding codons in late‐expressing genes of HIV‐1 including gag and pol 73, 74. Leu‐tRNALeu(UAA) is dramatically underrepresented in CD4+ T‐lymphocytes, the primary target cells for HIV‐1 infection in humans, that is, it is 20‐fold less abundant than the major Leu isoacceptor Leu‐tRNALeu(CAG) 69. The high demand for Leu‐tRNALeu(UAA) to achieve efficient gag and gag‐pol translation may additionally deplete the pool of free Leu‐tRNALeu(UAA) in the cell, thus further decreasing its availability for decoding at the gag‐pol frameshifting site. Furthermore, tRNA levels are known to fluctuate in response to interferon activation and to changes in gene expression triggered by viral infection 75, 76. In light of these observations, FFR and FLR –1‐frame products resulting from two different routes in HIV‐1 may represent an adaptation strategy of the virus to the changing availability of the crucial Leu‐tRNALeu(UAA). When the concentration of tRNALeu(UAA) decreases, the ribosome switches to the FFR route leading to robust –1PRF, thereby maintaining a stationary frameshifting level independent of small tRNALeu(UAA) fluctuations that are realistic in vivo. We also note that unlike in dnaX, where the ‘hungry’ frameshifting pathway is activated only under starvation conditions, HIV‐1 can use both ‘hungry’ (FFR) and translocation‐dependent (FLR) pathways constitutively to achieve a constant –1PRF efficiency. Unlike HIV‐1, SFV primarily infects neuronal cells (neurons and oligodendrocytes) 77, in which the level of tRNALeu(UAA) is not known; thus the physiological relevance of the two potential –1PRF pathways in SFV remains to be elucidated.
Given the low level of tRNALeu(UAA) in human T‐lymphocytes, the question remains how HIV‐1 can satisfy its high demand for this tRNA to achieve an efficient translation of its late‐expressing genes. HIV‐1 can package some cellular tRNAs, among them tRNALys, tRNAIle and to a lesser extent tRNALeu(UAA), during virion assembly 78. Because tRNA packaging happens passively governed by the concentration gradient, these tRNAs must be present in the cell at significant concentrations. There are multiple indirect indications that HIV‐1 itself can affect the tRNA pools by yet unknown mechanisms 74 and that HIV infection can change the cellular localization of individual aa‐tRNA synthetases from the multi‐aa‐tRNA synthetase complex 79, which may affect their aminoacylation activity. Other viruses whose genomes have a codon usage different from their host can alter the free tRNA pools by changing polysome‐associated tRNA levels (vaccinia and influenza A) or by tRNA misacylation (influenza A and adenovirus) 76, 80, but the mechanism of how HIV‐1 could modulate tRNA concentrations remains unknown.
Along with HIV‐1 and SFV, –1PRF in the extended CAG repeats in the human huntingtin mRNA is also modulated by the availability of the aa‐tRNA 81. Expansion of CAG‐encoded poly‐glutamine (polyQ) stretches beyond a certain threshold leads to the development of progressive neurodegenerative diseases including Huntington's disease and spinocerebellar ataxia 82. Translation of the extended CAG repeats results in the depletion of tRNAGln(CUG) which recognizes the CAG codon. The depletion of the pool of free tRNA, in turn, causes –1PRF, the efficiency of which depends on the length of polyQ stretches 81. Here –1PRF likely proceeds via the one‐ tRNA (or ‘hungry’) slippage mechanism, as discussed above for dnaX, HIV‐1 gag‐pol (FFR) and SFV 6K (FFS). Interestingly, tRNAGln(CUG) shows tissue‐specific expression levels in humans, and the lowest concentration of this tRNA was detected in the brain region called striatum, which is the primary site for Huntington's disease 81. In summary, the low level of the target tRNA is linked to the increased frameshifting levels and accumulation of the mutated huntingtin protein, thereby contributing to the disease severity in Huntington's patients.
The strong inhibition of –1PRF in HIV‐1 by excess amounts of tRNALeu(UAA) offers a potential new approach in antiviral therapy. Based on codon usage differences between retroviruses and the human host, multiple tRNA species were predicted that are critical for retroviral protein synthesis but dispensable for human translation, laying the foundation for the hypothetical tRNA Inhibition Therapy (TRIT) 83. Inactivation of these tRNAs should drastically reduce the elongation rate of viral protein synthesis leaving the host translation unaffected. One of the best targets of TRIT, which could be exploited in HIV‐1 and other retroviruses (HIV‐2, HTLV‐1 and 2), is tRNALeu(UAG) reading the CUA Leu codon 83. Furthermore, interferon‐induced protein Schlafen 11 (SLFN11) was shown to selectively inhibit the expression of late HIV‐1 proteins in a codon‐dependent manner 84. SLFN11 induces a selective cleavage of tRNAs with a long variable loop, such as tRNASer and tRNALeu 85. Because HIV‐1 gag‐pol mRNA harbors multiple UUA codons, its translation is susceptible to the action of SLFN11 overexpression due to the tRNALeu(UAA) depletion 85. Recently, another interferon‐induced protein, Shiftless, was shown to dysregulate –1PRF and inhibit replication of HIV‐1 35. However, its potential use in antiviral therapy remains unclear because Shiftless binds to the ribosome and frameshifting sequences of many viral and even human cellular mRNAs 35.
Alternative slippages: examples and mechanisms
Frameshifting events are not limited to −1‐ and +1‐slipagges: ribosomes can slip by −2, +2, +4, +5, or +6 nucleotides 13, 14, 15, 16, 17. For example, the SS of dnaX in E. coli supports not only –1PRF, but also −2, +2, and even +4 frameshifting 14, 15. In dsDNA tailed phages, the tail assembly is typically regulated by a highly conserved –1PRF event; however, in Mu phage a −2 slippage is required instead 86. The synthesis of the full‐length human antizyme requires +1PRF, which can proceed via +1PRF (predominant) and –2PRF in fission yeast, but results from –2PRF in budding yeast; here –2PRF occurs via a one‐tRNA slippage in the presence of the empty A site, as described for ‘hungry’ –1PRF 17. Porcine reproductive and respiratory syndrome virus utilizes both –1 and –2 slippage on the same SS G1 GUU4 UUU7 to produce different replicase polyproteins 87. This is the only described example of –2PRF where the –2 slippage requires the presence of a special mRNA stimulatory sequence downstream of the SS, similar to enhancers known to regulate +1PRF 87, 88. We note that a small amount of +1 and −2 products in HIV‐1 may also result from the transcriptional slippage of RNA polymerase on the gag‐pol SS1 89.
The SS1 of HIV‐1 was also reported to support –2PRF 16. Because in those experiments, the frameshifting sequence was placed into an unnatural context followed by an antisense oligonucleotide‐binding site, we were prompted to study the extent of alternative slippages on the native gag‐pol mRNA frameshifting site 69. HIV‐1 gag‐pol SS1 indeed allows –2 and even +1 slippage, but their contribution to the overall frameshifting efficiency becomes significant only when certain aa‐tRNAs are omitted. Thus, alternative slippages on SS1 must follow the ‘hungry’ route of frameshifting.
In HIV‐1, ribosomes that continue translation in the +1‐ or –2‐frame, soon encounter one of the multiple downstream stop codons, which leads to premature termination. Premature termination is often used upon slippage on nonprogrammed tetra‐ and heptanucleotide slippery sites to abort production of nonfunctional peptides, especially under conditions of aa‐tRNA limitation 90. Premature termination upon frameshifting can also result in the production of functional proteins. One example is E. coli gene copA encoding a copper ion transporter 91. Here –1PRF causes premature termination and formation of a truncated peptide CopA(Z), which turned out to be a copper chaperone protecting cells from excessive copper concentrations in the environment 91. On the other hand, premature termination upon –1PRF in human CCR5 mRNA leads to mRNA degradation by the nonsense‐mediated decay pathway, thus, regulating mRNA stability and gene expression 32.
Appearance of potential slippery sites in viruses upon antiviral therapy
A hallmark of HIV‐1 is its vast genetic diversity and rapid evolution, in particular in response to antiviral therapies. Some of the most potent antiviral drugs target the protease that cleaves the Gag‐Pol polyprotein to yield mature functional viral proteins 92, 93. One of the Gag protease cleavage sites, p1/p6, is located in the part of the protein encoded by the UUU‐CUU codons in pSS2 downstream of SS1; the cleavage is between Phe and Leu 94, 95. In response to antiviral treatment with protease inhibitors, the protease gene of HIV‐1 accumulates mutations that reduce the affinity of the protease for the inhibitor, but simultaneously impair the recognition of its canonical cleavage sites 92, 94, 95. This defect is partially rescued by a compensatory mutation of the CUU to the UUU in pSS2, because the resulting Leu to Phe substitution creates a new functional cleavage site by enhancing van der Waals interactions between the substrate and the mutant protease, thereby allowing Gag‐Pol processing 96. The mutation also turns the U1 UUU4 CUU7 sequence into U1 UUU4 UUU7, which can support ribosome slippage (Fig. 1A) 97, 98, 99, 100, 101. The mutated slippery pSS2 supports both –1 and –2 slippage, but its contribution to the overall frameshifting efficiency depends on the availability of SS1 (Fig. 4) 69. In case of mutated SS1, the native pSS2 cannot rescue the –1PRF efficiency suggesting that its contribution to frameshifting is negligible. When SS1 is functional and pSS2 harbors the C5U mutation, the overall –1 frameshifting efficiency remains unchanged but a significant amount of –2 frameshifting is observed. Most importantly, when SS1 is dysfunctional, the C5U mutation in pSS2 can restore about 70% of the wild‐type –1PRF efficiency, which might be enough to sustain the viral lifecycle 69.
Figure 4.
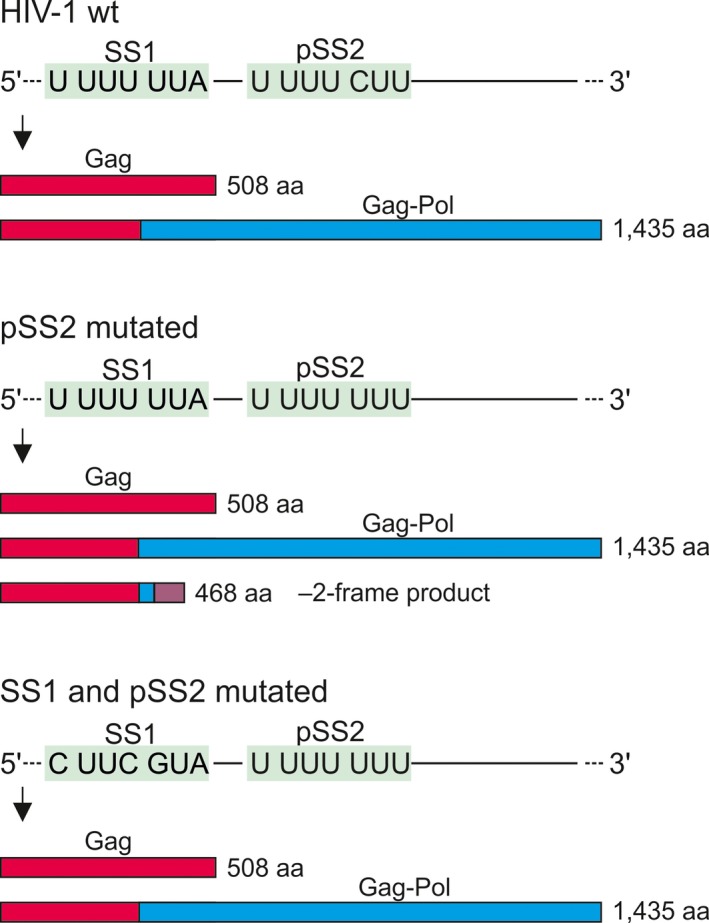
The role of the pSS2 in maintaining the permissive Gag to Gag‐Pol ratio. Top, in the wt gag‐pol mRNA, –1PRF on the SS1 accounts for most of the Gag‐Pol product and pSS2 is silent. Middle, a compensatory mutation in pSS2 that emerges in response to treatment with inhibitors targeting the viral protease makes pSS2 slippery. The production of Gag and Gag‐Pol is unchanged, but a small fraction of ribosomes slips into –2‐frame, resulting in synthesis of a truncated protein. Bottom, when SS1 is mutated, –1‐frameshifting on pSS2 restores Gag‐Pol production to about 70% of that on the wt sequence.
Interestingly, HIV‐1 is not the only virus which develops an alternative slippery site in response to antiviral treatment. Herpes simplex viruses resistant to acyclovir treatment accumulate mutations in their thymidine kinase (TK) gene turning previously silent repeats of G (G‐string) or C (C‐cord) nucleotides into functional slippery sites, supporting +1 and –1 slippage, respectively 102, 103. –1PRF in the C‐cord is thought to proceed via single P‐site tRNA slippage and is stimulated by a nonstop mRNA, presumably, by stalling the ribosome on the polyA‐tail 102. These frameshifting events provide a sufficient level of TK and thus represent a rescue mechanism for the virus to ensure its viability despite the damage caused by the antiviral therapy 102, 103.
Conclusions and future perspectives
Frameshifting sites utilize a great variety of the mRNA secondary structure elements. Despite this diversity, –1PRF seems to operate via two major evolutionary conserved kinetic pathways: through two‐tRNA slippage during translocation or through one‐tRNA P‐site slippage at limited supply of the tRNA that decodes the 0‐frame A‐site codon. The choice of the frameshifting pathway is defined by the availability of the tRNA reading the slippery site codons, which is of crucial importance for pathogenic viruses whose codon usage is different from that of their hosts. Pathogenic viruses have developed different strategies, such as utilizing alternative frameshifting routes, newly emerging slippery sites or alternative slippages, to ensure robust frameshifting efficiencies and thus robust viral cycle progression, regardless of environmental or therapy‐induced changes.
Dependence of –1PRF on cis‐acting elements—the slippery sites and the mRNA secondary structures—is well understood, whereas the list of trans‐acting factors modulating frameshifting is still growing. Recent evidence suggests that multiple proteins of the human immune system can influence the –1PRF efficiency in infectious viruses by acting directly on the frameshifting sites or competing for the translation resources 84, 85. Small interfering RNAs represent another group of important players in modulating frameshifting 32. Better understanding of trans‐acting factors will provide not only novel insights into the mechanism of frameshifting, but will also yield the necessary understanding of the host–pathogen–host interaction and coadaptation, which could lay a foundation for future therapeutic approaches.
Classical antiviral therapies targeting key components of the viral life cycle lead to the emergence of resistant virus isolates due to selection pressure and high mutation rates in viral genomes 104. Frameshifting sites and especially slippery heptamers represent attractive targets for antiviral drug design because their sequences are highly conserved among virus subtypes 20, 105 and frameshifting often determines virus viability. Attempts to develop antiframeshifting therapeutics against HIV‐1 using both natural and synthetic molecules has so far not succeed in clinical trials because of their cytotoxicity and off‐target effects 106, 107, 108. This is because –1PRF relies on highly conserved elements of the translation machinery, which makes it likely that both the viral and host components are targeted. Our recent understanding of frameshifting modulation suggests that exploitation of the host cellular resources could become an alternative approach to dysregulate frameshifting in pathogenic viruses. Because of the differences in codon usage between HIV‐1 and its human host, a strategy worth exploring would be to change the levels of the tRNAs crucial for the decoding of the viral frameshifting motif but dispensable for the host translation, such as tRNALeu(UAA); or overexpression of human genes with virus‐like codon usage to deplete the available translation resources. Thus, frameshifting—which is a remarkable recoding event—remains an interesting target for future medical research.
Edited by Michael Ibba
References
- 1. Demeshkina N, Jenner L, Westhof E, Yusupov M and Yusupova G (2013) New structural insights into the decoding mechanism: translation infidelity via a G.U pair with Watson‐Crick geometry. FEBS Lett 587, 1848–1857. [DOI] [PubMed] [Google Scholar]
- 2. Rodnina MV (2018) Translation in prokaryotes. Cold Spring Harb Perspect Biol 10, a032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodnina MV (2012) Quality control of mRNA decoding on the bacterial ribosome. Adv Protein Chem Struct Biol 86, 95–128. [DOI] [PubMed] [Google Scholar]
- 4. Rodnina MV, Fischer N, Maracci C and Stark H (2017) Ribosome dynamics during decoding. Philos Trans R Soc Lond B Biol Sci 372 10.1098/rstb.2016.0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramakrishnan V (2014) The ribosome emerges from a black box. Cell 159, 979–984. [DOI] [PubMed] [Google Scholar]
- 6. Voorhees RM and Ramakrishnan V (2013) Structural basis of the translational elongation cycle. Annu Rev Biochem 82, 203–236. [DOI] [PubMed] [Google Scholar]
- 7. Garofalo R, Wohlgemuth I, Pearson M, Lenz C, Urlaub H and Rodnina MV (2019) Broad range of missense error frequencies in cellular proteins. Nucleic Acids Res 47, 2932–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kurland CG (1992) Translational accuracy and the fitness of bacteria. Annu Rev Genet 26, 29–50. [DOI] [PubMed] [Google Scholar]
- 9. Atkins JF, Loughran G, Bhatt PR, Firth AE and Baranov PV (2016) Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res 44, 7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Advani VM and Dinman JD (2016) Reprogramming the genetic code: the emerging role of ribosomal frameshifting in regulating cellular gene expression. BioEssays 38, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caliskan N, Peske F and Rodnina MV (2015) Changed in translation: mRNA recoding by ‐1 programmed ribosomal frameshifting. Trends Biochem Sci 40, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brierley I, Gilbert RJC & Pennell S (2010) Pseudoknot‐dependent programmed —1 ribosomal frameshifting: structures, mechanisms and models In Recoding: Expansion of Decoding Rules Enriches Gene Expression (Atkins JF. & Gesteland RF, eds), pp. 149–174. Springer New York, New York, NY. [Google Scholar]
- 13. Weiss RB, Dunn DM, Atkins JF and Gesteland RF (1987) Slippery runs, shifty stops, backward steps, and forward hops: ‐2, ‐1, +1, +2, +5, and +6 ribosomal frameshifting. Cold Spring Harb Symp Quant Biol 52, 687–693. [DOI] [PubMed] [Google Scholar]
- 14. Yan S, Wen JD, Bustamante C and Tinoco I Jr (2015) Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caliskan N, Wohlgemuth I, Korniy N, Pearson M, Peske F and Rodnina MV (2017) Conditional switch between frameshifting regimes upon translation of dnaX mRNA. Mol Cell 66, 558–567. [DOI] [PubMed] [Google Scholar]
- 16. Lin Z, Gilbert RJ and Brierley I (2012) Spacer‐length dependence of programmed ‐1 or ‐2 ribosomal frameshifting on a U6A heptamer supports a role for messenger RNA (mRNA) tension in frameshifting. Nucleic Acids Res 40, 8674–8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivanov IP, Gesteland RF, Matsufuji S and Atkins JF (1998) Programmed frameshifting in the synthesis of mammalian antizyme is +1 in mammals, predominantly +1 in fission yeast, but ‐2 in budding yeast. RNA 4, 1230–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brierley I (1995) Ribosomal frameshifting viral RNAs. J Gen Virol 76 (Pt 8), 1885–1892. [DOI] [PubMed] [Google Scholar]
- 19. Plant EP (2012) Ribosomal frameshift signals in viral genomes In Viral Genomes – Molecular Structure, Diversity, Gene Expression Mechanisms and Host‐Virus Interactions (Garcia PM, ed.). pp. 91–122 InTech; Rijeka, Croatia. [Google Scholar]
- 20. Firth AE, Chung BY, Fleeton MN and Atkins JF (2008) Discovery of frameshifting in Alphavirus 6K resolves a 20‐year enigma. Virol J 5, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ and Varmus HE (1988) Characterization of ribosomal frameshifting in HIV‐1 gag‐pol expression. Nature 331, 280–283. [DOI] [PubMed] [Google Scholar]
- 22. Melian EB, Hinzman E, Nagasaki T, Firth AE, Wills NM, Nouwens AS, Blitvich BJ, Leung J, Funk A, Atkins JF et al (2010) NS1’ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J Virol 84, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snyder JE et al (2013) Functional characterization of the alphavirus TF protein. J Virol 87, 8511–8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blinkowa AL and Walker JR (1990) Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res 18, 1725–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishimaru D et al (2013) RNA dimerization plays a role in ribosomal frameshifting of the SARS coronavirus. Nucleic Acids Res 41, 2594–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brierley I, Gilbert R and Pennell S (2010) Pseudoknot‐dependent programmed –1 ribosomal frameshifting: structures, mechanisms and models In Recoding: Expansion of Decoding Rules Enrichesgene Expression (Atkins JF. and Gesteland RF, eds), pp. 149–174. Springer, New York, Dordrecht, Heidelberg, London. [Google Scholar]
- 27. Chung BY, Firth AE and Atkins JF (2010) Frameshifting in alphaviruses: a diversity of 3’ stimulatory structures. J Mol Biol 397, 448–456. [DOI] [PubMed] [Google Scholar]
- 28. Giedroc D and Cornish P (2009) Frameshifting RNA pseudoknots: structure and mechanism. Virus Res 139, 193–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caliskan N, Katunin VI, Belardinelli R, Peske F and Rodnina MV (2014) Programmed ‐1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim HK, Liu F, Fei J, Bustamante C, Gonzalez RL Jr and Tinoco I Jr (2014) A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc Natl Acad Sci USA 111, 5538–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Larsen B, Wills N, Gesteland R and Atkins J (1994) rRNA‐mRNA base pairing stimulates a programmed ‐1 ribosomal frameshift. J Bacteriol 176, 6842–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, Kasprzak WK, Shapiro BA and Dinman JD (2014) Ribosomal frameshifting in the CCR32 mRNA is regulated by miRNAs and the NMD pathway. Nature 512, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Napthine S, Ling R, Finch LK, Jones JD, Bell S, Brierley I and Firth AE (2017) Protein‐directed ribosomal frameshifting temporally regulates gene expression. Nat Commun 8, 15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kobayashi Y, Zhuang J, Peltz S and Dougherty J (2010) Identification of a cellular factor that modulates HIV‐1 programmed ribosomal frameshifting. J Biol Chem 285, 19776–19784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X, Xuan Y, Han Y, Ding X, Ye K, Yang F, Gao P, Goff SP and Gao G (2019) Regulation of HIV‐1 Gag‐Pol expression by Shiftless, an inhibitor of programmed ‐1 ribosomal frameshifting. Cell 176, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Endoh T and Sugimoto N (2013) Unusual ‐1 ribosomal frameshift caused by stable RNA G‐quadruplex in open reading frame. Anal Chem 85, 11435–11439. [DOI] [PubMed] [Google Scholar]
- 37. Yu CH, Teulade‐Fichou MP and Olsthoorn RC (2014) Stimulation of ribosomal frameshifting by RNA G‐quadruplex structures. Nucleic Acids Res 42, 1887–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brierley I, Digard P and Inglis S (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57, 537–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larsen B, Gesteland RF and Atkins JF (1997) Structural probing and mutagenic analysis of the stem‐loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50%. J Mol Biol 271, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Hong S, Ruangprasert A, Skiniotis G and Dunham CM (2018) Alternative mode of E‐Site tRNA binding in the presence of a downstream mRNA stem loop at the entrance channel. Structure 26, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dulude D, Baril M and Brakier‐Gingras L (2002) Characterization of the frameshift stimulatory signal controlling a programmed ‐1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res 30, 5094–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Staple DW and Butcher SE (2005) Solution structure and thermodynamic investigation of the HIV‐1 frameshift inducing element. J Mol Biol 349, 1011–1023. [DOI] [PubMed] [Google Scholar]
- 43. Staple DW and Butcher SE (2003) Solution structure of the HIV‐1 frameshift inducing stem‐loop RNA. Nucleic Acids Res 31, 4326–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mazauric M‐H, Seol Y, Yoshizawa S, Visscher K and Fourmy D (2009) Interaction of the HIV‐1 frameshift signal with the ribosome. Nucleic Acids Res 37, 7654–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen C, Zhang H, Broitman SL, Reiche M, Farrell I, Cooperman BS and Goldman YE (2013) Dynamics of translation by single ribosomes through mRNA secondary structures. Nat Struct Mol Biol 20, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen J, Petrov A, Johansson M, Tsai A, O'Leary SE and Puglisi JD (2014) Dynamic pathways of ‐1 translational frameshifting. Nature 512, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adio S, Senyushkina T, Peske F, Fischer N, Wintermeyer W and Rodnina MV (2015) Fluctuations between multiple EF‐G‐induced chimeric tRNA states during translocation on the ribosome. Nat Commun 6, 7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wasserman MR, Alejo JL, Altman RB and Blanchard SC (2016) Multiperspective smFRET reveals rate‐determining late intermediates of ribosomal translocation. Nat Struct Mol Biol 23, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou J, Lancaster L, Donohue JP and Noller HF (2013) Crystal structures of EF‐G‐ribosome complexes trapped in intermediate states of translocation. Science 340, 1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou J, Lancaster L, Donohue JP and Noller HF (2014) How the ribosome hands the A‐site tRNA to the P site during EF‐G‐catalyzed translocation. Science 345, 1188–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agirrezabala X, Liao HY, Schreiner E, Fu J, Ortiz‐Meoz RF, Schulten K, Green R and Frank J (2012) Structural characterization of mRNA‐tRNA translocation intermediates. Proc Natl Acad Sci USA 109, 6094–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Belardinelli R, Sharma H, Caliskan N, Cunha CE, Peske F, Wintermeyer W and Rodnina MV (2016) Choreography of molecular movements during ribosome progression along mRNA. Nat Struct Mol Biol 23, 342–348. [DOI] [PubMed] [Google Scholar]
- 53. Zhang W, Dunkle JA and Cate JH (2009) Structures of the ribosome in intermediate states of ratcheting. Science 325, 1014–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou J, Lancaster L, Donohue JP and Noller HF (2019) Spontaneous ribosomal translocation of mRNA and tRNAs into a chimeric hybrid state. Proc Natl Acad Sci USA 116, 7813–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tsuchihashi Z and Brown PO (1992) Sequence requirements for efficient translational frameshifting in the Escherichia coli dnaX gene and the role of an unstable interaction between tRNA(Lys) and an AAG lysine codon. Genes Dev 6, 511–519. [DOI] [PubMed] [Google Scholar]
- 56. Tsuchihashi Z and Kornberg A (1990) Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci USA 87, 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Namy O, Moran SJ, Stuart DI, Gilbert RJ and Brierley I (2006) A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Farabaugh PJ (1996) Programmed translational frameshifting. Microbiol Rev 60, 103–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Horsfield J, Wilson D, Mannering S, Adamski F and Tate W (1995) Prokaryotic ribosomes recode the HIV‐1 gag‐pol‐1 frameshift sequence by an E/P site post‐translocation simultaneous slippage mechanism. Nucl Acids Res 23, 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weiss RB, Dunn DM, Shuh M, Atkins JF and Gesteland RF (1989) E. coli ribosomes re‐phase on retroviral frameshift signals at rates ranging from 2 to 50 percent. New Biol 1, 159–169. [PubMed] [Google Scholar]
- 61. Yelverton E, Lindsley D, Yamauchi P and Gallant JA (1994) The function of a ribosomal frameshifting signal from human immunodeficiency virus‐1 in Escherichia coli . Mol Microbiol 11, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liao PY, Choi YS, Dinman JD and Lee KH (2011) The many paths to frameshifting: kinetic modelling and analysis of the effects of different elongation steps on programmed ‐1 ribosomal frameshifting. Nucleic Acids Res 39, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Laine S, Thouard A, Komar AA and Rossignol JM (2008) Ribosome can resume the translation in both +1 or ‐1 frames after encountering an AGA cluster in Escherichia coli. Gene 412, 95–101. [DOI] [PubMed] [Google Scholar]
- 64. Cardno TS, Shimaki Y, Sleebs BE, Lackovic K, Parisot JP, Moss RM, Crowe‐McAuliffe C, Mathew SF, Edgar CD, Kleffmann T et al (2015) HIV‐1 and human PEG10 frameshift elements are functionally distinct and distinguished by novel small molecule modulators. PLoS ONE 10, e0139036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Atkins JF and Bjork GR (2009) A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P‐site realignment. Microbiol Mol Biol Rev 73, 178–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leipuviene R and Bjork GR (2005) A reduced level of charged tRNAArgmnm5UCU triggers the wild‐type peptidyl‐tRNA to frameshift. RNA 11, 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karacostas V, Wolffe EJ, Nagashima K, Gonda MA and Moss B (1993) Overexpression of the HIV‐1 gag‐pol polyprotein results in intracellular activation of HIV‐1 protease and inhibition of assembly and budding of virus‐like particles. Virology 193, 661–671. [DOI] [PubMed] [Google Scholar]
- 68. Brunelle M, Payant C, Lemay G and Brakier‐Gingras L (1999) Expression of the human immunodeficiency virus frameshift signal in a bacterial cell‐free system: influence of an interaction between the ribosome and a stem‐loop structure downstream from the slippery site. Nucleic Acids Res 27, 4783–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Korniy N, Goyal A, Hoffmann M, Samatova E, Peske F, Pöhlmann S and Rodnina MV (2019) Modulation of HIV‐1 Gag/Gag‐Pol Frameshifting by tRNA abundance. Nucleic Acids Res 47, 5210–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grentzmann G, Ingram J, Kelly P, Gesteland R and Atkins J (1998) A dual‐luciferase reporter system for studying recoding signals. RNA 4, 479–565. [PMC free article] [PubMed] [Google Scholar]
- 71. Mathew SF, Crowe‐McAuliffe C, Graves R, Cardno TS, McKinney C, Poole ES and Tate WP (2015) The highly conserved codon following the slippery sequence supports ‐1 frameshift efficiency at the HIV‐1 frameshift site. PLoS ONE 10, e0122176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Plant EP and Dinman JD (2006) Comparative study of the effects of heptameric slippery site composition on ‐1 frameshifting among different eukaryotic systems. RNA 12, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sharp PM, Cowe E, Higgins DG, Shields DC, Wolfe KH and Wright F (1988) Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within‐species diversity. Nucleic Acids Res 16, 8207–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van Weringh A, Ragonnet‐Cronin M, Pranckeviciene E, Pavon‐Eternod M, Kleiman L and Xia X (2011) HIV‐1 modulates the tRNA pool to improve translation efficiency. Mol Biol Evol 28, 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smith BL, Chen G, Wilke CO and Krug RM (2018) Avian influenza virus PB1 gene in H3N2 viruses evolved in humans to reduce interferon inhibition by skewing codon usage toward interferon‐altered tRNA. Pools MBio, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pavon‐Eternod M, David A, Dittmar K, Berglund P, Pan T, Bennink JR and Yewdell JW (2013) Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res 41, 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fragkoudis R, Tamberg N, Siu R, Kiiver K, Kohl A, Merits A and Fazakerley JK (2009) Neurons and oligodendrocytes in the mouse brain differ in their ability to replicate Semliki Forest virus. J Neurovirol 15, 57–70. [DOI] [PubMed] [Google Scholar]
- 78. Pavon‐Eternod M, Wei M, Pan T and Kleiman L (2010) Profiling non‐lysyl tRNAs in HIV‐1. RNA 16, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Duchon AA, St Gelais C, Titkemeier N, Hatterschide J, Wu L and Musier‐Forsyth K (2017) HIV‐1 exploits a dynamic multi‐aminoacyl‐tRNA synthetase complex to enhance viral replication. J Virol 91 10.1128/JVI.01240-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Netzer N, Goodenbour JM, David A, Dittmar KA, Jones RB, Schneider JR, Boone D, Eves EM, Rosner MR, Gibbs JS et al (2009) Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 462, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Girstmair H, Saffert P, Rode S, Czech A, Holland G, Bannert N and Ignatova Z (2013) Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep 3, 148–159. [DOI] [PubMed] [Google Scholar]
- 82. Orr HT and Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30, 575–621. [DOI] [PubMed] [Google Scholar]
- 83. Frias D, Monteiro‐Cunha JP, Mota‐Miranda AC, Fonseca VS, de Oliveira T, Galvao‐Castro B and Alcantara LC (2013) Human retrovirus codon usage from tRNA point of view: therapeutic insights. Bioinform Biol Insights 7, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon‐Eternod M, Jones TE, Landry S, Pan T, Weitzman MD et al (2012) Codon‐usage‐based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li M, Kao E, Malone D, Gao X, Wang JYJ and David M (2018) DNA damage‐induced cell death relies on SLFN11‐dependent cleavage of distinct type II tRNAs. Nat Struct Mol Biol 25, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu J, Hendrix RW and Duda RL (2004) Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell 16, 11–21. [DOI] [PubMed] [Google Scholar]
- 87. Fang Y, Treffers EE, Li Y, Tas A, Sun Z, van der Meer Y, de Ru AH, van Veelen PA, Atkins JF, Snijder EJ et al (2012) Efficient −2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc Natl Acad Sci USA 109, E2920–E2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guarraia C, Norris L, Raman A and Farabaugh PJ (2007) Saturation mutagenesis of a +1 programmed frameshift‐inducing mRNA sequence derived from a yeast retrotransposon. RNA 13, 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Penno C, Kumari R, Baranov PV, van Sinderen D and Atkins JF (2017) Specific reverse transcriptase slippage at the HIV ribosomal frameshift sequence: potential implications for modulation of GagPol synthesis. Nucleic Acids Res 45, 10156–10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ketteler R (2012) On programmed ribosomal frameshifting: the alternative proteomes. Front Genet 3, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Meydan SK, Klepacki D, Karthikeyan S, Margus T, Thomas P, Jones JE, Khan Y, Briggs J, Dinman JD, Vazquez‐Laslop N et al (2017) Programmed ribosomal frameshifting generates a copper transporter and a copper chaperone from the same gene. Mol Cell 65, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ghosh AK, Osswald HL and Prato G (2016) Recent progress in the development of HIV‐1 protease inhibitors for the treatment of HIV/AIDS. J Med Chem 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lv Z, Chu Y and Wang Y (2015) HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl) 7, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bally F, Martinez R, Peters S, Sudre P and Telenti A (2000) Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res Hum Retroviruses 16, 1209–1213. [DOI] [PubMed] [Google Scholar]
- 95. Banke S, Lillemark MR, Gerstoft J, Obel N and Jorgensen LB (2009) Positive selection pressure introduces secondary mutations at Gag cleavage sites in human immunodeficiency virus type 1 harboring major protease resistance mutations. J Virol 83, 8916–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ozen A, Lin KH, Kurt Yilmaz N and Schiffer CA (2014) Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV‐1 protease. Proc Natl Acad Sci USA 111, 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Doyon L, Payant C, Brakier‐Gingras L and Lamarre D (1998) Novel Gag‐Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J Virol 72, 6146–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Girnary R, King L, Robinson L, Elston R and Brierley I (2007) Structure‐function analysis of the ribosomal frameshifting signal of two human immunodeficiency virus type 1 isolates with increased resistance to viral protease inhibitors. J Gen Virol 88, 226–235. [DOI] [PubMed] [Google Scholar]
- 99. Knops E, Brakier‐Gingras L, Schulter E, Pfister H, Kaiser R and Verheyen J (2012) Mutational patterns in the frameshift‐regulating site of HIV‐1 selected by protease inhibitors. Med Microbiol Immunol 201, 213–218. [DOI] [PubMed] [Google Scholar]
- 100. Brierley I and Dos Ramos FJ (2006) Programmed ribosomal frameshifting in HIV‐1 and the SARS‐CoV. Virus Res 119, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Garcia‐Miranda P, Becker JT, Benner BE, Blume A, Sherer NM and Butcher SE (2016) Stability of HIV frameshift site RNA correlates with frameshift efficiency and decreased virus infectivity. J Virol 90, 6906–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pan D and Coen DM (2012) Net ‐1 frameshifting on a noncanonical sequence in a herpes simplex virus drug‐resistant mutant is stimulated by nonstop mRNA. Proc Natl Acad Sci USA 109, 14852–14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Griffiths A (2011) Slipping and sliding: frameshift mutations in herpes simplex virus thymidine kinase and drug‐resistance. Drug Resist Updat 14, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Saini S, Bhalla P, Gautam H, Baveja UK, Pasha ST and Dewan R (2012) Resistance‐associated mutations in HIV‐1 among patients failing first‐line antiretroviral therapy. J Int Assoc Physicians AIDS Care (Chic) 11, 203–209. [DOI] [PubMed] [Google Scholar]
- 105. Baril M, Dulude D, Gendron K, Lemay G and Brakier‐Gingras L (2003) Efficiency of a programmed ‐1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA 9, 1246–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Brakier‐Gingras L, Charbonneau J and Butcher SE (2012) Targeting frameshifting in the human immunodeficiency virus. Expert Opin Ther Targets 16, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ofori LO, Hilimire TA, Bennett RP, Brown NW Jr, Smith HC and Miller BL (2014) High‐affinity recognition of HIV‐1 frameshift‐stimulating RNA alters frameshifting in vitro and interferes with HIV‐1 infectivity. J Med Chem 57, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hung M, Patel P, Davis S and Green SR (1998) Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol 72, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hartmuth K, Raker VA, Huber J, Branlant C and Luhrmann R (1999) An unusual chemical reactivity of Sm site adenosines strongly correlates with proper assembly of core U snRNP particles. J Mol Biol 285, 133–147. [DOI] [PubMed] [Google Scholar]