

Abstract
As one of the most prevalent post‐translational modifications in eukaryotic cells, ubiquitylation plays vital roles in many cellular processes, such as protein degradation, DNA metabolism, and cell differentiation. Substrate proteins can be tagged by distinct types of polymeric ubiquitin (Ub) chains, which determine the eventual fate of the modified protein. A facile, click chemistry based approach for the efficient generation of linkage‐defined Ub chains, including Ub dimers, was recently established. Within these chains, individual Ub moieties are connected through a triazole linkage, rather than the natural isopeptide bond. Herein, it is reported that the conformation of an artificially K48‐linked Ub dimer resembles that of the natively linked dimer, with respect to structural and dynamic characteristics, as demonstrated by means of high‐resolution NMR spectroscopy. Thus, it is proposed that artificially linked Ub dimers, as generated by this approach, represent potent tools for studying the inherently different properties and functions of distinct Ub chains.
Keywords: click chemistry, dimerization, NMR spectroscopy, proteins, ubiquitylation
Ubiquitylation is one of the most prevalent post‐translational modifications (PTMs) in eukaryotic cells and plays vital roles in many cellular processes, including protein degradation, DNA metabolism, signal transduction, and cell proliferation and differentiation.1 Substrate proteins can be modified with a single moiety of ubiquitin (Ub), which consists of 76 amino acids, or a polymeric Ub chain. The individual Ub moieties within a chain are linked through isopeptide bonds formed between a distinct lysine residue (i.e., K6, K11, K27, K29, K33, K48, K63) of one Ub molecule and the C‐terminal glycine of the adjacent Ub.2 Distinct types of Ub chains are assumed to mark substrate proteins for different outcomes.3 For example, K48‐linked Ub chains target substrates for proteasomal degradation,4 whereas K11‐linked chains appear to play a role in both proteolytic and non‐proteolytic signalling events.5 Furthermore, ubiquitylation is reversible by the action of a superfamily of isopeptidases termed deubiquitylating enzymes (DUBs).6 DUBs can trim or completely remove Ub chains in a linkage‐specific manner, and thereby, affect the subcellular localisation and function of a modified protein.7 Collectively, the type of Ub chain and dynamic modulation by DUBs determine the eventual fate of a protein in a precise spatiotemporal manner.8
Numerous efforts have been made to prepare linkage‐defined Ub chains. Enzymatic approaches frequently employ Ub mutants (in vitro many E2 Ub‐conjugating enzymes or E3 Ub ligases use more than one lysine for chain formation) or a combination of E2/E3 and DUBs to assemble homogenous populations of Ub chains.9 Thus, such approaches are laborious and time‐consuming. Therefore, a number of chemical methods,10 for example, silver‐catalysed chemical condensation,11 thiol–ene coupling,12 and native chemical ligation,13 have been explored for the preparation of naturally or artificially linked Ub chains. Ub chains linked by an isopeptide bond or a thioether bond have been widely used in determining the linkage preference of DUB enzymes. Yet, the susceptibility of these Ub species to DUB‐mediated hydrolysis severely limits their application in studies involving tissue or cell lysates to identify linkage‐specific interaction partners.
To overcome this obstacle, we and others have developed click chemistry based approaches for the generation of Ub dimers (Figure 1 A and Figure S1 in the Supporting Information), as well as longer Ub oligomers, in which individual Ub moieties are connected through a non‐hydrolysable triazole linkage.14 These artificially linked Ub chains exhibited great potency in identifying Ub‐interacting proteins from whole‐cell extracts.15 Although triazole‐linked Ub chains are assumed to be reliable surrogates of the natural isopeptide‐linked chains (Figure S1), structural evidence for this assumption is missing. Ub dimers, the shortest form of a Ub chain, have previously been used to study structure–function relationships of respective Ub chain types.16 Thus, we characterised Ub dimers that were triazole‐linked at position 48 ( dimer) by means of high‐resolution NMR spectroscopy, and showed that the structure and dynamics of the artificially linked dimer closely resembled those of the natively linked dimer.
Figure 1.
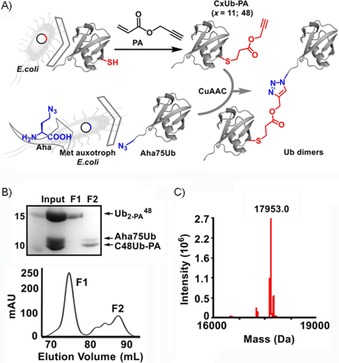
A) Scheme for the synthesis of artificially linked Ub dimers based on alkyne‐ (e.g., CxUb‐PA; x=11 or 48, PA: propargyl acrylate) and azide‐functionalised (Aha75Ub) Ub monomers through copper(I)‐catalysed azide–alkyne cycloaddition (CuAAC). B) Top: SDS‐PAGE analysis of the initial preparation (input) of and fractions thereof obtained by means of size‐exclusion chromatography (F1, F2). Bottom: Chromatogram (determined at λ=214 nm) indicating separation between the Ub dimer (F1) and Ub monomers (F2). C) Mass spectrum obtained for (calculated molecular mass M w=17 953.4 Da).
Firstly, we synthesised the artificially linked dimer based on a previously described approach.15a, 17 Briefly, we bacterially expressed the monomeric C48Ub mutant (replacement of Lys48 by Cys) and, upon purification, functionalised it with an alkyne group through a Michael reaction with PA (C48Ub‐PA; Figures 1 A and S2). For the preparation of azide‐functionalised Ub, we deleted the C‐terminal Gly76 of Ub and replaced Gly75 with the unnatural amino acid azidohomoalanine (Aha) through selective pressure incorporation to yield monomeric Aha75Ub (for further details, see Figures 1 A and S3).18 Finally, C48Ub‐PA and Aha75Ub were mixed, and dimers were formed through CuAAC‐based protein conjugation19 and subsequent purification by means of size‐exclusion chromatography (Figure 1 B). Notably, we managed to obtain the linkage‐defined dimer in milligram quantities from 1 L of bacterial culture for the expression of each Ub monomer. We determined the molecular mass of to be 17 953.0 Da by means of ESI‐MS; this value fitted well with the calculated molecular mass of 17 953.4 Da (Figure 1 C). This demonstrates that no or little copper‐induced protein oxidation or other damage occurred during the click reaction.
We next characterised by means of western blot analysis with an antibody that specifically recognised native K48‐linked Ub chains.20 As a specificity control, we employed , which was similarly prepared through the above approach (Figure S4). The result obtained indicates that the triazole linkage does not alter the structure or conformation of the synthesised Ub dimers, insofar as the antibody recognises , but not (Figure S5 A). Furthermore, the synthesised Ub dimers were used by the Ub conjugation machinery in so‐called autoubiquitylation assays21 (Figure S5 B); thus further proving that our method used for dimer formation does not distort the overall conformation of Ub.
To obtain structural insights into the artificially linked Ub dimer on a residue‐by‐residue level, we analysed by means of 2D heteronuclear 1H,15N HSQC NMR spectroscopy (Figures 2 and S6). To this end, 15N‐labelled C48Ub was generated in Escherichia coli grown in M9 minimal medium supplemented with 15NH4Cl as the only nitrogen source. Subsequently, purified 15N‐labelled monomeric C48Ub was modified with PA, and 15N‐ was generated by conjugation with non‐isotopically labelled monomeric Aha75Ub. We structurally characterised the 15N‐ dimer and the monomeric forms 15N‐C48Ub and 15N‐C48Ub‐PA by performing a sequential assignment procedure with 3D 15N‐edited NOESY‐HSQC and TOCSY‐HSQC methodology.22
Figure 2.
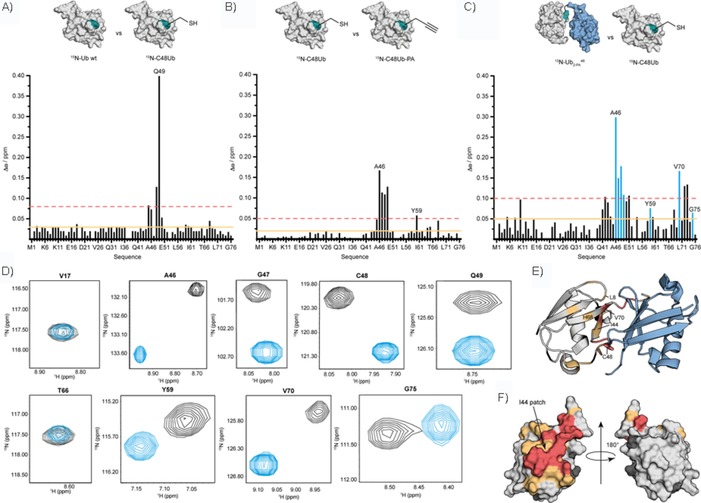
Structural properties of artificially linked . A) Structural impact of Lys‐to‐Cys mutation by comparing monomeric wild‐type Ub with the monomeric C48Ub variant. B) Structural impact of linking PA to Ub by comparing monomeric C48Ub with monomeric C48Ub‐PA. C) Structural impact of the distal Ub moiety on the proximal moiety by comparing dimeric with monomeric C48Ub. Comparisons of surface representations of Ub variants are shown at the top of A)–C); structures from PDB IDs 2BGF (isopeptide‐linked dimeric K48 Ub) and 1D3Z (wild‐type monomeric Ub) were used. Horizontal lines shown in A)–C) indicate chemical shift perturbations (CSPs, Δω) that are larger than the mean (orange solid line) and larger than the mean plus one standard deviation (red dotted line). CSPs of residues (blue) are shown in D) separately. D) Close‐up views of selected 1H,15N HSQC cross signals acquired for the 15N‐labelled proximal unit within dimeric (black) and 15N‐labelled monomeric C48Ub (blue) at pH 6.8, T=298 K. The entire 1H,15N HSQC spectrum is shown in Figure S6 D. E) Residues comprising the 15N‐labelled proximal Ub moiety within are coloured according to CSPs shown in C). Red: Δω>0.1 ppm, orange: 0.05<Δω<0.1 ppm. The non‐isotopically labelled distal Ub moiety is coloured in blue. The site used for artificial linkage in 15N‐ ; Cys48; and the residues of the hydrophobic patch, Leu8, Ile44, His68, and Val70, are indicated. F) Surface representation of monomeric wild‐type Ub (PDB ID: 1D3Z) is coloured according to Δω values presented in C) and is shown in two orientations.
After completion of the backbone assignment of the 2D 1H,15N HSQC spectra of 15N‐ , 15N‐C48Ub, wild‐type 15N‐Ub, and 15N‐C48Ub‐PA (Figure S6), we were able to identify the effect of Lys‐to‐Cys replacement in monomeric Ub (Figure 2 A). This single‐point mutation causes changes in chemical shifts that are locally limited to residues close to the mutation site (Figures 2 A, S6 A); thus making C48Ub an ideal template for Ub conjugation. Furthermore, we unravelled the amino acids in the 15N‐labelled proximal Ub moiety of dimeric 15N‐ , which was affected by the presence of the non‐isotopically labelled distal Ub moiety, and PA, which was used for conjugation (Figure S6 B–D). To do so, we evaluated the differences in chemical shifts, Δω, for all backbone 1H and 15N resonances by comparing 15N‐C48Ub‐PA with 15N‐C48Ub and 15N‐ with 15N‐C48Ub (Figure 2 B, C).23 By analysing the pattern of CSPs, we noticed the most significant CSP effects on residues close to the Ile44 hydrophobic patch (i.e., Leu8, Ile44, His68, and Val70), which had the largest CSP values (Figure 2 C–F). This patch serves as a hydrophobic interaction surface in natively linked K48 Ub dimers, in which the proximal and distal Ub moieties are close in space.24 Thus, our CSP data are highly consistent with the idea that, similar to the natively linked dimer, the artificially linked 15N‐ dimer adopts a predominantly closed conformation through Ile44 patch interaction.25 In addition, residues Ala46, Gly47, Cys48, and Gln49 experienced substantial CSPs (Figure 2 C). These perturbations are limited to those residues located in close proximity to the triazole‐linkage site and have no significant influence on the overall structural properties of , since the dispersion seen in the 2D 1H,15N HSQC spectrum resembles that of an overall Ub‐like fold (Figure S6 C, D).
In addition to structural investigations, we also highlight the backbone dynamics of 15N‐ by means of high‐resolution NMR spectroscopy. Heteronuclear NOE (hetNOE) spectroscopy, which is sensitive on the picosecond to nanosecond timescale, reveals that amide protons comprising the proximal unit of dimeric 15N‐ resemble the wild‐type pattern, with only one exception (Figure 3 A). The backbone mobility of residues close to the C48‐PA conjugation site is decreased compared with that of monomeric wild‐type Ub, since the hetNOE values of these residues increase from 0.6 to about 0.8 (Figure 3 A). Analysis of the longitudinal and transversal relaxation rate constants, R 1 and R 2, confirmed that, on this timescale, backbone mobility is conserved between artificially linked 15N‐ and natively linked (Figure S7). In the case of 15N‐ , the average values for R 1 and R 2 are (1.27±0.08), and (11±7) s−1, respectively, whereas the values for the natively linked counterpart are R 1≈1.4 s−1 and R 2≈11 s−1.26 Finally, we extended the dynamic characterisation of 15N‐ by using a modified MEXICO sequence to analyse the ability of amide protons to exchange with solvent protons, which takes place on a millisecond timescale.27 The residues Leu8, Ala46, and Gly75 of the proximal moiety of 15N‐ had the largest values of k HX; thus characterising the accessibility of amide protons to the solvent (Figure 3 B). Comparing the rate constants determined for proximal amide protons of the 15N‐ dimer with those for the corresponding amide protons of monomeric wild‐type Ub revealed that the exchange of Leu8, Ala46, Arg74, and Gly75 with solvent protons was significantly reduced in the dimeric state (Figure 3 C). The observation that the accessibility of amide protons is reduced at the surface between the proximal and distal moieties is highly consistent with the data obtained above concerning the structural properties of (Figure 2 C). In other words, the distal and proximal moieties of the dimer come into close proximity, as is the case in the overall closed conformation of natively linked K48 Ub dimers.
Figure 3.
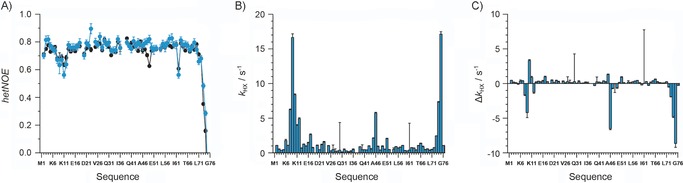
Dynamic properties of artificially linked . A) 1H,15N hetNOEs of backbone residues comprising the proximal unit of (blue) compared with monomeric wild‐type Ub (black). The relaxation parameters R 1 and R 2 are shown in Figure S7. B) Rate constants (k HX) for the exchange of amide protons of the proximal unit of versus protons of the solvent detected in the modified MEXICO experiment. C) Difference in exchange rate constants (Δk HX) observed for amide protons presented in B) and amide protons comprising wild‐type monomeric Ub.
Taken together, the high‐resolution NMR spectroscopy data reported herein strongly indicate that the triazole‐linked dimer maintains both structural and dynamic properties of the natural isopeptide‐linked K48 Ub dimer.
Ub dimers represent the smallest possible Ub chain and, thus, the availability of linkage‐defined Ub dimers is of great value for studying the structural characteristics and biological functions of distinct Ub chain types. Here, we adapted a previously established approach for the synthesis of linkage‐defined Ub chains to the facile generation of K48‐based Ub dimers, , with yields in the milligram range. Notably, this approach can be readily extended to the preparation of any possible Ub dimer, as shown herein for K11 Ub dimers. A potential concern of this methodology is that the two Ub moieties are connected through a triazole linkage and not by the natural isopeptide bond. To prove that the artificially linked Ub dimer resembles the native isopeptide‐linked counterpart, we performed a comprehensive set of biochemical and biophysical experiments. Most notably, high‐resolution NMR spectroscopy analysis revealed that the artificially linked dimers adopted an overall conformation and possessed dynamic features very similar to those of the natively linked counterparts; thus demonstrating that the artificial triazole linkage constituted a reliable surrogate for the isopeptide linkage. This indicates that artificially linked Ub dimers represent potent tools for studying the diverse properties and functions of differently linked Ub chains.
Experimental Section
Synthesis of Ub dimers through click chemistry: A sample containing C11Ub‐PA or C48Ub‐PA (100 μm) was mixed with Aha75Ub (100 μm) in Tris⋅HCl (20 mm, pH 7.0) buffer. Then, SDS and tris‐hydroxypropyltriazolylmethylamine (THPTA), with final concentrations of 0.5 and 5 mm, were added sequentially, followed by argon flushing. The click reaction was initiated by adding Cu(MeCN)4BF4 (2.5 mm), and samples were incubated on ice for 1 h. Samples of the reaction mixture (20 μL) were withdrawn for SDS‐PAGE analysis. The remainder was applied directly to size‐exclusion chromatography (HiLoad 16/600 Superdex 75 PG, ÄKTA purifier FPLC system) with Tris⋅HCl (25 mm, pH 7.5), NaCl (300 mm) as the elution buffer. The elution fractions were collected every 1 mL per tube and all collected fractions were analysed by means of SDS‐PAGE. Fractions containing Ub dimers were combined, concentrated by using an Amicon Ultra Centrifugal Filter (10 kDa MWCO), and quantified by means of a BCA protein assay. Detailed protocols for the expression and purification of monomeric proximal CxUb, CxUb‐PA, and distal Aha75Ub moieties, as well as analysis by means of mass spectrometry are provided in the Supporting Information. The 15N‐isotopically labelled wild‐type monomeric Ub was purchased from Giotto Biotech (Italy).
NMR spectroscopy analysis: NMR spectroscopy data acquisition was performed with 600 μL of 15N‐labelled wild‐type Ub monomer, 15N‐labelled C48Ub, 15N‐labelled C48Ub‐PA and 15N‐proximally labelled (concentration 100–250 μm in 20 mm Na3PO4 (pH 6.8), 5 % (v/v) D2O) on a Bruker Avance III 600 MHz spectrometer equipped with a TCI‐H/C/N triple resonance cryoprobe at T=298 K. The water signal was suppressed by using WATERGATE (water suppression by gradient tailored excitation) and presaturation methodologies. The 2D 1H,15N HSQC spectra were recorded with 1024 data points in the 1H dimension, 128–256 data points in the 15N dimension, and 2–16 scans. The 3D 1H,15N NOESY‐HSQC spectra were recorded with 1024 data points in the direct 1H dimension, 72–84 data points in the 15N dimension, 232–256 data points in the indirect 1H dimension, and 4–8 scans. The NOESY mixing time was set to 60 ms. The 3D 1H,15N TOCSY‐HSQC spectra were recorded with 1024 data points in the direct 1H dimension, 80–108 data points in the 15N dimension, 238–256 data points in the indirect 1H dimension, and 4–8 scans. The TOCSY mixing time was set to 40 ms. CSPs, Δω, obtained for 15N‐ , compared with the monomeric unit 15N‐C48Ub, were calculated by using Equation (1):
| (1) |
A modified version of the MEXICO experiment (measurement of fast proton exchange rates in isotopically labelled compounds) based on 1H,15N HSQC spectra was used to obtain exchange processes in the millisecond time regime27 at T=298 K. Rate constants of hydrogen exchange with the solvent, k HX, were individually determined for each amide proton comprising the proximal moiety in . Signal intensities, I, were determined at different exchange periods, ranging from 10 to 250 ms, and were used in Equation (2).28
| (2) |
in which R 1 is the relaxation rate constant of the amide protons. The relaxation rate constant of water protons, R 1w, was separately determined to be 0.31 s−1. Error values were estimated from the mean standard deviation of replicate measurements at two different exchange periods and were included in weighted curve fitting. Backbone amide 15N longitudinal (R 1) and transversal relaxation experiments (R 2) were applied at T=298 K, according to a previous study.29 Delay times were in a range between 10 and 3000 and 8 and 296 ms, respectively, to identify signal intensities, I, for the determination of R 1 and R 2 relaxation rate constants. Values for R 1 and R 2 were determined by using the single exponential function I=I 0exp(−R 1,2 t), in which I 0 represents the signal intensity at t=0.
The software TopSpin 3.5 (Bruker) was used for data acquisition. NMRPipe30 was used to process and NMRViewJ 8.031 (Bruce Johnson, One Moon Scientific) was used to visualise and analyse the 2D and 3D NMR spectra. To represent the 3D structure of Ub, the software PyMOL Molecular Graphics System 1.8 (Schrödinger LLC) was used.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the DFG (SFB969, projects B03, B09). X.Z. thanks the DAAD and the Konstanz Research School Chemical Biology for support by fellowships. We further thank Dr. Anna Śladewska‐Marquardt and Dr. Andreas Marquardt of the Proteomics Center for MS measurements. We thank Tobias Lange for support at the initial state of the project.
X. Zhao, M. Mißun, T. Schneider, F. Müller, J. Lutz, M. Scheffner, A. Marx, M. Kovermann, ChemBioChem 2019, 20, 1772.
Contributor Information
Prof. Dr. Martin Scheffner, Email: martin.scheffner@uni-konstanz.de.
Prof. Dr. Andreas Marx, Email: andreas.marx@uni-konstanz.de.
Prof. Dr. Michael Kovermann, Email: michael.kovermann@uni-konstanz.de.
References
- 1.
- 1a. Rape M., Nat. Rev. Mol. Cell Biol. 2017, 19, 59–70; [DOI] [PubMed] [Google Scholar]
- 1b. Popovic D., Vucic D., Dikic I., Nat. Med. 2014, 20, 1242–1253. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Komander D., Rape M., Annu. Rev. Biochem. 2012, 81, 203–229; [DOI] [PubMed] [Google Scholar]
- 2b. Alfano C., Faggiano S., Pastore A., Trends Biochem. Sci. 2016, 41, 371–385. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Yau R., Rape M., Nat. Cell Biol. 2016, 18, 579–586; [DOI] [PubMed] [Google Scholar]
- 3b. Ulrich H. D., Walden H., Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Glickman M. H., Ciechanover A., Physiol. Rev. 2002, 82, 373–428; [DOI] [PubMed] [Google Scholar]
- 4b. Grice G. L., Nathan J. A., Cell. Mol. Life Sci. 2016, 73, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Bremm A., Komander D., Trends Biochem. Sci. 2011, 36, 355–363; [DOI] [PubMed] [Google Scholar]
- 5b. Wickliffe K. E., Williamson A., Meyer H.-J., Kelly A., Rape M., Trends Cell Biol. 2011, 21, 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Komander D., Clague M. J., Urbe S., Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563; [DOI] [PubMed] [Google Scholar]
- 6b. Nijman S. M. B., Luna-Vargas M. P. A., Velds A., Brummelkamp T. R., Dirac A. M. G., Sixma T. K., Bernards R., Cell 2005, 123, 773–786. [DOI] [PubMed] [Google Scholar]
- 7. Mevissen T. E. T., Komander D., Annu. Rev. Biochem. 2017, 86, 159–192. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Grabbe C., Husnjak K., Dikic I., Nat. Rev. Mol. Cell Biol. 2011, 12, 295–307; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Kravtsova-Ivantsiv Y., Sommer T., Ciechanover A., Angew. Chem. Int. Ed. 2013, 52, 192–198; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 202–209. [Google Scholar]
- 9.
- 9a. Michel M. A., Komander D., Elliott P. R., Methods Mol. Biol. 2018, 1844, 73–84; [DOI] [PubMed] [Google Scholar]
- 9b. Faggiano S., Alfano C., Pastore A., Anal. Biochem. 2016, 492, 82–90. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Abeywardana T., Pratt M. R., ChemBioChem 2014, 15, 1547–1554; [DOI] [PubMed] [Google Scholar]
- 10b. Strieter E. R., Korasick D. A., ACS Chem. Biol. 2012, 7, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Dixon E. K., Castañeda C. A., Kashyap T. R., Wang Y., Fushman D., Bioorg. Med. Chem. 2013, 21, 3421–3429; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Nakasone M. A., Livnat-Levanon N., Glickman M. H., Cohen R. E., Fushman D., Structure 2013, 21, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Valkevich E. M., Guenette R. G., Sanchez N. A., Chen Y.-c., Ge Y., Strieter E. R., J. Am. Chem. Soc. 2012, 134, 6916–6919; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Valkevich E. M., Sanchez N. A., Ge Y., Strieter E. R., Biochemistry 2014, 53, 4979–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Kumar K. S. A., Spasser L., Erlich L. A., Bavikar S. N., Brik A., Angew. Chem. Int. Ed. 2010, 49, 9126–9131; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9312–9317; [Google Scholar]
- 13b. Castañeda C. A., Spasser L., Bavikar S. N., Brik A., Fushman D., Angew. Chem. Int. Ed. 2011, 50, 11210–11214; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11406–11410; [Google Scholar]
- 13c. Haj-Yahya N., Haj-Yahya M., Castañeda C. A., Spasser L., Hemantha H. P., Jbara M., Penner M., Ciechanover A., Fushman D., Brik A., Angew. Chem. Int. Ed. 2013, 52, 11149–11153; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11355–11359; [Google Scholar]
- 13d. Virdee S., Kapadnis P. B., Elliott T., Lang K., Madrzak J., Nguyen D. P., Riechmann L., Chin J. W., J. Am. Chem. Soc. 2011, 133, 10708–10711; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13e. Pan M., Gao S., Zheng Y., Tan X., Lan H., Tan X., Sun D., Lu L., Wang T., Zheng Q., Huang Y., Wang J., Liu L., J. Am. Chem. Soc. 2016, 138, 7429–7435. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Eger S., Scheffner M., Marx A., Rubini M., J. Am. Chem. Soc. 2010, 132, 16337–16339; [DOI] [PubMed] [Google Scholar]
- 14b. Schneider T., Schneider D., Rösner D., Malhotra S., Mortensen F., Mayer T. U., Scheffner M., Marx A., Angew. Chem. Int. Ed. 2014, 53, 12925–12929; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13139–13143; [Google Scholar]
- 14c. Weikart N. D., Mootz H. D., ChemBioChem 2010, 11, 774–777; [DOI] [PubMed] [Google Scholar]
- 14d. Sommer S., Weikart N. D., Brockmeyer A., Janning P., Mootz H. D., Angew. Chem. Int. Ed. 2011, 50, 9888–9892; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10062–10066; [Google Scholar]
- 14e. McGouran J. F., Gaertner S. R., Altun M., Kramer H. B., Kessler B. M., Chem. Biol. 2013, 20, 1447–1455; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14f. Flierman D., van der Heden van Noort G. J., Ekkebus R., Geurink P. P., Mevissen T. E. T., Hospenthal M. K., Komander D., Ovaa H., Cell Chem. Biol. 2016, 23, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Zhao X., Lutz J., Hoellmueller E., Scheffner M., Marx A., Stengel F., Angew. Chem. Int. Ed. 2017, 56, 15764–15768; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15972–15976; [Google Scholar]
- 15b. Zhang X., Smits A. H., van Tilburg G. B. A., Jansen P. W. T. C., Makowski M. M., Ovaa H., Vermeulen M., Mol. Cell 2017, 65, 941–955. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Békés M., van der Heden van Noort G. J., Ekkebus R., Ovaa H., Huang T. T., Lima C. D., Mol. Cell 2016, 62, 572–585; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Mali S. M., Singh S. K., Eid E., Brik A., J. Am. Chem. Soc. 2017, 139, 4971–4986; [DOI] [PubMed] [Google Scholar]
- 16c. van Tilburg G. B. A., Elhebieshy A. F., Ovaa H., Curr. Opin. Struct. Biol. 2016, 38, 92–101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Michel M. A., Swatek K. N., Hospenthal M. K., Komander D., Mol. Cell 2017, 68, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rösner D., Schneider T., Schneider D., Scheffner M., Marx A., Nat. Protoc. 2015, 10, 1594–1611. [DOI] [PubMed] [Google Scholar]
- 18. Kiick K. L., Saxon E., Tirrell D. A., Bertozzi C. R., Proc. Natl. Acad. Sci. USA 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Schneider D., Schneider T., Aschenbrenner J., Mortensen F., Scheffner M., Marx A., Bioorg. Med. Chem. 2016, 24, 995–1001; [DOI] [PubMed] [Google Scholar]
- 19b. Schneider D., Schneider T., Rösner D., Scheffner M., Marx A., Bioorg. Med. Chem. 2013, 21, 3430–3435. [DOI] [PubMed] [Google Scholar]
- 20. Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M., Cell 2008, 134, 668–678. [DOI] [PubMed] [Google Scholar]
- 21. Mortensen F., Schneider D., Barbic T., Sladewska-Marquardt A., Kühnle S., Marx A., Scheffner M., Proc. Natl. Acad. Sci. USA 2015, 112, 9872–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sattler M., Schleucher J., Griesinger C., Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar]
- 23.
- 23a. Zuiderweg E. R. P., Biochemistry 2002, 41, 1–7; [DOI] [PubMed] [Google Scholar]
- 23b. Swanson K. A., Kang R. S., Stamenova S. D., Hicke L., Radhakrishnan I., EMBO J. 2003, 22, 4597–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pickart C. M., Fushman D., Curr. Opin. Chem. Biol. 2004, 8, 610–616. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Varadan R., Walker O., Pickart C., Fushman D., J. Mol. Biol. 2002, 324, 637–647; [DOI] [PubMed] [Google Scholar]
- 25b. Takeshi T., Kenichiro F., Hidehito T., Kazuhiro I., Hayato M. E., Hidenori H., Shigeo M., Hidekazu H., Mamoru S., Keiji T., Masahiro S., Genes Cells 2004, 9, 865–875;15461659 [Google Scholar]
- 25c. Castañeda C. A., Liu J., Kashyap T. R., Singh R. K., Fushman D., Cropp T. A., Chem. Commun. 2011, 47, 2026–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castañeda C. A., Chaturvedi A., Camara C. M., Curtis J. E., Krueger S., Fushman D., Phys. Chem. Chem. Phys. 2016, 18, 5771–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gemmecker G., Jahnke W., Kessler H., J. Am. Chem. Soc. 1993, 115, 11620–11621. [Google Scholar]
- 28. Hofmann H., Weininger U., Low C., Golbik R. P., Balbach J., Ulbrich-Hofmann R., J. Am. Chem. Soc. 2009, 131, 140–146. [DOI] [PubMed] [Google Scholar]
- 29. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E., Biochemistry 1994, 33, 5984–6003. [DOI] [PubMed] [Google Scholar]
- 30. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 31. Johnson B. A., Blevins R. A., J. Biomol. NMR 1994, 4, 603–614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary