

Abstract
Background
We performed a randomized phase 3 study of trabectedin versus dacarbazine in previously‐treated patients with liposarcoma/leiomyosarcoma (LPS/LMS).
Methods
Patients were randomized 2:1 to trabectedin (n = 384) or dacarbazine (n = 193) administered intravenously every 3 weeks. The primary objective was overall survival (OS). Secondary objectives were progression‐free survival, objective response rate, safety, and patient‐reported outcomes, all previously reported and demonstrating superior disease control with trabectedin. Results of the final OS analysis in preplanned subgroups of patients with LPS/LMS are presented.
Results
At the time of the final OS analysis, 577 patients had been assigned randomly, including 423 (73%) with LMS and 154 (27%) with LPS. The median duration of treatment exposure was higher in the trabectedin arm compared with the dacarbazine arm (4 vs 2 cycles), as was the proportion of patients receiving an extended number of therapy courses (≥6 cycles: 42% vs 22%). This pattern was consistent across histological subgroups: the median number of treatment cycles (4 vs 2 for both subgroups) and proportion of patients with ≥6 treatment cycles (LMS, 43% vs 24%; LPS, 40% vs 16%). Despite improved disease control by trabectedin, no improvement in OS was observed; the final median OS for trabectedin versus dacarbazine was 13.7 versus 13.1 months (P = .49). Sensitivity analyses of OS suggest confounding by post‐study anticancer therapies, which were utilized in most patients in both treatment arms (71% vs 69%, respectively).
Conclusion
The final OS results demonstrated comparable survival between LPS/LMS patients receiving trabectedin or dacarbazine, which is consistent with the interim analysis results. Both LPS and LMS demonstrated improved disease control with trabectedin.
Keywords: dacarbazine, trabectedin, leiomyosarcoma, liposarcoma, soft tissue sarcoma
Short abstract
A final analysis of overall survival demonstrates comparable results between patients with liposarcoma/leiomyosarcoma receiving trabectedin or dacarbazine, which is consistent with interim analysis results. Both liposarcoma and leiomyosarcoma show improved disease control with trabectedin.
Introduction
Effective treatment for patients with advanced soft‐tissue sarcoma (STS) remains an area of unmet need. First‐line chemotherapy for advanced STS is evolving, with anthracycline‐based treatment1, 2 or gemcitabine plus docetaxel3, 4, 5 as commonly used regimens.6 The optimal approach for patients following failure of first‐line chemotherapy is less well‐defined.
Trabectedin is an antineoplastic alkaloid agent with a complex, multimodal mechanism of action,7 binding the minor groove of the DNA double helix, bending it toward the major groove, and initiating a cascade of events that may interfere with transcription and inhibit activities of DNA binding factors and DNA repair processes downstream.8, 9, 10 In vitro, trabectedin can inhibit tumor growth by modulating production of certain chemokines, cytokines, and other factors involved in inflammation8, 9 in the tumor microenvironment.
Trabectedin was approved in the European Union in 2007 for the treatment of advanced, unresectable STS in patients following failure of anthracyclines and ifosfamide or who were unfit to receive those agents.9, 11 In 2015, trabectedin was approved in the United States for patients with unresectable or metastatic liposarcoma (LPS) or leiomyosarcoma (LMS) who had received a prior anthracycline‐containing regimen.12 United States approval was based on progression‐free survival (PFS) and objective response rate (ORR) results and safety data from a phase 3 trial (ET743‐SAR‐3007)13 that was limited to patients with LPS/LMS and was based upon the previously demonstrated superior efficacy in LPS/LMS compared with other STS subtypes.14 Trabectedin demonstrated a 45% reduction in risk of disease progression or death (hazard ratio [HR], 0.55; P < .001) with a median PFS of 4.2 versus 1.5 months13 and a safety profile consistent with the well‐characterized toxicities observed in previous trabectedin studies.13, 14, 15 Herein, we report the final overall survival (OS) results and preplanned histology‐specific subgroup analyses from this phase 3, randomized controlled trial.
Materials and Methods
Patients
Inclusion and exclusion criteria have been previously reported.13 Eligible patients were aged ≥15 years with unresectable, locally advanced, or metastatic LPS (pleomorphic, dedifferentiated, or myxoid/round cell) or LMS (uterine vs nonuterine) who were previously treated with at least a regimen containing an anthracycline and ifosfamide or an anthracycline and ≥1 additional cytotoxic chemotherapy regimens. Review boards at all participating institutions approved the study, which was conducted in accordance with the Declaration of Helsinki. Prior to any study‐related procedures, all patients provided written informed consent.
Study Design
This was a phase 3, randomized, multicenter, active‐controlled, open‐label, parallel‐group study conducted between May 2011 and January 2015. Per protocol, a final analysis of OS—defined as time between randomization and death due to any cause—was to be conducted after 376 death events had occurred, with a protocol‐specified interim analysis after 50% of those deaths had occurred. At clinical cutoff for the interim analysis (September 16, 2013), 63 of 173 (36%) patients (dacarbazine) and 126 of 345 (37%) patients (trabectedin) had died. The final OS analysis was conducted following a clinical cutoff of January 5, 2015, after 123 of 193 (64%) patients (dacarbazine) and 258 of 384 (67%) patients (trabectedin) had died.
Efficacy Evaluations
The primary endpoint was OS. Investigator assessment of response data was used to evaluate secondary endpoints related to disease control, including PFS, ORR, time to progression, and duration of response. Clinical benefit rate (CBR, defined as complete + partial responses + stable disease with duration of ≥18 weeks) and duration of stable disease were analyzed as parameters for prolonged disease control. Investigator assessments of PFS were validated through an audit of approximately 60% of radiological scans by blinded, independent radiologists’ review.13 Following clinical cutoff for final OS, prespecified subgroup analyses were conducted for patients with LMS compared with patients with LPS.
The time to initiation of post‐study anticancer therapy was performed as an exploratory analysis. This was defined as time between the date of randomization and the initiation date of any post‐study surgery, radiation, or drug therapy. Four post hoc analyses were conducted to investigate the potential confounding effect of post‐study anticancer therapies on the OS endpoint. Two of these analyses were also conducted for the interim analysis. Because greater proportions of patients in both arms received post‐study anticancer therapy in the final OS analysis compared with the interim analysis, two additional sensitivity analyses were performed to adjust for the potential confounding effect of treatment change (Supporting Information).
Safety Evaluations
Detailed safety assessments have been reported previously.13
Statistical Analyses
The primary statistical methodology for comparing treatment impact on OS was the unstratified log‐rank test. To detect a difference between a median OS of 10.0 and 13.5 months, respectively, in the dacarbazine and trabectedin arms (HR, 0.74) at an overall 2‐sided significance level of 0.05 with an 80% power required 376 death events. Employing a group sequential method with the O'Brien‐Fleming boundaries, as implemented by the Lan‐DeMets alpha spending function, 1 interim analysis and 1 final analysis were planned for OS. The cumulative alpha spent was 0.003 and 0.047 for the interim and final OS analyses, respectively. The PFS and ORR for the histological subgroups were tested using the Hochberg test procedure to control an overall type I error rate at the 2‐tailed 0.05 level. Employing a Cox proportional hazards model, efficacy analyses were also conducted to include covariates of baseline Eastern Cooperative Oncology Group (ECOG) performance status score (0, 1), number of lines of prior chemotherapy (1, ≥2), L‐sarcoma subtype (LPS, LMS), and age. Descriptive statistics were used in safety data analysis.
Results
Patient Disposition and Baseline Demographics
After the interim analysis of OS, an additional 59 patients had enrolled, reflecting a total randomized population of 577 patients (384 trabectedin, 193 dacarbazine). Of these, 550 patients (378 trabectedin, 172 dacarbazine) received the study drug. The most frequent reason for nontreatment was withdrawal of consent for both the trabectedin and dacarbazine arms (0.8% vs 9.8%, respectively). At the time of final OS analysis, 370 patients (trabectedin) and 170 patients (dacarbazine) had discontinued study treatment, with disease progression most commonly cited as the reason for discontinuation in each arm (71% and 80%, respectively) (Fig. 1).
Figure 1.
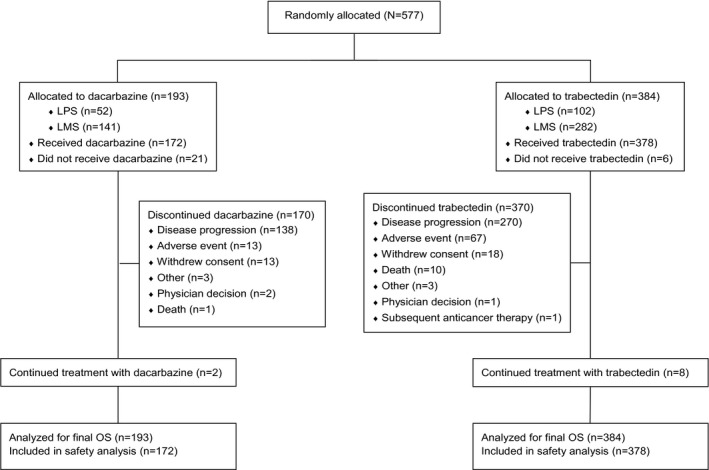
CONSORT diagram. LMS, leiomyosarcoma; LPS, liposarcoma; OS, overall survival.
Of the 577 randomized patients, 423 (73%; 282 trabectedin, 141 dacarbazine) and 154 (27%; 102 trabectedin, 52 dacarbazine) had LMS and LPS, respectively. Patient demographics were generally well‐balanced across both treatment arms (Table 1). Demographics with mild imbalances (≥5% frequency difference in trabectedin vs dacarbazine) were age ≥65 years (24% vs 19%), body mass index ≥30 (41% vs 36%), and female sex (68% vs 73%), which reflected the increased proportion of patients with uterine LMS histology (51% vs 62%) randomized to dacarbazine. Approximately 88% had ≥2 prior lines of chemotherapy, and the median time from last disease progression to randomization was <1 month in both treatment arms (0.85 months each).
Table 1.
Patient Demographics and Baseline Disease Characteristics by Histologic Subgroup
| Liposarcoma (n = 154) | Leiomyosarcoma (n = 423) | Total (n = 577) | ||||
|---|---|---|---|---|---|---|
|
Dacarbazine (n = 52) |
Trabectedin (n = 102) |
Dacarbazine (n = 141) |
Trabectedin (n = 282) |
Dacarbazine (n = 193) |
Trabectedin (n = 384) |
|
| Age, y | ||||||
| <65 | 42 (81) | 79 (78) | 114 (81) | 211 (75) | 156 (81) | 290 (76) |
| ≥65 | 10 (19) | 23 (23) | 27 (19) | 71 (25) | 37 (19) | 94 (24) |
| Median (range) | 53 (17‐74) | 57 (18‐81) | 56 (19‐79) | 57 (26‐81) | 56 (17‐79) | 57 (18‐81) |
| Sex | ||||||
| Men | 31 (60) | 71 (70) | 22 (16) | 51 (18) | 53 (28) | 122 (32) |
| Women | 21 (40) | 31 (30) | 119 (84) | 231 (82) | 140 (73) | 262 (68) |
| Race | ||||||
| American Indian or Alaska Native | 2 (4) | 0 | 2 (1) | 1 (0.4) | 4 (2) | 1 (0.3) |
| Asian | 5 (10) | 6 (6) | 6 (4) | 5 (2) | 11 (6) | 11 (3) |
| Black or African American | 0 | 10 (10) | 22 (16) | 38 (14) | 22 (11) | 48 (13) |
| White | 42 (81) | 79 (78) | 102 (72) | 221 (78) | 144 (75) | 300 (78) |
| Other | 0 | 1 (1) | 0 | 4 (1) | 0 | 5 (1) |
| Unknown | 1 (2) | 1 (1) | 4 (3) | 7 (3) | 5 (3) | 8 (2) |
| Not reported | 2 (4) | 5 (5) | 5 (4) | 6 (2) | 7 (4) | 11 (3) |
| Ethnicity | ||||||
| Hispanic or Latino | 9 (17) | 9 (9) | 14 (10) | 25 (9) | 23 (12) | 34 (9) |
| Not Hispanic or Latino | 40 (77) | 91 (89) | 113 (80) | 245 (87) | 153 (79) | 336 (88) |
| Unknown | 1 (2) | 2 (2) | 8 (6) | 2 (1) | 9 (5) | 4 (1) |
| Not reported | 2 (4) | 0 | 6 (4) | 10 (4) | 8 (4) | 10 (3) |
| Baseline BMI, kg/m2 | ||||||
| <30 | 38 (73) | 60 (59) | 86 (61) | 167 (59) | 124 (64) | 227 (59) |
| ≥30 | 14 (27) | 42 (41) | 55 (39) | 115 (41) | 69 (36) | 157 (41) |
| Median (range) | 26.17 (13.3‐40.5) | 28.44 (18.8‐58.7) | 28.13 (15.4‐66.7) | 28.03 (14.5‐78.1) | 27.51 (13.3‐66.7) | 28.11 (14.5‐78.1) |
| Baseline BSA, m2 | 45 (87) | 102 (100) | 127 (90) | 276 (98) | 172 (89) | 378 (98) |
| Median (range) | 1.85 (1.3‐2.3) | 1.91 (1.3‐2.6) | 1.73 (1.4‐2.4) | 1.74 (1.3‐2.4) | 1.77 (1.3‐2.4) | 1.78 (1.3‐2.6) |
| Histology | ||||||
| Leiomyosarcoma | 0 | 0 | 141 (100) | 282 (100) | 141 (73) | 282 (73) |
| Uterine | 0 | 0 | 88 (62) | 144 (51) | 88 (46) | 144 (38) |
| Nonuterine | 0 | 0 | 53 (38) | 138 (49) | 53 (27) | 138 (36) |
| Liposarcoma | 52 (100) | 102 (100) | 0 | 0 | 52 (27) | 102 (27) |
| Myxoid +/‐ round cell | 19 (37) | 42 (41) | 0 | 0 | 19 (10) | 42 (11) |
| Pleomorphic | 5 (10) | 11 (11) | 0 | 0 | 5 (3) | 11 (3) |
| Dedifferentiated | 28 (54) | 49 (48) | 0 | 0 | 28 (15) | 49 (13) |
| Baseline ECOG PS score | ||||||
| 0 | 26 (50) | 51 (50) | 67 (48) | 133 (47) | 93 (48) | 184 (48) |
| 1 | 26 (50) | 51 (50) | 74 (53) | 149 (53) | 100 (52) | 200 (52) |
| No. of lines of prior chemotherapy | ||||||
| 1 | 15 (29) | 24 (24) | 11 (8) | 22 (8) | 26 (14) | 46 (12) |
| 2 | 26 (50) | 47 (46) | 57 (40) | 129 (46) | 83 (43) | 176 (46) |
| 3 | 2 (4) | 21 (21) | 46 (33) | 78 (28) | 48 (25) | 99 (26) |
| 4 | 5 (10) | 7 (7) | 17 (12) | 32 (11) | 22 (11) | 39 (10) |
| >4 | 4 (8) | 3 (3) | 10 (7) | 21 (7) | 14 (7) | 24 (6) |
| Time from last disease progression to randomization, months | ||||||
| Median (range) | 0.67 (0.1‐4.1) | 0.92 (0.1‐8.5) | 0.92 (0.1‐9.8) | 0.85 (0.0‐13.7) | 0.85 (0.1‐9.8) | 0.85 (0.0‐13.7) |
Abbreviations: BMI, body mass index; BSA, body surface area; ECOG, Eastern Cooperative Oncology Group; PS, performance status.
Data are presented as n (%) unless specified otherwise.
Exposure to Study Treatment
Patients in both treatment arms received inpatient or outpatient study treatment according to local preference. Approximately 73% of trabectedin patients and 98% of dacarbazine patients received study treatment as outpatients. Consistent with the interim analysis, the median number of treatment cycles in the trabectedin arm was twice that of the dacarbazine arm (4 vs 2 cycles, respectively). The proportion of patients receiving extended courses of therapy increased in both treatment groups, relative to the interim analysis, with more trabectedin‐treated patients receiving ≥6 cycles (42% vs 22%), ≥9 cycles (26% vs 10%), and ≥12 cycles (18% vs 5%). At clinical cutoff, 8 (2%) and 2 (1%) patients had ongoing treatment with trabectedin and dacarbazine, respectively, and the maximum number of cycles received was 44 (ongoing) for trabectedin and 30 (ongoing) for dacarbazine. Treatment exposure for each agent was similar within the LPS and LMS patient subgroups (Table 2). Dose reductions and cycle delays, in accordance with protocol guidelines, were reported in 42% and 63% of trabectedin patients, respectively, compared with 12% and 42% of dacarbazine patients, respectively (Supporting Table 1). Dose reductions occurred most often in response to transaminase elevations, whereas dose delays were most often a result of hematologic toxicity (data not shown). Dose modifications did not appear to impact treatment efficacy, as 62% of trabectedin‐treated patients achieving long‐term exposure (≥6 cycles) underwent ≥1 dose reduction.
Table 2.
Cumulative Treatment Exposure
| Liposarcoma (n = 147) | Leiomyosarcoma (n = 403) | Total (n = 550) | ||||
|---|---|---|---|---|---|---|
|
Dacarbazine (n = 45) |
Trabectedin (n = 102) |
Dacarbazine (n = 127) |
Trabectedin (n = 276) |
Dacarbazine (n = 172) |
Trabectedin (n = 378) |
|
| Total treatment cycles | 2 (1‐21) | 4 (1‐41) | 2 (1‐30) | 4 (1‐44) | 2 (1‐≥30)a | 4 (1‐≥44)a |
| Cumulative treatment cycles, n (%) | ||||||
| ≥6 | 7 (16) | 41 (40) | 30 (24) | 118 (43) | 37 (22) | 159 (42) |
| ≥9 | 3 (7) | 28 (28) | 14 (11) | 70 (25) | 17 (10) | 98 (26) |
| ≥12 | 1 (2) | 22 (22) | 7 (6) | 45 (16) | 8 (5) | 67 (18) |
| Cumulative doseb | 2.0 (1.0‐19.4) | 4.2 (1.5‐61.5) | 2.1 (0.8‐30.0) | 6.0 (1.5‐54.5) | 2.0 (0.8‐30.0) | 5.9 (1.5‐61.5) |
| Dose intensity, per cycleb | 0.99 (0.5‐1.0) | 1.3 (0.7‐1.6) | 0.98 (0.4‐1.0) | 1.3 (0.7‐1.6) | 0.98 (0.4‐1.0) | 1.3 (0.7‐1.6) |
| Relative dose intensity, % | 0.99 (0.5‐1.0) | 0.89 (0.4‐1.1) | 0.98 (0.4‐1.0) | 0.87 (0.5‐1.0) | 0.98 (0.40‐1.0) | 0.88 (0.4‐1.1) |
Data are presented as median (range) unless specified otherwise.
Patients with 44 and 30 cycles in the trabectedin and dacarbazine arms, respectively, were ongoing at the time of clinical cutoff for final overall survival analysis.
Units of measure are g/m2 for dacarbazine and mg/m2 for trabectedin.
Efficacy
At the time of the final OS analysis, the median duration of survival follow‐up was 21.2 months. Data were censored for 126 (33%) patients in the trabectedin arm and 70 (36%) in the dacarbazine arm. A total of 381 deaths had occurred: 258 (67%) and 123 (64%) in the trabectedin and dacarbazine arms, respectively. The median OS was 13.7 months (95% confidence interval [CI], 12.2‐16.0) for the trabectedin arm and 13.1 months (95% CI, 9.1‐16.2) for the dacarbazine arm. The unstratified final OS analysis showed no statistically significant improvement, with an overall reduction in risk of death by 7.3% in the trabectedin arm compared with the dacarbazine arm (HR, 0.93; 95% CI, 0.75‐1.15; P = .49) (Fig. 2A).
Figure 2.
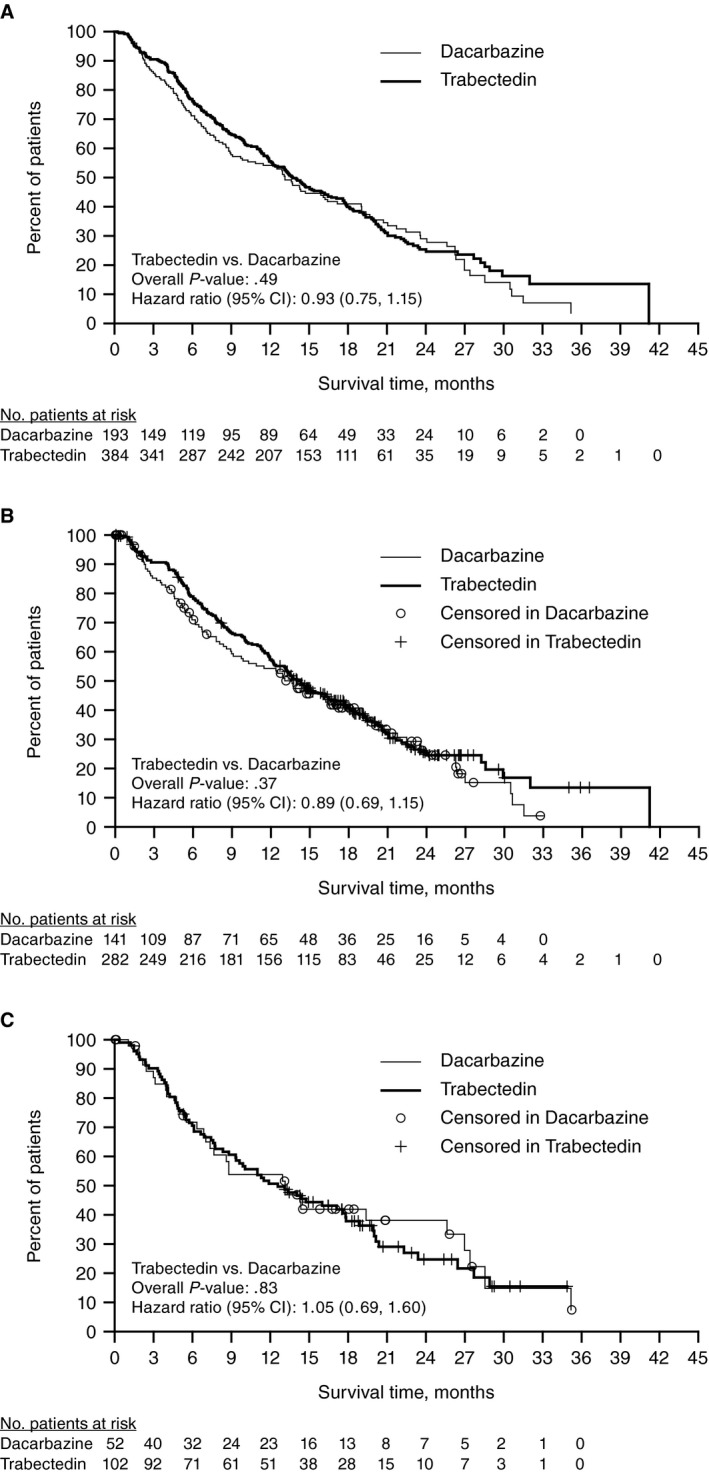
Kaplan‐Meier estimate of overall survival (OS) at the final analysis. (A) OS for total patient population. (B) OS for patients with leiomyosarcoma. (C) OS for patients with liposarcoma. The median OS (95% confidence interval [CI]) for patients with leiomyosarcoma was 14.1 (12.2‐16.5) versus 13.6 (9.1‐17.2) months (P = .37) for patients in the trabectedin versus dacarbazine arms; for those with liposarcoma, the median OS was 13.1 (7.0‐25.6) versus 12.6 (9.3‐17.8) months (P = .83).
A multivariate analysis of OS assessing age, subtype (LPS vs LMS), number of lines of prior chemotherapy (≥2 vs 1), and ECOG performance status score (1 vs 0) was performed. The number of lines of prior chemotherapy and ECOG performance status score were identified as parameters that independently predicted improved OS. Survival outcomes were better for patients with 1 prior lines of chemotherapy and an ECOG performance status score of 0 (Supporting Table 2). Subgroup analysis of 19 different demographic and baseline characteristics showed estimated HRs for OS consistent with the HR for the overall study population (0.93) (Supporting Fig. 1).
The use of post‐study anticancer therapy was more balanced between treatment arms at the time of final OS analysis (71% vs 69% for trabectedin and dacarbazine, respectively) than at the time of interim analysis (47% vs 56%); however, qualitative differences were observed in patterns of therapies received (Supporting Table 3). Although post‐study anticancer therapies were used at similar rates in both treatment arms, improved disease control observed in patients treated with trabectedin resulted in a correspondingly prolonged time to starting any post‐study anticancer therapy in the trabectedin arm compared with the dacarbazine arm (median 6.8 vs 3.5 months; HR, 0.53; 95% CI, 0.43, 0.65; P < .001) (Fig. 3) (Supporting Table 3).
Figure 3.
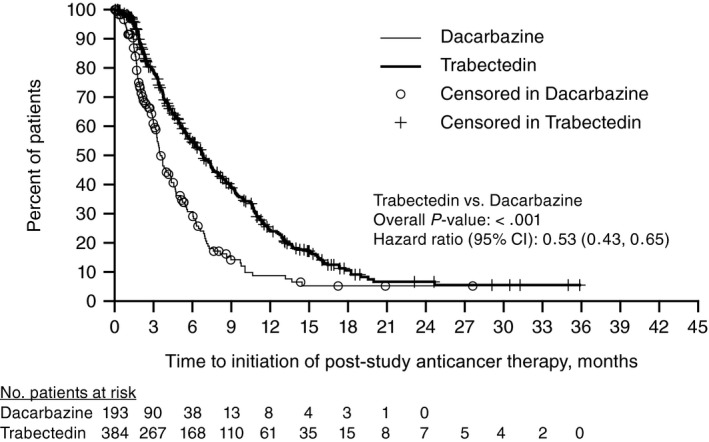
Time to post‐study anticancer therapy utilization in trabectedin versus dacarbazine. CI, confidence interval.
Given the prevalence of post‐study anticancer therapy use, post hoc sensitivity analyses were conducted to investigate a potential confounding effect of those therapies on OS. A stronger trend toward improved survival was observed when OS analyses excluded patients with post‐study anticancer therapy or censored them at initiation of that therapy (Supporting Fig. 2). An analysis with post‐study anticancer therapy as a time‐dependent covariate and an inverse probability of censoring weighted analysis were conducted. At the final OS analysis, the HR prior to receiving post‐study anticancer therapy was 0.67 (95% CI, 0.45‐1.01; P = .06) versus after receiving post‐study anticancer therapy (HR, 1.23), which reflected earlier initiation of post‐study anticancer therapy within the dacarbazine arm (95% CI, 0.95‐1.59; P = .11) (Table 3).
Table 3.
Sensitivity Analyses for Effect of Post‐study Anticancer Therapy on Overall Survival
| HR (95% CI) | P | |
|---|---|---|
| No post‐study anticancer therapy | 0.74 (0.49‐1.11) | .14 |
| Censored at post‐study anticancer therapy | 0.70 (0.47‐1.06) | .09 |
| Subsequent therapy as time‐dependent covariate | ||
| Prior to receiving post‐study anticancer therapy | 0.67 (0.45‐1.01) | .06 |
| After receiving post‐study anticancer therapy | 1.23 (0.95‐1.59) | .11 |
| Inverse probability of censoring weighted analysis | 0.68 (0.45‐1.02) | .06 |
Abbreviations: CI, confidence interval; HR, hazard ratio.
Preplanned Histology‐Specific Subgroup Analyses
In both LMS/LPS histologic subgroups, improvements in PFS, assessed at the time of the interim OS analysis, were of equal magnitude for patients treated with trabectedin vs dacarbazine (Fig. 4). For patients with LMS, the median PFS was 4.3 months (95% CI, 3.6‐5.0 months) in the trabectedin arm versus 1.6 months (95% CI, 1.5‐2.8 months) in the dacarbazine arm (P < .001). For patients with LPS, the median PFS (95% CI) was 3.0 months (95% CI, 1.5‐4.8 months) in the trabectedin arm vs 1.5 months (95% CI, 1.4‐2.7 months) in the dacarbazine arm (P = .009). Partial responses were observed in similar proportions for the trabectedin versus dacarbazine arms in both histologic subgroups (LMS, 10% vs 7%; LPS, 9% vs 6%). Among patients with LMS, the larger of the 2 subgroups, CBR was significantly higher in the trabectedin arm versus the dacarbazine arm (37% vs 20%; P < .001). The CBR among LPS patients, although similar in magnitude, did not reach statistical significance (28% vs 15% for trabectedin vs dacarbazine; P = .096) (Supporting Table 4). Results for OS were similar between trabectedin and dacarbazine, across both histologic subgroups, and consistent with the total study population OS, demonstrating no significant improvement (Fig. 2B,C).
Figure 4.
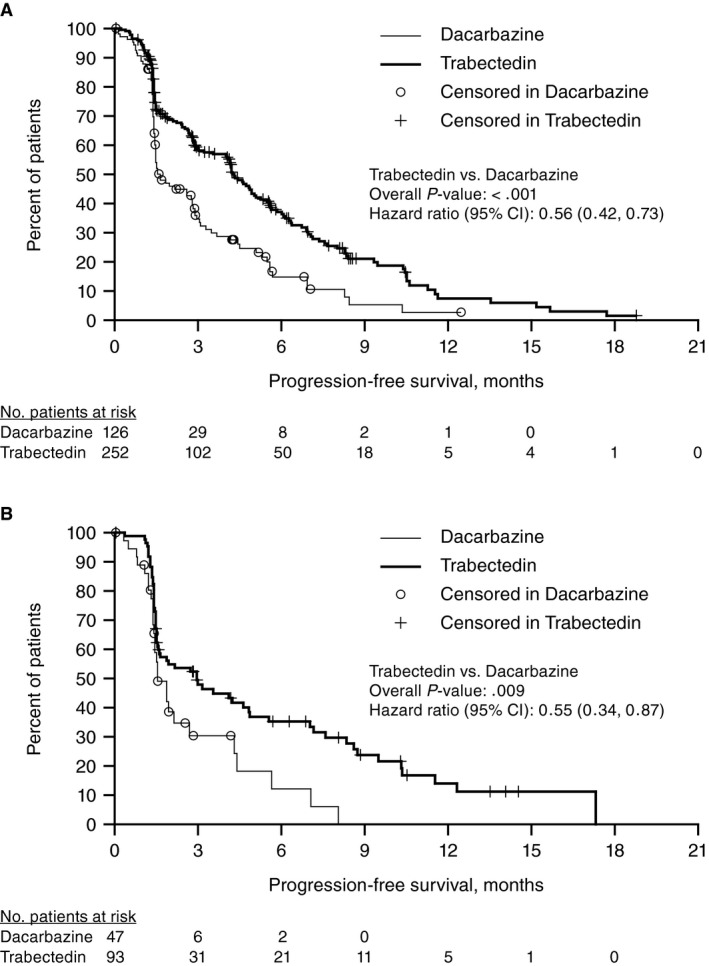
Kaplan‐Meier estimate of progression‐free survival (PFS) at final analysis. (A) PFS for patients with leiomyosarcoma. (B) PFS for patients with liposarcoma. CI, confidence interval.
Safety
The safety results from ET743‐SAR‐3007 have been reported previously and are consistent with the previously described safety profiles for each agent.12, 13 At the time of final OS analysis, no new or additional safety findings were noted.13 The incidence of grade 3‐4 toxicities were generally consistent for both treatment arms when assessed across the 2 histologic subgroups, with the most frequently observed severe toxicities reflecting laboratory‐related parameters of hematologic toxicity and transient elevations in hepatic alanine aminotransferase and aspartate aminotransferase. Febrile neutropenia was increased in the trabectedin arm for both histologic subgroups (4% for LMS and 7% for LPS) when compared with dacarbazine‐treated patients (2% each for LMS or LPS) (Supporting Table 5). Although the incidence of patients who died within 60 days of first dose of study drug was similar in both treatment arms (7% each), deaths due to treatment‐related adverse events were reported for 9 (2%) patients in the trabectedin arm and none in the dacarbazine arm. These deaths were primarily related to infections, rhabdomyolysis, and renal failure.
Discussion
With the earliest phase 1 studies of trabectedin having been conducted in the late 1990s,16, 17, 18 trabectedin is one of the most extensively studied therapeutics for treatment of STS. The reported PFS results of the randomized phase 3 ET743‐SAR‐3007 trial directly demonstrated the superior disease control of trabectedin over dacarbazine, confirming the results of the phase 2 STS‐201 study15 and historical comparisons that led to its initial approval in most countries outside the United States.19 The corresponding safety profiles of trabectedin and dacarbazine, observed in ET743‐SAR‐3007, were consistent with the safety profiles of both agents, with serious adverse events of grade 3‐4 severity in the trabectedin arm characterized by transient elevations in hepatic transaminases and hematological toxicities. Toxicities were managed proactively in ET743‐SAR‐3007 through protocol‐mandated monitoring and appropriate dose delays or dose reductions, minimizing toxicity and allowing patients to achieve prolonged disease control while maintaining their quality of life.20
The final OS analysis presented here from the ET743‐SAR‐3007 study reflected a median follow‐up of 21.2 months and included 381 deaths (trabectedin, n = 258; dacarbazine, n = 123). Consistent with interim analysis findings, the final OS analysis did not demonstrate a statistically significant improvement in survival after treatment with trabectedin compared with dacarbazine (HR, 0.93; 95% CI, 0.75‐1.15; P = .49) and was similar across both histologic subgroups.
Most patients in both treatment arms had received post‐study anticancer therapies (71% and 69% for trabectedin and dacarbazine arms, respectively), which were initiated 3.3 months earlier in the dacarbazine arm, consistent with the earlier failure of this agent compared with trabectedin. Four different exploratory analyses evaluating influence of post‐study anticancer therapy support a potential confounding effect of post‐study anticancer therapy on OS results. Of note, the study was powered to detect a median survival difference of 10.0 and 13.5 months for dacarbazine and trabectedin, respectively. The improved performance of dacarbazine may reflect contribution of post‐study anticancer therapies. The performance of the dacarbazine control arm in ET743‐SAR‐3007 stands in contrast to that observed in the recently reported eribulin phase 3 study, which enrolled a similar patient population contemporaneously with ET743‐SAR‐3007 and showed a median OS of 11.5 months in the dacarbazine control arm within the entire cohort and a median OS of 8.4 months within the LPS subgroup.21, 22, 23 Differences in dacarbazine treatment dose, geographic distribution of patients, and baseline characteristics and stratification strategies may have contributed to the difference in survival outcomes in these 2 control arms.22
Although OS was not improved in this phase 3 trial, the previously described benefit in disease control with trabectedin—in terms of PFS, ORR, and CBR—was improved relative to dacarbazine in both histological subtypes studied. Similarly, greater proportions of trabectedin patients in both subgroups received extended treatment courses, with 40% of LPS and 43% of LMS patients receiving ≥6 cycles (range, 1‐41 and 1‐44 cycles, respectively), versus 16% and 24% (range, 1‐21 and 1‐30 cycles, respectively) in the dacarbazine arm, reflecting both the efficacy and tolerability of trabectedin in these patients.
The results of this study distinguish trabectedin from 2 recently approved therapies, in which regulatory approvals were supported by data limited to either the LMS or LPS subtype: the pazopanib phase 3 PALETTE study included no patients with LPS based on results of a prior phase 2 study,24 and the eribulin phase 3 study failed to demonstrate activity superior to dacarbazine in patients with LMS.21, 23 The findings highlight an unmet need in these histologic subtypes of STS and further support a role for trabectedin as a clinically meaningful treatment option for patients with unresectable, locally advanced, or metastatic LPS/LMS.
In conclusion, the final OS analysis of this phase 3 randomized trial demonstrated no statistically significant improvement in OS of trabectedin over dacarbazine, despite a significant and clinically relevant improvement in PFS with trabectedin that was observed equally in both histological subsets. Prevalent use of active post‐study anticancer therapies may have confounded findings of the OS analysis. The improved OS in both arms may reflect an increasing number and variety of active therapies that are gradually improving the outcome for patients with metastatic sarcoma. The safety and efficacy results from this large prospective trial confirm and expand the favorable risk/benefit profile of trabectedin as a treatment option for LPS/LMS patients who have received prior anthracycline‐based chemotherapy.
Funding Support
This study was supported by Janssen Research & Development, LLC. Martee L. Hensley was supported in part by the Memorial Sloan Kettering Cancer Center Support Grant P30 CA008748. George D. Demetri was supported in part by a grant from the Ludwig Center at Harvard and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Conflict of Interest Disclosures
Shreyaskumar Patel has received grants and personal fees from Janssen; personal fees from PharmaMar and M.J. Hennessey/OncLive; grants from Blueprint Medicines; and personal fees from CytRx, Eli Lilly, EMD Serono, Epizyme, Immune Design, and Novartis Oncology. Margaret von Mehren received support paid to Fox Chase Cancer Center for conduct of the study and received consultant fees from Janssen and personal fees for advisory board participation; was a member of the trial's scientific steering committee; and has been a paid consultant and data and safety monitoring board member for Eisai. Damon R. Reed has served on advisory boards for Epizyme, Janssen, Loxo, and Shire Pharmaceuticals; has been a web site consultant for Epizyme; has been on a speaker's bureau for OncLive; and has received clinical trial support from Janssen associated with the ET743‐SAR‐3007 trial. Pamela Kaiser reports contracting (per patient placed on trial) between Johnson & Johnson (Janssen) and Lutheran General/Oncology Specialists. John Charlson has received clinical trial support for the ET743‐SAR‐3007 and ET743‐SAR‐3002 trials (institutional support to Medical College of Wisconsin) as well as from Blueprint, CytRx, Deciphera, Lilly, Novartis, and Threshold and has participated on advisory boards for CytRx, Immune Design, Lilly, and Threshold. Christopher W. Ryan has received clinical trial research funding from Janssen Research & Development, LLC; has received grants from Argos Therapeutics, Bayer, Bristol‐Myers Squibb, CytRx Corporation, Daiichi Sankyo, Eisai, Exelixis, Genentech, GlaxoSmithKline, Janssen, Karyopharm Therapeutics, MabVax, Merck, Morphotek, Novartis, OSI Pharmaceuticals, Pfizer, Threshold Pharmaceuticals, and TRACON Pharmaceuticals; and has received personal consulting fees from Eisai, EMD Serono, Exelixis, Genentech, Novartis, and Pfizer. Daniel Rushing has received grant funding from Sarcoma Alliance for Research through Collaboration and OncLive; has received personal fees from Janssen; and has received institutional payments from Janssen for the conduct of the trial as well as possible reimbursement from Eli Lilly and Eisai for a physician advisory panel. Michael Livingston has received clinical trial support (institution received money from Janssen to defray the cost of the study) through Blumenthal Cancer Center (now Levine Cancer Center). Arun Singh has served on advisory boards for Eli Lilly, Daiichi Sankyo, and Roche; has served on speaker's bureaus for Eisai, Eli Lilly, Novartis, and OncLive; has received consulting fees from Eisai; has been on the Board of Directors for Certis Oncology Solutions and also holds stock in the company; and has received clinical trial support from Janssen associated with the ET743‐SAR‐3007 trial. Rahul Seth has received clinical trial support (ie, institutional fees) from Janssen. Charles Forscher has received research support from Janssen and Karyopharm and has served on a medical advisory board for Janssen, Epizyme, and Blueprint. Gina D'Amato has served on speaker's bureaus and advisory boards for Eisai, Eli Lilly, Janssen, Novartis, and OncLive and has served on an advisory board for Epizyme. Sant P. Chawla has served as a consultant and/or advisor for, has served on speakers’ bureaus for, and has received honoraria and research funding from Amgen, CytRx Corporation, GlaxoSmithKline, Ignyta, Immune Design, Janssen, Karyopharm Therapeutics, Roche, Sarcoma Alliance for Research through Collaboration, Threshold Pharmaceuticals, and TRACON Pharma. George Wang, Trilok Parekh, Roland Knoblauch, and Sharon McCarthy are all employees of Janssen (a subsidiary of Johnson & Johnson). Martee L. Hensley has received institutional research support from Janssen for the conduct of the study; was a faculty speaker for Research to Practice; served on a faculty expert panel for OncLive; has served on advisory boards for Janssen, Lilly/Lilly Oncology, and Tesaro; and reports chapter author royalties for UpToDate, advisory and market research roles for Tesaro, ongoing research support to Memorial Sloan Kettering Cancer Center from Bristol‐Myers Squibb, research honoraria from Comsort, and a spouse's ongoing employment with Sanofi. Robert G. Maki has received grants from PharmaMar; has received personal consulting fees and clinical trial support (ie, institutional grants) from Janssen while conducting trabectedin studies between 1998 and 2017; has received consultant fees from Arcus, Bayer, Foundation Medicine, Janssen/PharmaMar, Presage Biosciences, and Tracon Pharmaceuticals; has received honoraria from the American Association for Cancer Research and the American Society of Clinical Oncology; has served on the data safety monitoring board for AADi, Deciphera, and Karyopharm; is a member of the Medical Oncology Exam Committee for the American Board of Internal Medicine; has received clinical trial support from Daiichi‐Sankyo, Genentech, Immune Design, Immunocor, Janssen/PharmaMar, Lilly/Imclone, Presage Biosciences, Regeneron, Sarcoma Alliance for Research through Collaboration, and Tracon Pharmaceuticals; has received royalties from Springer, Wiley, and UpToDate; and has received a grant from Fondazione Enrico Pallazzo. George D. Demetri has received grants, personal fees, and nonfinancial support from PharmaMar—as well as travel expenses for a research meeting—and from Janssen in relation to the submitted work. In addition, he has received grants from AbbVie, Adaptimmune, Bayer, Daiichi‐Sankyo, Epizyme, GlaxoSmithKline, Ignyta, Loxo Oncology, Novartis, Pfizer, and Roche; has received personal fees from AbbVie, Adaptimmune, Bayer, Blueprint Medicines, Daiichi‐Sankyo, Caris Life Sciences, Champions Oncology, EMD‐Serono, Epizyme, G1 Therapeutics, Ignyta, Loxo Oncology, Mirati Therpeutics, Merrimack Pharmaceuticals, M.J. Hennessey/OncLive, Novartis, Pfizer, Polaris Pharmaceuticals, Roche, Sanofi, WIRB Copernicus Group, ZioPharm; has received nonfinancial support from AbbVie, Daiichi‐Sankyo, Epizyme, Novartis, and Roche; has received travel expenses for an educational meeting for M.J. Hennessey/OncLive, Novartis, and Pfizer; has received travel expenses for an advisory board meeting for Bayer, Caris Life Sciences, Daiichi‐Sankyo, EMD‐Serono, Loxo Oncology, Roche, and WIRB Copernicus Group; has received travel expenses for an FDA meeting for Epizyme; has received travel expenses for a research meeting for study for Adaptimmune; has received travel expenses for board meetings for Blueprint Medicines and Merrimack Pharmaceuticals; has equity for Blueprint Medicines and G1 Therapeutics; has equity options for Bessor Pharmaceuticals, Caris Life Sciences, Champions Oncology, Erasca Pharmaceuticals, G1 Therapeutics, and Merrimack Pharmaceuticals; reports a patent that is issued and licensed to PharmaMar for trabectedin use for cancer (patent from PharmaMar [for which he receives no funds and no license to Dana‐Farber Cancer Center or to Dr. Demetri]); and reports a patent that is issued and licensed to Novartis for imatinib use for gastrointestinal stromal tumors as well as royalties stemming from the patent.
Author Contributions
All authors were involved in making substantial contributions to the conception or design of the work or the acquisition, analysis, or interpretation of data; drafting the manuscript or revising it critically for important intellectual content; final approval of the version to be submitted; and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of it are appropriately investigated and resolved. Shreyaskumar Patel: study conceptualization; design and conduct of the trial; data analysis and interpretation; writing of the manuscript; approval of final version submitted. Margaret von Mehren: study conceptualization; design and conduct of the trial; contributions of data collection/patients for study; data interpretation; writing and approval of final manuscript; scientific leadership of the study. Damon R. Reed: data collection; data analysis; data interpretation; writing. Pamela Kaiser: critical review of the manuscript with suggestions/edits; data collection of patients placed on protocol/treatment; review of interpretative data. John Charlson: data collection; manuscript review. Christopher W. Ryan: accrual of patients; data collection; writing and final approval of manuscript. Daniel Rushing: enrollment and management of patients on the trial while following all protocol requirements; review of manuscript and all revisions; comments toward publication; agreement with the decision to submit manuscript for publication; vouching for the accuracy and completeness of the data and analysis. Michael Livingston: patient evaluation and manuscript review. Arun Singh: investigator on the trial; put patients on the trial; review of manuscript. Rahul Seth: data collection, analysis, and interpretation; writing and editing the manuscript; review of data after comments. Charles Forscher: writing and editing of the manuscript. Gina D'Amato: principal investigator on the trial; patient enrollment; submission of patient survival data per protocol; review of paper in detail and provision of comments on first‐line therapy. Sant P. Chawla: substantial contribution to drafting of the paper and intellectual contributions to its contents; approval of final draft. Sharon McCarthy: contribution to protocol development; data collection; review, interpretation, and analysis. George Wang: study design; data collection; data analysis; data interpretation. Trilok Parekh: study design; data analysis; data interpretation. Roland Knoblauch: oversight of conduct of the study as clinical leader of trabectedin; data collection, analysis, and interpretation; preparation of the manuscript. Martee L. Hensley: data collection; data interpretation; review of manuscript. Robert Maki: provision of patients; study conduct; data collection; writing and final approval of manuscript. George Demetri: study concept and design; chairing of steering committee; oversight of study implementation/data collection; extensive data analysis and interpretation; writing and approval of final version of manuscript.
Supporting information
We thank all of the patients who participated in the study and the site investigators for their contributions to the ET743‐SAR‐3007 study (Supporting Table 6). We also thank Chu Ri Shin, MD, for critical review of the manuscript and Gianna Paone, MS, MPH, for providing medical writing, editorial, and submission support (both of Janssen Scientific Affairs, LLC) and Sajeve Thomas, MD, (University of Florida Health Cancer Center–Orlando Health) and Nushmia Khokhar, MD, (Janssen Research & Development, LLC) for their involvement in the study.
This study is connected with clinical trial NCT01343277 (ClinicalTrials.gov).
References
- 1. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft‐tissue sarcoma: an open‐label phase 1b and randomised phase 2 trial. Lancet. 2016;388:488‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first‐line treatment of advanced or metastatic soft‐tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15:415‐423. [DOI] [PubMed] [Google Scholar]
- 3. Lee EM, Rha SY, Lee J, Park KH, Ahn JH. Phase II study of weekly docetaxel and fixed dose rate gemcitabine in patients with previously treated advanced soft tissue and bone sarcoma. Cancer Chemother Pharmacol. 2012;69:635‐642. [DOI] [PubMed] [Google Scholar]
- 4. Hensley ML, Miller A, O'Malley DM, et al. Randomized phase III trial of gemcitabine plus docetaxel plus bevacizumab or placebo as first‐line treatment for metastatic uterine leiomyosarcoma: an NRG Oncology/Gynecologic Oncology Group study. J Clin Oncol. 2015;33:1180‐1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hensley ML, Blessing JA, Mannel R, Rose PG. Fixed‐dose rate gemcitabine plus docetaxel as first‐line therapy for metastatic uterine leiomyosarcoma: a Gynecologic Oncology Group phase II trial. Gynecol Oncol. 2008;109:329‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis EJ, Chugh R, Zhao L, et al. A randomised, open‐label, phase II study of neo/adjuvant doxorubicin and ifosfamide versus gemcitabine and docetaxel in patients with localised, high‐risk, soft tissue sarcoma. Eur J Cancer. 2015;51:1794‐1802. [DOI] [PubMed] [Google Scholar]
- 7. Petek BJ, Loggers ET, Pollack SM, Jones RL. Trabectedin in soft tissue sarcomas. Mar Drugs. 2015;13:974‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puyo S, Montaudon D, Pourquier P. From old alkylating agents to new minor groove binders. Crit Rev Oncol Hematol. 2014;89:43‐61. [DOI] [PubMed] [Google Scholar]
- 9. D'Incalci M, Galmarini CM. A review of trabectedin (ET‐743): a unique mechanism of action. Mol Cancer Ther. 2010;9:2157‐2163. [DOI] [PubMed] [Google Scholar]
- 10. Laroche‐Clary A, Chaire V, Le Morvan V, et al. BRCA1 haplotype and clinical benefit of trabectedin in soft‐tissue sarcoma patients. Br J Cancer. 2015;112:688‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yondelis . Summary of product characteristics. PharmaMar S.A.: Madrid, Spain; 2016. [Google Scholar]
- 12. Yondelis . Prescribing information. Horsham, PA: Janssen Products, LP; 2015. [Google Scholar]
- 13. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34:786‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol. 2013;24:1703‐1709. [DOI] [PubMed] [Google Scholar]
- 15. Demetri GD, Chawla SP, von Mehren M, et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol. 2009;27:4188‐4196. [DOI] [PubMed] [Google Scholar]
- 16. Taamma A, Misset JL, Riofrio M, et al. Phase I and pharmacokinetic study of ecteinascidin‐743, a new marine compound, administered as a 24‐hour continuous infusion in patients with solid tumors. J Clin Oncol. 2001;19:1256‐1265. [DOI] [PubMed] [Google Scholar]
- 17. Delaloge S, Yovine A, Taamma A, et al. Ecteinascidin‐743: a marine‐derived compound in advanced, pretreated sarcoma patients—preliminary evidence of activity. J Clin Oncol. 2001;19:1248‐1255. [DOI] [PubMed] [Google Scholar]
- 18. Ryan DP, Supko JG, Eder JP, et al. Phase I and pharmacokinetic study of ecteinascidin 743 administered as a 72‐hour continuous intravenous infusion in patients with solid malignancies. Clin Cancer Res. 2001;7:231‐242. [PubMed] [Google Scholar]
- 19. Van Glabbeke M, Verweij J, Judson I, et al; on behalf of the EORTC Soft Tissue and Bone Sarcoma Group . Progression‐free rate as the principal end‐point for phase II trials in soft‐tissue sarcomas. Eur J Cancer. 2002;38:543‐549. [DOI] [PubMed] [Google Scholar]
- 20. Demetri GD, von Mehren M, Jones RL, et al. Patient‐reported outcomes from randomized, phase‐3 study of trabectedin (T) vs. dacarbazine (D) in advanced leiomyosarcoma (LMS) or liposarcoma (LPS). Poster presented at: 2016 American Society of Clinical Oncology Annual Meeting; June 3‐7, 2016; Chicago, IL.
- 21. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open‐label, multicentre, phase 3 trial. Lancet. 2016;387:1629‐1637. [DOI] [PubMed] [Google Scholar]
- 22. Chawla S, Schöffski P, Grignani G, et al. Subtype‐specific activity in liposarcoma patients from a phase 3, open‐label, randomized study of eribulin versus dacarbazine in patients with advanced liposarcoma or leiomyosarcoma. Poster presented at: 2016 American Society of Clinical Oncology Annual Meeting; June 3‐7, 2016; Chicago, IL.
- 23. Halaven . Prescribing information. Woodcliff Lake, NJ: Eisai, Inc; 2016. [Google Scholar]
- 24. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft‐tissue sarcoma (PALETTE): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2012;379:1879‐1886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials