

Abstract
A 1,3‐aminothiolation was realized by reacting 2‐substituted cyclopropane 1,1‐dicarboxylates with sulfonamides and N‐(arylthio)succinimides. Under Sn(OTf)2 catalysis the transformation proceeded smoothly to the corresponding ring‐opened products bearing the sulfonamide in the 1‐position next to the donor and the arylthio residue in the 3‐position next to the acceptor. The procedure was extended to the corresponding selenium analogues by employing N‐(phenylseleno)succinimides as an electrophilic selenium source.
Keywords: 1,3-aminochalcogenation; bisfunctionalization; cyclopropanes; donor–acceptor compounds; ring-opening reactions
The cyclopropane molecule is the smallest and most strained carbocyclic ring system; astonishingly, it is kinetically relatively stable. However, its reactivity is dramatically increased when the cyclic structure is decorated with donor and acceptor substitutents in vicinal positions. These molecular entities, so‐called donor–acceptor (D–A) cyclopropanes, were introduced by Wenkert and Reissig in the late 1970s and 1980s.1 After a quiet time at the end of the 20th century, D–A cyclopropanes have enjoyed a renaissance during the last two decades in synthetic organic chemistry and have been widely exploited in methodology, and also in natural product synthesis. These “spring‐loaded” systems benefit from a highly polarized bond, caused by a vicinial arrangement of electron‐donating and ‐withdrawing substitutents, and further assisted by the high ring strain of about 115 kJ mol−1.2 The resulting special reactivity paves the way for a plethora of unusual transformations.3
Along these lines, rearrangement reactions, which are atom‐economic transformations, have been developed with D–A cyclopropanes, leading to ring‐enlarged hetero‐ and carbocyclic structures by embedding the acceptor moiety into the ring system.4 While rearrangement reactions are intramolecular transformations, that is, only one component is required, cycloadditions as intermolecular reactions need a second component. Depending on the number of atoms to be incorporated in the emerging cyclic structure, such (3+n)‐cycloadditions lead to five‐,5 six‐,6 or seven‐membered7 ring systems. Even complex bicyclic structures, which are prominent structural motifs in natural product chemistry, are easily obtained by these cycloadditions when the two reactive moieties are tethered to each other.8 While formal cycloaddition reactions are well established in D–A cyclopropane chemistry, ring‐opening reactions, especially 1,3‐bisfunctionalizations, seem to be underexplored. Whereas in previous years the focus was on monofunctionalization with heteroatom nucleophiles (e.g. phenols,9 naphthols,10 amines,11 azides,12 or thiols)13 or carbon nucleophiles14 (Scheme 1 a), nowadays more challenging 1,3‐bisfunctionalizations are under investigation by several groups.15 Seminal work in this field was performed by Sparr and Gilmour with an enantioselective 1,3‐dichlorination protocol of meso‐cyclopropyl carbaldehydes under organocatalytic conditions, as depicted in Scheme 1 b.16
Scheme 1.
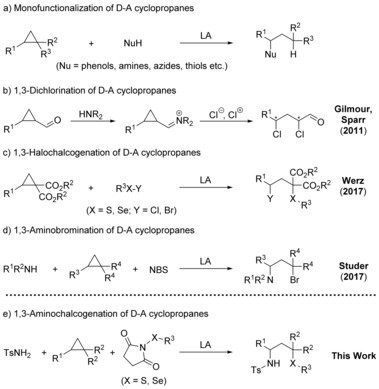
Ring‐opening reactions of D–A cyclopropanes.
Recently, we investigated a ring‐opening reaction of cyclopropane dicarboxylates with chalcogenyl chlorides and bromides to afford 1,3‐halochalcogenated products (Scheme 1 c),17 whereas Studer and co‐workers presented an elegant 1,3‐aminobromination of D–A cyclopropanes by using N‐bromosuccinimide and electron‐deficient anilines or sulfonamides (Scheme 1 d).18 Based on these results, we envisioned that N‐(arylthio)succinimides might be captured by the intermediate carbanion emerging in the reaction of cyclopropane dicarboxylates and sulfonamides (Scheme 1 e). Herein, we report the first 1,3‐aminochalcogenation of D–A cyclopropanes by such a three‐component approach using Lewis acid catalysis.
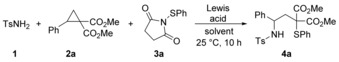
To test our notion, we first investigated a variety of reaction conditions along with various Lewis acids commonly used for the activation of D–A cyclopropanes (Table 1). Tosylamide 1, cyclopropane 2 a and succinimide derivative 3 a were chosen as model substrates. In 1,2‐dichloroethane at room temperature, Sn(OTf)2 proved to be the most suitable Lewis acid for our anticipated transformation and delivered our desired product 4 a in 69 % yield. Interestingly, Lewis acids such as AlCl3, MgI2 or Sc(OTf)3, which had shown excellent results for many other reactions in the field of D–A cyclopropane chemistry, afforded not even a trace of the product (Table 1, entries 1–3). During our thorough screening and the subsequent optimization studies, we realized that the transformation is very sensitive to moisture; therefore, the reactions were set up and conducted in a glovebox. As major side product the monofunctionalized product 8 was isolated (see the Supporting Information). Decreasing the amount of the Lewis acid to 10 mol % was invaluable and resulted in an increased yield of 86 % (entry 7). The choice of the solvent proved to be crucial for a successful outcome; changing the solvent to dioxane or dichloromethane shut down the reaction completely or afforded a greatly decreased yield of only 29 %, respectively (entries 8–9). In contrast, increasing the amount of succinimide 3 a resulted in an improved yield of 93 % (entry 10), whereas using the phthalimide analogue strongly impaired the outcome of the reaction (entry 12).
Table 1.
Optimization of the reaction conditions.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Lewis acid | [mol %] | 3 a [equiv] | Solvent | Yield [%] |
| 1 | AlCl3 | 20 | 1.3 | DCE | 0 |
| 2 | MgI2 | 20 | 1.3 | DCE | 0 |
| 3 | Sc(OTf)3 | 20 | 1.3 | DCE | 0 |
| 4 | Sn(OTf)2 | 20 | 1.3 | DCE | 69 |
| 5 | Y(OTf)3 | 20 | 1.3 | DCE | 0 |
| 6 | Sn(OTf)2 | 5 | 1.3 | DCE | 0 |
| 7 | Sn(OTf)2 | 10 | 1.3 | DCE | 86 |
| 8 | Sn(OTf)2 | 10 | 1.3 | CH2Cl2 | 29 |
| 9 | Sn(OTf)2 | 10 | 1.3 | dioxane | 0 |
| 10 | Sn(OTf)2 | 10 | 1.7 | DCE | 93 |
| 11 | Sn(OTf)2 | 10 | 2.3 | DCE | 72 |
| 12[b] | Sn(OTf)2 | 10 | 1.3 | DCE | 35 |
[a] Reaction conditions: 1 (165 μmol), Lewis acid, solvent (1.5 mL), 2 a (150 μmol), 3 a, 25 °C, 10 h, Ar atmosphere; yields represent isolated and purified products; [b] The corresponding phthalimide derivative was used instead of 3 a.
With optimized conditions in hand, we examined the scope of the 1,3‐aminochalcogenation. Thus, a broad variety of D–A cyclopropanes were tested. We started our exploration by using the diethyl ester to trigger the ring‐opening process and obtained compound 4 b in 77 % yield. Next, we probed the variation of the aryl unit of model substrate 2 a. Halogens in para‐position delivered the products 4 c–e in excellent yields (91–94 %); the same holds true for an acetoxy (4 f, 88 %) and a trifluoromethyl substitution (4 g, 86 %). Methyl‐substitution in the ortho‐, meta‐ and para‐position delivered the desired products in good yields (66–89 %). Whereas nitro groups in meta‐ and para‐position (4 k and 4 l) proved to be uncritical (82–84 %), the highly electron‐withdrawing pentafluorophenyl residue (4 m) showed a significant drop in yield to 54 %. The naphthyl residue (4 n), an extended π‐system, allowed the transformation again in good yield. Finally, upscaling the reaction to the 1.5 mmol scale with respect to D–A cyclopropane 2 a delivered compound 4 a in 69 % yield. In conclusion, a high functional‐group tolerance with respect to the employed 14 D–A cyclopropanes was observed. To unequivocally prove our anticipated 1,3‐aminothiolation, we were able to grow single crystals of 4 a suitable for X‐ray crystallographic analysis. The structure shows the sulfonamide in the 1‐position next to the donor and the thiophenyl residue in the 3‐position adjacent to the two carboxylates (Scheme 2).19
Scheme 2.
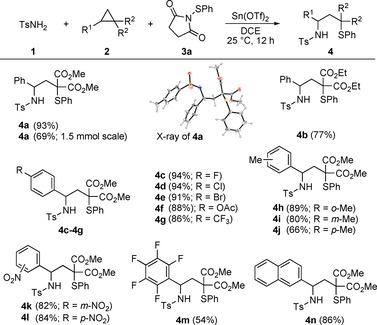
Scope of the 1,3‐aminothiolation with respect to various D–A cyclopropanes 2. Reaction conditions: 1 (220 μmol), 2 (200 μmol), 3 a (340 μmol), Sn(OTf)2 (10 mol %), DCE (1.5 mL), 25 °C, 12 h. Yields represent isolated and purified products. DCE=1,2‐dichloroethane.
To test the generality of our protocol, we next subjected various N‐(arylthio)succinimide derivatives 3 to our reaction system (Scheme 3). In the first instance, we tested different substitution patterns of the transferred thiophenyl moiety. Thus, methyl groups in the ortho‐, meta‐ and para‐position of the phenyl residue were installed and the respective derivatives 3 employed in the reaction. The formation proceeded smoothly for all three variants (5 a–c) in moderate to good yields. Transfer of the p‐methoxyphenylthio residue (5 d) proceeded astonishingly well in 95 % yield, whereas N‐(naphthylthio)succinimde delivered compound 5 e in good yield.
Scheme 3.
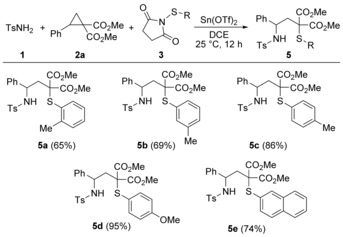
Scope of the 1,3‐aminothiolation with respect to various N‐(arylthio)succinimide derivatives 3. Reaction conditions: 1 (220 μmol), 2 a (200 μmol), 3 (340 μmol), Sn(OTf)2 (10 mol %), DCE (1.5 mL), 25 °C, 12 h. Yields represent isolated and purified products.
Inspired by these results, we were keen to test whether a 1,3‐aminoselenation is able to deliver similar selenium analogues. Therefore, N‐(phenylseleno)succinimide 6 was prepared and subjected to our standard reaction conditions. The transformation proceeded smoothly and delivered compound 7 a in 88 % yield (Scheme 4), whereas the respective ethyl diester gave 77 % yield (7 b). The extension of the π‐system to a naphthyl donor provided the respective product in a yield of only 39 % (7 c). Substitution of the phenyl ring in para‐position with halogen atoms furnished 7 d and 7 e in moderate yields; the same applied for the decoration in ortho‐position with a methyl group leading to 7 f. X‐ray structural elucidation of compound 7 d unambiguously confirmed the anticipated 1,3‐aminoselenolation.19
Scheme 4.
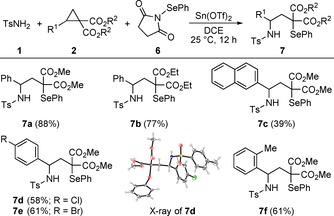
Scope of the 1,3‐aminoselenation with respect to various D–A cyclopropanes 2. Reaction conditions: 1 (220 μmol), 2 (200 μmol), 6 (340 μmol), Sn(OTf)2 (10 mol %), DCE (1.5 mL), 25 °C, 12 h. Yields represent isolated and purified products.
To shed light onto the reaction mechanism, we subjected enantioenriched D–A cyclopropane (S)‐2 a′ (>99 % ee) to our standard conditions in order to test the stereospecificity of our transformation. To our surprise, the reaction proceeded with a significant loss of stereoinformation (4 a, 10 % ee). Further investigations clearly revealed that D–A cyclopropane (S)‐2 a′ undergoes racemization when treated with Sn(OTf)2 only. Based on these observations and literature evidance,20 we propose the following reaction mechanism as depicted in Scheme 5. Initially, cyclopropane dicarboxylate (S)‐2 a′ is activated by Sn(OTf)2, chelating the geminal diesters, whereby fast racemization occurs. One might speculate that the redox ability of SnII is the reason for this unexpected behavior, which is not often observed with non‐redox‐active Lewis acids. The activated three‐membered ring allows a nucleophilic attack of tosyl amide 1. Under the given conditions the formation of side product 8 is diminished. The emerging carbanion is trapped by succinimide derivative 3 a; succinimide is released and the desired product 4 a is formed. It seems that proton transfer from NH2 of the sulfonamide to the anionic malonate is relatively slow in DCE. This is the prerequisite so that the third component, the thiosuccinimide, is able to come into play and the decisive nucleophilic substitution at the chalcogen occurs. Traces of water are detrimental because water immediately leads to a proton transfer and thus to the monofunctionalized product.
Scheme 5.
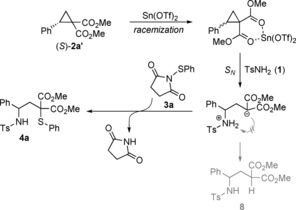
Proposed reaction mechanism.
In conclusion, we have developed a novel 1,3‐aminothiolation and 1,3‐aminoselenation protocol by ring‐opening of D–A cyclopropanes. Sn(OTf)2 proved to be the Lewis acid of choice for this three‐component approach using tosyl amides as nucleophiles, the cyclopropane as a masked zwitterion and chalcogenosuccinimides as electrophilic components. The catalytic 1,3‐bisfunctionalization proceeded smoothly in yields up to 95 %, whereby the transformation tolerates various donors including electron‐rich and ‐deficient aryl residues. This approach nicely complements previously developed methods to exploit cyclopropanes as a formally zwitterionic synthon for open‐chain 1,3‐bisfunctionalized compounds.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the European Research Council (ERC Consolidator Grant “GAINBYSTRAIN” to D.B.W.). A.U.A. thanks A. Bauschke (TU Braunschweig) for his kind support of this work.
A. U. Augustin, P. G. Jones, D. B. Werz, Chem. Eur. J. 2019, 25, 11620.
Contributor Information
André U. Augustin, http://www.werzlab.de
Prof. Dr. Daniel B. Werz, Email: d.werz@tu-braunschweig.de.
References
- 1.
- 1a. Wenkert E., Alonso M. E., Buckwalter B. L., Chou K. J., J. Am. Chem. Soc. 1977, 99, 4778; [Google Scholar]
- 1b. Piers E., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1979, 18, 791; [Google Scholar]; Angew. Chem. 1979, 91, 857; [Google Scholar]
- 1c. Reissig H.-U., Hirsch E., Angew. Chem. Int. Ed. Engl. 1980, 19, 813; [Google Scholar]; Angew. Chem. 1980, 92, 839; [Google Scholar]
- 1d. Brückner C., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1985, 24, 588; [Google Scholar]; Angew. Chem. 1985, 97, 578. [Google Scholar]
- 2. Gordon M. S., J. Am. Chem. Soc. 1980, 102, 7419. [Google Scholar]
- 3.
- 3a. Reissig H.-U., Zimmer R., Chem. Rev. 2003, 103, 1151; [DOI] [PubMed] [Google Scholar]
- 3b. Yu M., Pagenkopf B. L., Tetrahedron 2005, 61, 321; [Google Scholar]
- 3c. Agrawal D., Yadav V. K., Chem. Commun. 2008, 6471; [DOI] [PubMed] [Google Scholar]
- 3d. Carson C. A., Kerr M. A., Chem. Soc. Rev. 2009, 38, 3051; [DOI] [PubMed] [Google Scholar]
- 3e. De Simone F., Waser J., Synthesis 2009, 3353; [Google Scholar]
- 3f. Cavitt M. A., Phun L. H., France S., Chem. Soc. Rev. 2014, 43, 804; [DOI] [PubMed] [Google Scholar]
- 3g. Schneider T. F., Kaschel J., Werz D. B., Angew. Chem. Int. Ed. 2014, 53, 5504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5608; [Google Scholar]
- 3h. Grover H. K., Emmett M. R., Kerr M. A., Org. Biomol. Chem. 2015, 13, 655; [DOI] [PubMed] [Google Scholar]
- 3i. Novikov R. A., Tomilov Y. V., Mendeleev Commun. 2015, 25, 1; [Google Scholar]
- 3j. Rassadin V. A., Six Y., Tetrahedron 2016, 72, 4701. [Google Scholar]
- 4.
- 4a. Bowman R. K., Johnson J. S., Org. Lett. 2006, 8, 573; [DOI] [PubMed] [Google Scholar]
- 4b. Brand C., Rauch G., Zanoni M., Dittrich B., Werz D. B., J. Org. Chem. 2009, 74, 8779; [DOI] [PubMed] [Google Scholar]
- 4c. Gharpure S. J., Shukla M. K., Vijayasree U., Org. Lett. 2009, 11, 5466; [DOI] [PubMed] [Google Scholar]
- 4d. Schneider T. F., Kaschel J., Dittrich B., Werz D. B., Org. Lett. 2009, 11, 2317; [DOI] [PubMed] [Google Scholar]
- 4e. Schneider T. F., Kaschel J., Awan S. I., Dittrich B., Werz D. B., Chem. Eur. J. 2010, 16, 11276; [DOI] [PubMed] [Google Scholar]
- 4f. Schneider T. F., Werz D. B., Org. Lett. 2011, 13, 1848; [DOI] [PubMed] [Google Scholar]
- 4g. Kaschel J., Schneider T. F., Kratzert D., Stalke D., Werz D. B., Angew. Chem. Int. Ed. 2012, 51, 11153; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11315; [Google Scholar]
- 4h. Kaschel J., Schmidt C. D., Mumby M., Kratzert D., Stalke D., Werz D. B., Chem. Commun. 2013, 49, 4403; [DOI] [PubMed] [Google Scholar]
- 4i. Kaschel J., Schneider T. F., Kratzert D., Stalke D., Werz D. B., Org. Biomol. Chem. 2013, 11, 3494; [DOI] [PubMed] [Google Scholar]
- 4j. Kaschel J., Schneider T. F., Schirmer P., Maaß C., Stalke D., Werz D. B., Eur. J. Org. Chem. 2013, 4539; [Google Scholar]
- 4k. Schmidt C. D., Kaschel J., Schneider T. F., Kratzert D., Stalke D., Werz D. B., Org. Lett. 2013, 15, 6098; [DOI] [PubMed] [Google Scholar]
- 4l. Ivanova O. A., Chagarovskiy A. O., Shumsky A. N., Krasnobrov V. D., Levina I. I., Trushkov I. V., J. Org. Chem. 2018, 83, 543; [DOI] [PubMed] [Google Scholar]
- 4m. Ortega A., Manzano R., Uria U., Carrillo L., Reyes E., Tejero T., Merino P., Vicario J. L., Angew. Chem. Int. Ed. 2018, 57, 8225; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8357. [Google Scholar]
- 5.
- 5a. Parsons A. T., Campbell M. J., Johnson J. S., Org. Lett. 2008, 10, 2541; [DOI] [PubMed] [Google Scholar]
- 5b. Parsons A. T., Johnson J. S., J. Am. Chem. Soc. 2009, 131, 3122; [DOI] [PubMed] [Google Scholar]
- 5c. Smith A. G., Slade M. C., Johnson J. S., Org. Lett. 2011, 13, 1996; [DOI] [PubMed] [Google Scholar]
- 5d. Xu H., Qu J.-P., Liao S., Xiong H., Tang Y., Angew. Chem. Int. Ed. 2013, 52, 4004; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4096; [Google Scholar]
- 5e. Racine S., de Nanteuil F., Serrano E., Waser J., Angew. Chem. Int. Ed. 2014, 53, 8484; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8624; [Google Scholar]
- 5f. Racine S., Hegedüs B., Scopelliti R., Waser J., Chem. Eur. J. 2016, 22, 11997; [DOI] [PubMed] [Google Scholar]
- 5g. Sabbatani J., Maulide N., Angew. Chem. Int. Ed. 2016, 55, 6780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6892; [Google Scholar]
- 5h. Garve L. K. B., Kreft A., Jones P. G., Werz D. B., J. Org. Chem. 2017, 82, 9235; [DOI] [PubMed] [Google Scholar]
- 5i. Preindl J., Chakrabarty S., Waser J., Chem. Sci. 2017, 8, 7112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5j. Augustin A. U., Busse M., Jones P. G., Werz D. B., Org. Lett. 2018, 20, 820; [DOI] [PubMed] [Google Scholar]
- 5k. Kreft A., Jones P. G., Werz D. B., Org. Lett. 2018, 20, 2059; [DOI] [PubMed] [Google Scholar]
- 5l. Matsumoto Y., Nakatake D., Yazaki R., Ohshima T., Chem. Eur. J. 2018, 24, 6062; [DOI] [PubMed] [Google Scholar]
- 5m. Wang Z.-H., Zhang H.-H., Xu P.-F., Luo Y.-C., Chem. Commun. 2018, 54, 10128; [DOI] [PubMed] [Google Scholar]
- 5n. Zotova M. A., Novikov R. A., Shulishov E. V., Tomilov Y. V., J. Org. Chem. 2018, 83, 8193; [DOI] [PubMed] [Google Scholar]
- 5o. Akaev A. A., Bezzubov S. I., Desyatkin V. G., Vorobyeva N. S., Majouga A. G., Melnikov M. Y., Budynina E. M., J. Org. Chem. 2019, 84, 3340; [DOI] [PubMed] [Google Scholar]
- 5p. Xie M.-S., Zhao G.-F., Qin T., Suo Y.-B., Qu G.-R., Guo H.-M., Chem. Commun. 2019, 55, 1580; [DOI] [PubMed] [Google Scholar]
- 5q. Kreft A., Lücht A., Grunenberg J., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2019, 58, 1955; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1975. [Google Scholar]
- 6.
- 6a. Novikov R. A., Tarasova A. V., Korolev V. A., Shulishov E. V., Timofeev V. P., Tomilov Y. V., J. Org. Chem. 2015, 80, 8225; [DOI] [PubMed] [Google Scholar]
- 6b. Wang H.-P., Zhang H.-H., Hu X.-Q., Xu P.-F., Luo Y.-C., Eur. J. Org. Chem. 2015, 3486; [Google Scholar]
- 6c. Chidley T., Vemula N., Carson C. A., Kerr M. A., Pagenkopf B. L., Org. Lett. 2016, 18, 2922; [DOI] [PubMed] [Google Scholar]
- 6d. Das S., Chakrabarty S., Daniliuc C. G., Studer A., Org. Lett. 2016, 18, 2784; [DOI] [PubMed] [Google Scholar]
- 6e. Garve L. K. B., Petzold M., Jones P. G., Werz D. B., Org. Lett. 2016, 18, 564; [DOI] [PubMed] [Google Scholar]
- 6f. Sin S., Kim S.-G., Adv. Synth. Catal. 2016, 358, 2701; [Google Scholar]
- 6g. Mondal K., Pan S. C., Eur. J. Org. Chem. 2017, 534; [Google Scholar]
- 6h. Chagarovskiy A. O., Vasin V. S., Kuznetsov V. V., Ivanova O. A., Rybakov V. B., Shumsky A. N., Makhova N. N., Trushkov I. V., Angew. Chem. Int. Ed. 2018, 57, 10338; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10495; [Google Scholar]
- 6i. Petzold M., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2019, 58, 6225; [DOI] [PubMed] [Google Scholar]
- 6j. Varshnaya R. K., Banerjee P., J. Org. Chem. 2019, 84, 1614. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Ivanova O. A., Budynina E. M., Grishin Y. K., Trushkov I. V., Verteletskii P. V., Angew. Chem. Int. Ed. 2008, 47, 1107; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1123; [Google Scholar]
- 7b. Xu H., Hu J.-L., Wang L., Liao S., Tang Y., J. Am. Chem. Soc. 2015, 137, 8006; [DOI] [PubMed] [Google Scholar]
- 7c. Garve L. K. B., Pawliczek M., Wallbaum J., Jones P. G., Werz D. B., Chem. Eur. J. 2016, 22, 521. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Xing S., Pan W., Liu C., Ren J., Wang Z., Angew. Chem. Int. Ed. 2010, 49, 3215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3283; [Google Scholar]
- 8b. Xing S., Li Y., Li Z., Liu C., Ren J., Wang Z., Angew. Chem. Int. Ed. 2011, 50, 12605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12813; [Google Scholar]
- 8c. Bai Y., Tao W., Ren J., Wang Z., Angew. Chem. Int. Ed. 2012, 51, 4112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4188; [Google Scholar]
- 8d. Wang Z., Ren J., Wang Z., Org. Lett. 2013, 15, 5682; [DOI] [PubMed] [Google Scholar]
- 8e. Augustin A. U., Sensse M., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2017, 56, 14293; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14481; [Google Scholar]
- 8f. Zhang C., Tian J., Ren J., Wang Z., Chem. Eur. J. 2017, 23, 1231. [DOI] [PubMed] [Google Scholar]
- 9. Lifchits O., Alberico D., Zakharian I., Charette A. B., J. Org. Chem. 2008, 73, 6838. [DOI] [PubMed] [Google Scholar]
- 10. Kaicharla T., Roy T., Thangaraj M., Gonnade R. G., Biju A. T., Angew. Chem. Int. Ed. 2016, 55, 10061; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10215. [Google Scholar]
- 11.
- 11a. Blanchard L. A., Schneider J. A., J. Org. Chem. 1986, 51, 1372; [Google Scholar]
- 11b. Lifchits O., Charette A. B., Org. Lett. 2008, 10, 2809. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Emmett M. R., Grover H. K., Kerr M. A., J. Org. Chem. 2012, 77, 6634; [DOI] [PubMed] [Google Scholar]
- 12b. Ivanov K. L., Villemson E. V., Budynina E. M., Ivanova O. A., Trushkov I. V., Melnikov M. Y., Chem. Eur. J. 2015, 21, 4975. [DOI] [PubMed] [Google Scholar]
- 13. Braun C. M., Shema A. M., Dulin C. C., Nolin K. A., Tetrahedron Lett. 2013, 54, 5889. [Google Scholar]
- 14.
- 14a. de Nanteuil F., Loup J., Waser J., Org. Lett. 2013, 15, 3738; [DOI] [PubMed] [Google Scholar]
- 14b. Wales S. M., Walker M. M., Johnson J. S., Org. Lett. 2013, 15, 2558; [DOI] [PubMed] [Google Scholar]
- 14c. Irwin L. C., Renwick C. R., Kerr M. A., J. Org. Chem. 2018, 83, 6235; [DOI] [PubMed] [Google Scholar]
- 14d. Kilic H., Dalkilic O., ChemistrySelect 2019, 4, 3737. [Google Scholar]
- 15.
- 15a. Garve L. K. B., Barkawitz P., Jones P. G., Werz D. B., Org. Lett. 2014, 16, 5804; [DOI] [PubMed] [Google Scholar]
- 15b. Wallbaum J., Garve L. K. B., Jones P. G., Werz D. B., Chem. Eur. J. 2016, 22, 18756; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Garve L. K. B., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2017, 56, 9226; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9354; [Google Scholar]
- 15d. Das S., Daniliuc C. G., Studer A., Angew. Chem. Int. Ed. 2018, 57, 4053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4117; [Google Scholar]
- 15e.K. Singh, T. S. Bera, V. Jaiswal, S. Biswas, B. Mondal, D. Das, J. Saha, J. Org. Chem 2018; [DOI] [PubMed]
- 15f. Gregson C. H. U., Ganesh V., Aggarwal V. K., Org. Lett. 2019, 21, 3412. [DOI] [PubMed] [Google Scholar]
- 16. Sparr C., Gilmour R., Angew. Chem. Int. Ed. 2011, 50, 8391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8541. [Google Scholar]
- 17. Wallbaum J., Garve L. K. B., Jones P. G., Werz D. B., Org. Lett. 2017, 19, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das S., Daniliuc C. G., Studer A., Angew. Chem. Int. Ed. 2017, 56, 11554; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11712. [Google Scholar]
- 19.The CIF files have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1915047 (4 a) and 1915048 (7 d). These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- 20. Pohlhaus P. D., Sanders S. D., Parsons A. T., Li W., Johnson J. S., J. Am. Chem. Soc. 2008, 130, 8642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary