

Abstract
Objective
Results from anti‐CD20 therapies demonstrate that B‐ and T‐cell interaction is a major driver of multiple sclerosis (MS). The local presence of B‐cell follicle‐like structures and oligoclonal bands in MS patients indicates that certain B cells infiltrate the central nervous system (CNS) to mediate pathology. Which peripheral triggers underlie the development of CNS‐infiltrating B cells is not fully understood.
Methods
Ex vivo flow cytometry was used to assess chemokine receptor profiles of B cells in blood, cerebrospinal fluid, meningeal, and brain tissues of MS patients (n = 10). Similar analyses were performed for distinct memory subsets in the blood of untreated and natalizumab‐treated MS patients (n = 38). To assess T‐bet(CXCR3)+ B‐cell differentiation, we cultured B cells from MS patients (n = 21) and healthy individuals (n = 34) under T helper 1‐ and TLR9‐inducing conditions. Their CNS transmigration capacity was confirmed using brain endothelial monolayers.
Results
CXC chemokine receptor 3 (CXCR3)‐expressing B cells were enriched in different CNS compartments of MS patients. Treatment with the clinically effective drug natalizumab prevented the recruitment of CXCR3high IgG1+ subsets, corresponding to their increased ability to cross CNS barriers in vitro. Blocking of interferon‐γ (IFNγ) reduced the transmigration potential and antigen‐presenting function of these cells. IFNγ‐induced B cells from MS patients showed increased T‐bet expression and plasmablast development. Additional TLR9 triggering further upregulated T‐bet and CXCR3, and was essential for IgG1 switching.
Interpretation
This study demonstrates that T‐bethigh IgG1+ B cells are triggered by IFNγ and TLR9 signals, likely contributing to enhanced CXCR3‐mediated recruitment and local reactivity in the CNS of MS patients. ANN NEUROL 2019;86:264–278
B cells are one of the main contributors to chronic autoimmune pathology in multiple sclerosis (MS), as supported by results from large genome‐wide association studies.1 B‐cell repertoires in the central nervous system (CNS) and the periphery are closely connected, suggesting that disease‐relevant B‐cell networks interact at both sides of the blood–brain barrier.2, 3, 4, 5 There is evidence that the beneficial effects of anti‐CD20 monoclonal antibody therapy are related to the ablation of functional B cells interacting with T cells.6, 7 The meninges of MS patients contain tertiary lymphoid structures that are filled with B and T cells, close to cortical lesions.8 This strongly suggests that B‐ and T‐cell interaction is a major event in triggering and sustaining inflammation in the CNS.
In MS, autoreactive (naive) B cells escape peripheral selection and probably receive specific signals from CD4+ T cells within secondary lymphoid organs to differentiate into memory populations before entering the CNS.5, 9, 10 The presence of oligoclonal bands (OCBs) in the cerebrospinal fluid (CSF) of MS patients implies that these memory B cells undergo local reactivation (with the help of CD4+ T cells) to further develop into immunoglobulin (Ig)‐producing plasmablasts and plasma cells.8, 11 Although memory B cells have been recently shown to promote the differentiation of CNS‐infiltrating CD4+ T cells in MS, little is known about how and which functional B‐cell subsets are triggered in the periphery to infiltrate the CNS and contribute to MS pathology.
In mice, the T helper 1 (Th1) cytokine interferon‐γ (IFNγ) induces the interaction between autoreactive B cells and CD4+ T cells to form tertiary lymphoid structures and promote systemic autoimmune diseases such as systemic lupus erythematosus (SLE).12 In these cases, IFNγ induces the expression of the T‐box transcription factor T‐bet, resulting in enhanced Ig class switching and CXC chemokine receptor 3 (CXCR3) expression in murine B cells.13, 14 Interestingly, B‐cell–intrinsic T‐bet expression associates with increased pathogenic responses14, 15 and is induced by systemic infections,16 a major environmental trigger in MS.17 Toll‐like receptor 9 (TLR9), which binds to pathogen‐related CpG‐DNA, integrates with the B‐cell receptor (BCR), CD40, and cytokine signals to stimulate T‐bet+ B‐cell development.18, 19 Additionally, B cells from MS patients were previously reported to exhibit an enhanced proinflammatory phenotype when activated with IFNγ and TLR9 ligand CpG‐DNA.7
Here, we aimed to explore the CNS transmigration capacity of T‐bet(CXCR3)‐expressing B cells and which peripheral triggers are involved in the development of such populations in MS patients. We explored the recruitment of human CXCR3+ B cells to the CNS both ex vivo and in vitro. Furthermore, the susceptibility of blood‐derived B cells from MS patients and healthy individuals to T‐bet–inducing stimuli and how this influences their differentiation into CXCR3+ memory subsets was determined using different T‐cell–based culture systems.
Subjects and Methods
Patients
Postmortem CSF, meninges, brain tissues, and blood samples were freshly obtained from MS brain donors (Netherlands Brain Bank, Amsterdam, the Netherlands). The main cause of death was legally granted euthanasia (8 of 10 donors). The 2 other donors died from pneumonia or MS. These tissues had a very short postmortem delay of 8.92 hours (interquartile range [IQR] = 8.50 – 9.50 hours), and pH of the CSF was 6.59 (IQR = 6.44–6.87). All other MS patients and healthy controls were included at Erasmus Medical Center (Rotterdam, the Netherlands), which is a national tertiary referral center for MS patients (Multiple Sclerosis Center ErasMS). Patients and controls were age‐ and gender‐matched per study group. Patient characteristics are summarized in Table 1. Primary material was obtained between 2007 and 2018. All patients gave written informed consent, and study protocols were approved by the medical ethics committee of the Erasmus Medical Center (Rotterdam) and VU University Medical Center (Amsterdam, the Netherlands).
Table 1.
Characteristics of Patients and Controls Used in This Study
| Cohorts | Subject, n | Gender, Female, n (%) | Age, Median yr (IQR)a | Disease Duration, Median mo (IQR)b |
|---|---|---|---|---|
| Ex vivo B cells, CNS vs blood | ||||
| MS | 10 | 9 (90%) | 52 (50–65) | NA |
| Ex vivo B cells, blood subsets | ||||
| HC | 10 | 7 (70%) | 47 (32–54) | NA |
| MS, no Tx | 10 | 7 (70%) | 45 (43–53) | 48 (24–120) |
| MS, NAT Tx | ||||
| First cohort | 10 | 7 (70%) | 40 (29–46)c | 90 (31–124) |
| Second cohort | 9 | 6 (66%) | 36 (26–43)c | 46 (41–130) |
| Third cohortd | 9 | 5 (56%) | 28 (21–43)c | 28 (19–41) |
| In vitro–stimulated B cells | ||||
| HC | ||||
| Total | 10 | 8 (80%) | 44 (32–56) | NA |
| Naive | 8 | 5 (63%) | 39 (27–50) | NA |
| MS, no Tx | ||||
| Total | 9 | 7 (80%) | 41 (34–56) | 36 (36–73) |
| Naive | 12 | 8 (67%) | 38 (28–42) | 4 (3–15) |
At time of sampling.
Time from MS diagnosis to sampling.
At time of pretreatment sampling.
IgG subclass instead of total IgG analysis.
CNS = central nervous system; HC = healthy controls; IQR = interquartile range; MS = multiple sclerosis; NA = not applicable; NAT = natalizumab; Tx = treatment.
Mononuclear Cell Isolation from Blood and CNS Compartments
Peripheral blood mononuclear cells (PBMCs) were isolated according to the manufacturer's instructions from blood of MS patients and matched controls with the use of vacutainer CPT® tubes containing sodium heparin (BD Biosciences, Erembodegem, Belgium). PBMCs were frozen and stored in liquid nitrogen until use as previously described.20 Mononuclear cells were isolated from buffy coats using Ficoll‐Paque Plus (GE Healthcare, Freiburg, Germany) and density gradient centrifugation. Blood and CSF samples from MS brain donors were acquired postmortem through heart puncture and ventricle drainage, respectively.20 Heart blood mononuclear cells were isolated as described for buffy coat material. Collection tubes with CSF were centrifuged for 10 minutes at 500 × g. CSF and blood mononuclear cell fractions were resuspended in RPMI 1640 (Lonza, Verviers, Belgium) containing 10% heat‐inactivated human AB serum (Sanquin, Rotterdam, the Netherlands) and 1% penicillin/streptomycin (Lonza) and left to rest at 37°C until further use. Meninges were washed in phosphate‐buffered saline (PBS) containing 0.1% bovine serum albumin (BSA) 3 times, cut into pieces, and incubated with Liberase (Roche Applied Science, Penzberg, Germany) for 1 hour at 37°C, after which the meninges were filtered through a cell strainer (45μm) and cells were washed using Ficoll‐Paque Plus (GE Healthcare). Single‐cell suspensions from the meninges were resuspended in PBS containing 0.1% BSA until further use. Brain tissue samples were processed and single‐cell suspensions were obtained as previously reported.21
Antibodies and Flow Cytometry
Multicolor flow cytometric analysis was performed using fluorochrome‐labeled monoclonal anti‐human antibodies (mAbs; Table 2). PBMCs were stained extracellularly for 30 minutes at 4°C. Cultured B cells were stained with a fixable viability stain (FVS 700) for 15 minutes at 4°C and subsequently stained for either extracellular only or both extracellular and intracellular markers. For intracellular staining, cells were fixed with 2% paraformaldehyde (Merck, Schiphol‐Rijk, the Netherlands) and permeabilized with PBS pH7.4 containing 0.3% BSA and 0.5% saponin (Sigma‐Aldrich, St Louis, MO) and stained with T‐bet for 60 minutes at 4°C. All measurements were conducted with an LSRII‐Fortessa flow cytometer and analyzed using FACS Diva software, version 8.0.1 (both BD Biosciences). Ex vivo Th17.1 (IFNγhighIL‐17low) and Th17 (IFNγneg) cells in blood were defined as CCR6+CXCR3+CCR4− (Th17.1) and CCR6+CXCR3−CCR4+ (Th17), described recently.20
Table 2.
Monoclonal Antihuman Antibodies Used for Flow‐Activated Cell Sorting
| Antibody Marker | Fluorochrome | Clone | Company |
|---|---|---|---|
| CD3 | AF700 | SK7 | BioLegenda |
| CD3 | BV785 | SK7 | BD Biosciencesb |
| CD4 | BV510 | OKT4 | BioLegend |
| CD19 | BV785 | HIB19 | BD Biosciences |
| CD20 | AF700 | 2H7 | BD Biosciences |
| CD21 | BV711 | B‐Ly4 | BD Biosciences |
| CD27 | BV421 | M‐T271 | BD Biosciences |
| CD38 | PE‐Cy7 and PerCP‐Cy5.5 | HIT2 | BioLegend |
| CD49d (VLA‐4) | APC | 9F10 | BD Biosciences |
| CCR4 | PE‐Cy7 | L291H4 | BioLegend |
| CCR6 | PE | G024E3 | BioLegend |
| CXCR3 | BV605 and APC | G025H7 | BioLegend |
| CXCR5 | APC‐R700 | RF8B2 | BD Biosciences |
| IgA | FITC | IS11‐8E10 | Miltenyi Biotechc |
| IgD | PE and PE‐CF594 | IA6‐2 | BD Biosciences |
| IgG | ACP‐H7 | G18‐145 | BD Biosciences |
| IgG1 | PE | HP6001 | Southern Biotechd |
| IgG2 | AF488 | HP6002 | Southern Biotech |
| IgM | BV510 | MHM‐88 | BioLegend |
| T‐bet | PE‐Cy7 | 4B10 | BioLegend |
| Fixable viability dye (FVS) | AF700 | BD Biosciences |
BioLegend, London, UK.
BD Biosciences, Erembodegem, Belgium.
Miltenyi Biotech, Leiden, the Netherlands.
Southern Biotech via ITK Diagnostics, Uithoorn, the Netherlands.
Human B‐Cell Migration Assays
Flow‐activated cell sorting (FACS)‐sorted CD27− and CD27+ memory CD19+ B cells from buffy coat–derived PBMCs were placed on 96 permeable transwell plates with a 0.3μm pore size (2 × 105 cells/well; Corning, Amsterdam, the Netherlands). B‐cell migration toward medium or CXC chemokine ligand (CXCL)10 (900ng/ml; R&D Systems, Abingdon, UK) was analyzed after 3 hours at 37°C. In addition, 2.5–5 × 105 memory B cells were placed on confluent monolayers of human brain endothelial cells (hCMEC/D3) on 5μm pore size transwell plates (Corning) coated with collagen, and migration was analyzed after 5 hours.22 Percentages of memory B‐cell subsets were compared before and after transmigration using flow cytometry.
Antigen‐Primed Autologous B‐ and Th‐Cell Cocultures
BCR‐mediated uptake and presentation of Salmonella typhimurium SL1344 was used as a model for antigen presentation, as previously demonstrated.23 mAb anti‐human IgG (MH16‐1; Sanquin, Amsterdam, the Netherlands) was mixed with mAb against S. typhimurium lipopolysaccharide (LPS; 1E6; Invitrogen, Paisley, UK) and rat anti‐mouse IgG1 antibody (RM161.1, Sanquin) to generate BCR‐LPS tetrameric antibody complexes. Exponentially grown bacteria were washed twice with PBS, incubated with BCR‐LPS tetrameric antibody complexes for 30 minutes at room temperature, and washed twice to remove unbound antibodies. B cells were incubated with viable anti‐IgG–coated S. typhimurium 23 at 20 bacteria per cell for 45 minutes at 37°C without antibiotics. Next, cells were washed 3 times and cultured for 60 minutes in medium containing 100μg/ml gentamicin (Invitrogen) to eliminate nonphagocytosed bacteria. B cells were cocultured in RPMI supplemented with 5% fetal calf serum, 1% (100U/ml) penicillin, 1% (100μg/ml) streptomycin (Lonza), 1% (2mM) UltraGlutamine (Lonza), 0.1% (50μM) beta‐mercaptoethanol (Sigma‐Aldrich), 0.1% (20μg/ml) apotransferrin (depleted for human IgG with protein‐G sepharose; Sigma‐Aldrich; further referred to as B‐cell medium), and 10μg/ml gentamicin together with autologous CD4+ T cells (magnetic activated cell sorted). B cells (1 × 105) and T cells (0.5 × 105) were cultured in 200μl at 37°C in the presence of 5% CO2 in 96‐well round‐bottom plates (Greiner Bio‐One, Alphen aan den Rijn, the Netherlands) for 6 days. Cultures were performed in the presence of recombinant interleukin (IL)‐21 (50ng/ml; Thermo Fisher Scientific, Landsmeer, the Netherlands) and recombinant IL‐2 (50IU/ml, Miltenyi Biotec, Bergisch Gladbach, Germany), and with or without an anti‐IFNγ blocking antibody (MD‐1, 10μg/ml; U‐CyTech Biosciences, Utrecht, the Netherlands).
IL‐21/3T3‐CD40L Assay for Human B‐Cell Differentiation
To mimic B‐cell differentiation in vitro, murine NIH3T3 fibroblasts expressing human CD40L (3T3‐CD40L)23 were irradiated at 30Gy using a RS320 X‐ray (Beckhoff, Eindhoven, the Netherlands), taken up in B‐cell medium, and seeded on flat‐bottom 96‐well plates (10 × 103 cells per well; Greiner Bio‐One) to allow adherence overnight. CD19+ (total) B cells were isolated from buffy coat–derived PBMCs using CD19 microbeads and the autoMACS Pro Separator (both Miltenyi Biotec). Total, naive (CD38−/dimCD27−IgG−IgA−) and memory (CD38−/dimCD27+IgG+) B cells were isolated from healthy and MS blood using a BD FACSAria III cell sorter. These fractions were resuspended in B‐cell medium, and 20–25 × 103 cells were cocultured with irradiated 3T3‐CD40L cells and stimulated with a combination of IL‐21 (50ng/ml; Thermo Fisher Scientific), IFNγ (50ng/ml; Peprotech/Bio‐Connect, Huissen, the Netherlands), and CpG‐ODN (2006‐G5; 10μg/ml; InvivoGen/Bio‐Connect) at 37°C and 5% CO2. After 6 and 11 days of culture, supernatants were collected and stored at −80°C until use for enzyme‐linked immunosorbent assay (ELISA). The cells were stained and assessed using flow cytometry as described above.
IgG1 ELISA
Nunc MaxiSorp plates (Sanbio, Uden, the Netherlands) were coated overnight with antihuman IgG1 monoclonal capture antibody (1μg/ml; clone MH161‐1; Sanquin, Amsterdam, the Netherlands) in PBS. After washing with PBS–0.02% Tween‐20, the supernatants from in vitro B‐cell cultures (described above) were diluted in high‐performance ELISA buffer (HPE, Sanquin) and incubated for 60 minutes. Subsequently, plates were washed and incubated for 60 minutes with anti‐human IgG conjugated with horseradish peroxidase, a monoclonal detection antibody (1μg/ml; clone MH16‐1, Sanquin). After washing, the ELISA was developed with MQ containing 0.11M sodium‐acetate (pH 5.5), 100μg/ml tetramethylbenzidine, and 0.003% (vol/vol) H2O2 (all from Merck). The reaction was stopped by addition of 2M H2SO4 (Merck). Optical densities at 450nm were measured with a BioTek (Winooski, VT) Synergy 2. Background readings at 540nm were subtracted. Results were related to a titration curve of a serum sample of a healthy donor in each plate.
Statistical Analyses
All datasets were analyzed with GraphPad Prism Software, version 7 (GraphPad Software, San Diego, CA) and compared using 2‐sided Mann–Whitney U tests, Wilcoxon matched‐pairs signed rank test, 1‐ or 2‐way analysis of variance with Tukey post hoc test, Friedman paired test with Dunn post hoc test, and Spearman correlation coefficients (as indicated in each figure legend). Experimental data are depicted as the mean ± standard error of the mean. Prior to statistical analyses, datasets were tested for normal distribution. Probability values <0.05 were considered significant.
Results
CXCR3‐Expressing B Cells Are Selectively Enriched in Distinct CNS Compartments of MS Patients
Enhanced chemotaxis is one of the key mechanisms by which B cells can enter distinct CNS compartments of MS patients.11 Production of the chemoattractants CXCL10, CXCL13, and CCL20 in the CNS has been associated with B‐cell recruitment, distribution, and reactivity in MS.24, 25, 26 We compared the presence of B cells that express the chemokine receptors that correspond to these ligands, CXCR3+ (CXCL10), CXCR5+ (CXCL13), and CCR6+ (CCL20), between paired blood, CSF, and meningeal and brain tissues from 10 MS patients (see Table 1). To realize this, single‐cell suspensions were obtained from autopsied brain compartments using a standardized protocol.21 From these fractions, viable CD45+CD3−CD19+ B cells were gated and analyzed for chemokine receptor expression using flow cytometry (Fig 1A). We were able to measure sufficient numbers of viable B cells from each compartment for all donors (mean [range]: blood, 21,509 [610–98,562]; CSF, 12,629 [50–59,499]; meninges, 13,819 [91–36,644]; brain tissue, 2,889 [26–18,050]). The frequency of CXCR3+ but not CXCR5+ or CCR6+ B cells was strongly increased in ex vivo cell suspensions from MS brain tissues (p < 0.0001), meninges (p = 0.0003), and CSF (p < 0.0001) compared to blood (See Fig 1B). CXCR3‐expressing T cells, including Th17.1, were also enriched in the CNS compartments of these donors (data not shown), supporting our recent observations.20
Figure 1.
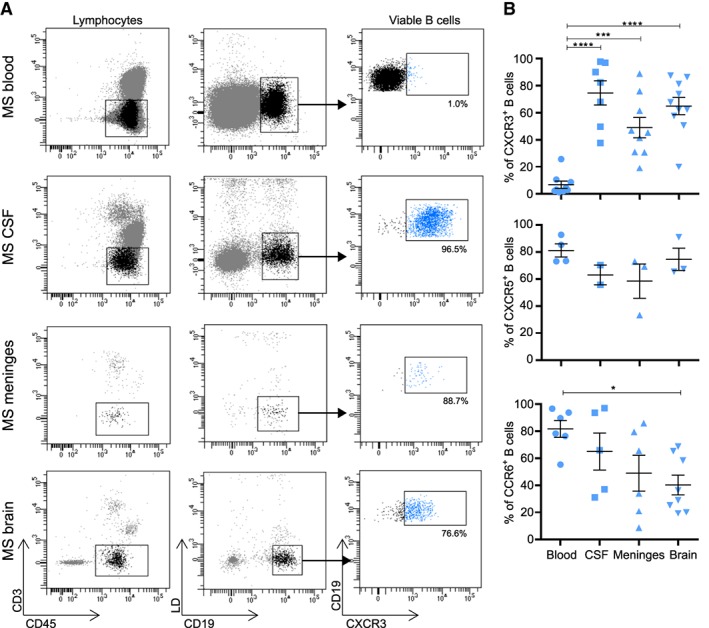
CXCR3+ B cells are abundant in the central nervous system compared to blood of multiple sclerosis (MS) patients. (A) Representative flow‐activated cell sorting (FACS) plots and gating of CXCR3‐expressing CD19+ B cells within viable CD45+CD3− lymphocyte fractions derived from the blood, cerebrospinal fluid (CSF), meninges, and brain tissue of an MS patient. (B) Frequencies of CXCR3+, CXCR5+, and CCR6+ B cells in distinct paired compartments from MS patients. For blood, CSF, and meningeal samples each dot represents a different patient. A total of 10 brain tissues from 7 different MS patients were used for the analysis of CXCR3+ B cells. Any samples with <25 viable B cells were excluded from these analyses. Data are presented as the mean ± standard error of the mean. *p < 0.05, ***p < 0.001, ****p < 0.0001. The p values for B were calculated by a 1‐way analysis of variance test. LD = live/dead (for detection of viable cells).
Reduced Frequencies of CXCR3+IgG(1)+ B Cells in the Blood of MS Patients Are Abrogated after Natalizumab Treatment
To determine how CXCR3 is involved in the local attraction of different B‐cell populations in MS, we assessed the proportion of CXCR3‐expressing naive (CD27−IgM+; IgMnaive) and both IgM+ memory (CD27+IgM+; IgMmem) and IgG+ memory (CD27+IgG+) B cells in the peripheral blood of untreated MS patients (n = 10) and age‐ and gender‐matched healthy controls (HCs; n = 10; see Table 1 and Fig 2A). CXCR3‐expressing IgG+ cells were reduced (p = 0.007), whereas no differences were seen for IgMnaive and IgMmem cells in MS versus HC blood (See Fig 2B).
Figure 2.
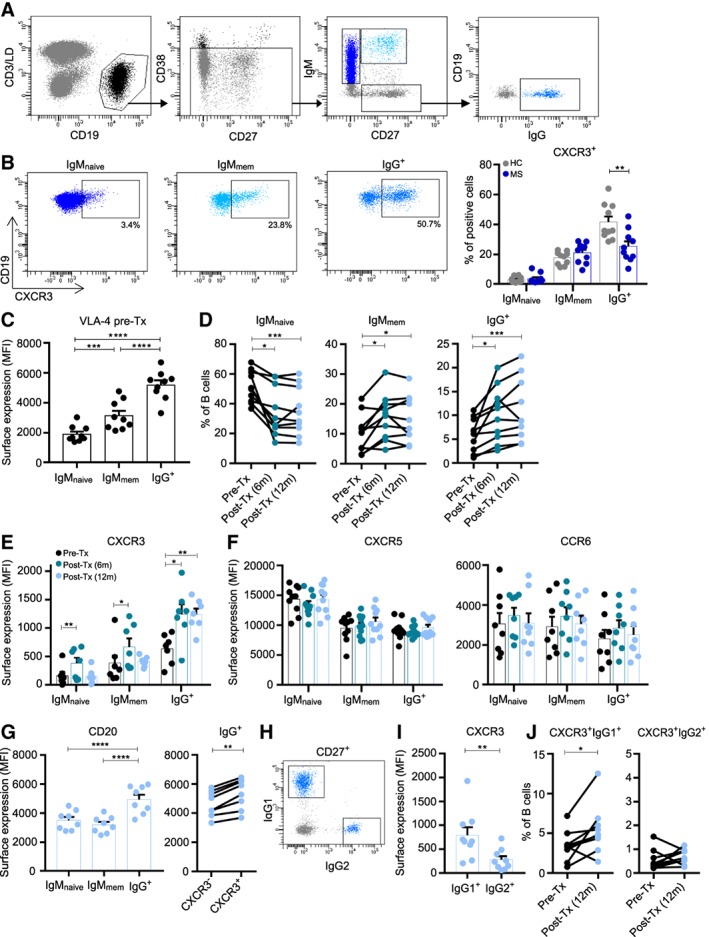
Reduced frequencies and natalizumab‐mediated accumulation of CXCR3+IgG(1)+ B cells in multiple sclerosis (MS) blood. (A) FACS gating strategy used to define IgMnaive (CD27−IgM+), IgMmem (CD27+IgM+), and IgG+ (CD27+IgG+) B‐cell subsets. (B) Gating and quantification of CXCR3‐expressing IgMnaive, IgMmem, and IgG+ B‐cell frequencies in the blood of untreated MS patients (n = 10; dark blue dots) and both age‐ and gender‐matched healthy controls (HC; n = 10; gray dots, see Table 1). (C) VLA‐4 surface expression on IgMnaive, IgMmem, and IgG+ B cells from blood of MS patients before natalizumab treatment (n = 9). (D) The percentage of IgMnaive, IgMmem, and IgG+ B cells in MS blood before (black dots) and both 6 months (marine blue dots) and 12 months (light blue dots) after natalizumab treatment (paired samples; n = 10; see Table 1). (E, F) Surface expression levels of CXCR3 (E), CXCR5 and CCR6 (F) on IgMnaive, IgMmem, and IgG+ B cells in MS patient blood before and after natalizumab treatment (n = 7–10). (G) CD20 expression on IgMnaive, IgMmem, and IgG+ B cells as well as paired CXCR3− and CXCR3+ IgG+ populations in blood of MS patients treated with natalizumab for 12 months (n = 8). (H–J) Gating example and quantifications of IgG1+ and IgG2+ B cells expressing CXCR3 in MS patients treated with natalizumab for 12 months (n = 9). Data are presented as the mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The p values were calculated by Mann–Whitney U (B), 2‐way analysis of variance (C, G), Friedman paired (D–F), and Wilcoxon matched‐pairs signed rank (G, I, and J) tests. MFI = mean fluorescence intensity; Tx = treatment; LD = live/dead (for detection of viable cells).
To address this potential migration of CXCR3+IgG+ memory B cells into the CNS, we analyzed the distribution of these B‐cell subsets in the blood of MS patients treated with the anti‐α4β1 integrin (VLA‐4) antibody natalizumab (see Table 1), a drug that effectively reduces MS disease activity by blocking lymphocyte recruitment to the CNS.27 VLA‐4 was most abundantly expressed on blood IgG+ B cells from MS patients prior to natalizumab treatment (see Fig 2). Elevated frequencies of both IgMmem (pretreatment vs 6 months post‐treatment p = 0.042 and 12 months post‐treatment p = 0.011) and IgG+ (pretreatment vs 6 months post‐treatment p = 0.022 and 12 months post‐treatment p < 0.001) B cells were found in the blood of MS patients both 6 and 12 months after versus before treatment. However, only IgG+ and not IgMnaive or IgMmem B cells from MS patients treated with natalizumab for 12 months showed increased expression levels of CXCR3 (p < 0.01), and not CXCR5 or CCR6. These findings were validated in a second cohort of 9 MS patients treated with natalizumab (see Table 1; data not shown). Notably, CD20 expression levels were increased on IgG+ B cells and higher on CXCR3+ compared to CXCR3− counterparts in the blood of natalizumab‐treated MS patients.
Because intrathecally synthesized OCBs are restricted to the IgG1 subclass in the CSF of MS patients,28 we also analyzed IgG1+ B cells for their frequencies and CXCR3 expression in the blood of a third cohort of natalizumab‐treated MS patients (see Fig 2). IgG1+ B cells not only expressed higher levels of CXCR3 (p = 0.007) but also showed increased frequencies in post‐treatment samples (p = 0.027) compared to IgG2+ B cells. The selective accumulation of CXCR3high IgG(1)+ B cells in the blood of natalizumab‐treated patients underlines the potency of this subset to transmigrate into the CNS to mediate MS disease activity.
CXCR3+IgG1+ B Cells Have an Enhanced Capacity to Transmigrate Across the Blood–Brain Barrier In Vitro
To functionally test the transmigration potential of CXCR3+IgG1+ B cells into the CNS, we sorted memory B cells from the blood and assessed in vitro migration of subsets toward CXCL10. Fractions of CXCR3‐expressing IgMmem, IgG1+, and IgG2+ B cells were assessed within the total memory pool before and after migration through transwell filters. In contrast to IgMmem and IgG2+ populations, IgG1+ B cells showed prominent recruitment to CXCL10 (p < 0.0001 before vs after migration; Fig 3). This was not seen using medium only. To mimic B‐cell transmigration across the blood–CNS barrier, these experiments were repeated using cultured confluent monolayers of human brain endothelial cells.22 We found a similar CXCL10‐mediated migratory advantage of IgG1+ B cells (p < 0.0001), which is consistent with the abundance of CXCR3 on IgG1+ compared to IgMmem and IgG2+ B cells in MS patients (see Fig 2G–I). These data demonstrate that CXCR3high IgG1+ B cells in the blood have a heightened ability to infiltrate the CNS, probably accounting for the local IgG1 subclass restriction of OCBs in MS patients.28
Figure 3.
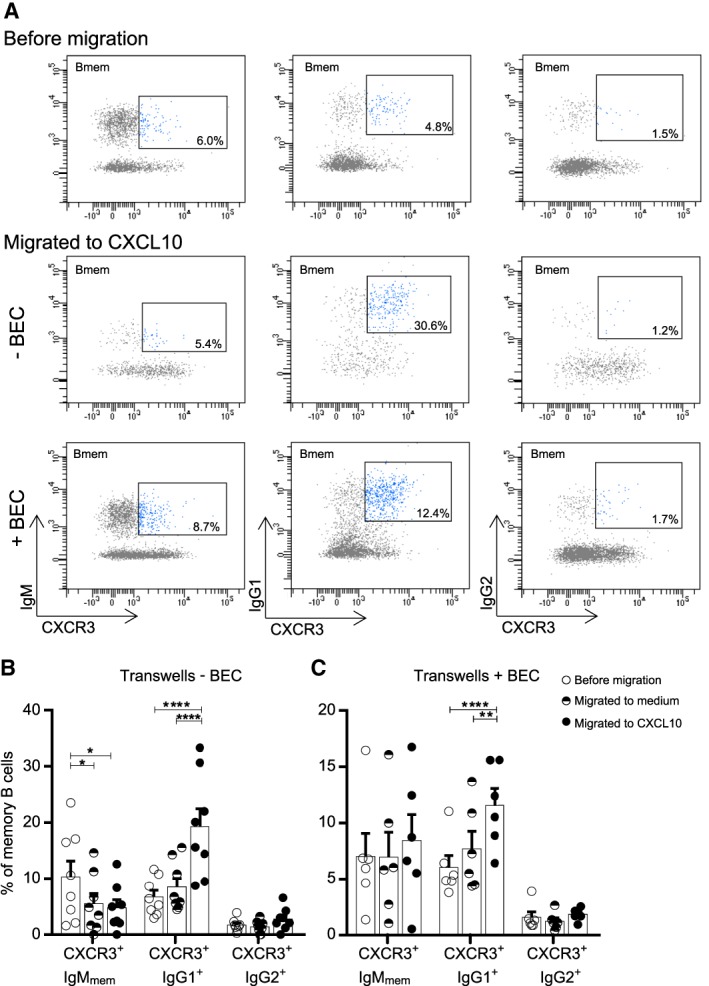
Enhanced migration of CXCR3+IgG1+ B cells across transwell filters and human brain endothelial monolayers in vitro. Sorted memory B cells from healthy donor blood were assessed for selective in vitro transmigration toward CXCL10. (A) Representative FACS plots and (B, C) quantifications of viable CXCR3‐expressing IgMmem, IgG1+ and IgG2+ B cells migrating across transwell filters with and without confluent monolayers of human brain endothelial cells (BEC). Percentages of subsets within the total memory pool were compared before and after migration, both to medium and to CXCL10 (−BEC, n = 8; +BEC, n = 6). These experiments were performed in duplicate for each donor for which the average is shown. Data are presented as the mean ± standard error of the mean. *p < 0.05, **p < 0.01, ****p < 0.0001. The p values were calculated by 2‐way analysis of variance (B, C).
IFNγ Promotes CXCR3 Expression and CD4+ T‐Cell Activation by Human T‐bet+ B Cells under T Follicular Helper–like Culture Conditions
In MS blood, the proportion of CXCR3+IgG+ B cells correlated to Th17.1 (IFNγhighIL‐17low; r = 0.566, p = 0.0003) and not to Th17 (IFNγneg) cells (Fig 4).20 Th‐cell–derived IFNγ is known as a central driver of autoreactive B cells in mice10 and also induces CXCR3 expression on human memory B cells.29 Therefore, we aimed to better understand how the differentiation and function of human CXCR3+ memory B cells is influenced by IFNγ before entering the CNS. To address this, we mimicked the effects of IFNγ‐producing T follicular helper (Tfh) cells on B‐cell subsets in vitro. First, an IL‐21–based human B‐ and T‐cell coculture system was used to assess whether Th1‐derived IFNγ influenced CXCR3 expression on IgG+ B cells in an antigen‐specific manner. Because antigen uptake by B cells is limited by the selectivity of the BCR, surface IgG was crosslinked with Th1‐associated pathogen S. typhimurium for efficient internalization, processing, and presentation to autologous Th cells.23, 30 After 6 days of coculture, we found that the induced expression of CXCR3 on Salmonella‐containing IgG+ and not IgMnaive or IgMmem B cells was abrogated by the addition of an IFNγ‐blocking antibody (p < 0.0001). Furthermore, blocking of IFNγ impaired B‐cell–induced T‐cell proliferation, activation, and effector memory formation. This implies that IFNγ additionally stimulates the antigen‐presenting function of CXCR3+IgG+ B cells, in parallel with previous findings in mice.31
Figure 4.
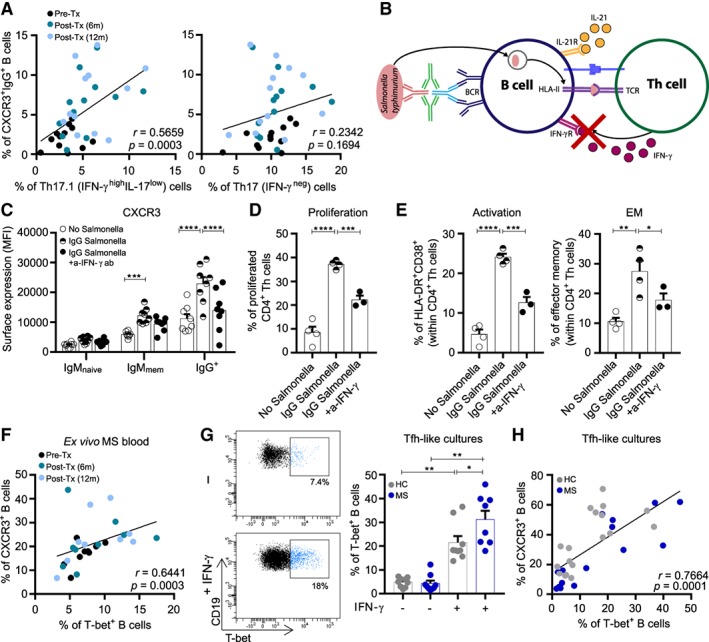
T helper (Th) 1 cytokine interferon‐γ (IFNγ) is a major trigger of CXCR3+(T‐bet+) B‐cell differentiation in multiple sclerosis (MS). (A) Correlation of ex vivo CXCR3+IgG+ B cells with Th17.1 (IFNγhighIL‐17low) and Th17 (IFNγneg) cells in MS blood before and after natalizumab treatment (pre‐Tx and post‐Tx; n = 12). (B) Experimental model of Salmonella‐primed autologous B‐ and T‐cell cocultures. (C–E) B cells from healthy donor blood were primed with S. typhimurium through B‐cell receptor (BCR) crosslinking using a tetrameric antibody complex, as described in Subjects and Methods. This allows BCR‐mediated Salmonella uptake, processing, and presentation on MHC II molecules to Th cells. IL‐21 was added with and without an IFNγ blocking antibody to analyze the effects on CXCR3 expression by B cells (C), and on the proliferation, activation, and effector memory (EM) phenotype of Th cells (D, E). These experiments were performed in 2 independent experiments and in duplicate for (C) 4 and (D, E) 2 different blood donors. (F) Correlation of surface CXCR3 and intracellular T‐bet expression in ex vivo B cells of MS patients before and after natalizumab treatment (pre‐Tx and post‐Tx; n = 9). (G, H) Total B cells from the blood of MS patients (n = 9) and both age‐ and gender‐matched healthy controls (HC; n = 10) were cultured in vitro under T follicular helper (Tfh)‐like conditions with IL‐21 and 3T3‐CD40L cells, and with or without IFNγ for 11 days. Representative FACS plots and quantification of in vitro–induced T‐bet+ B cells (G) and correlation of CXCR3 and T‐bet expression in these cultured B cells (H) are shown. Data are presented as the mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The p values were calculated by 2‐way analysis of variance (ANOVA; C), 1‐way ANOVA (D, E), and Wilcoxon matched‐pairs signed rank (G) tests. The correlation coefficients and p values for A, F, H were calculated by Spearman rank. m = months; MFI = mean fluorescence intensity; TCR = T‐cell receptor.
CXCR3 expression is under the direct control of IFNγ‐inducible transcription factor T‐bet, a critical regulator of memory B‐cell differentiation in mice.13, 14 Consistent with this, intracellular T‐bet positively correlated to surface CXCR3 expression in ex vivo B cells (r = 0.644, p = 0.0003) and showed a similar association with Th17.1 (IFNγhighIL‐17low) cells in MS patients (see Fig 4 and data not shown).20 To further explore the susceptibility of B cells to IFNγ in MS,7 we compared B cells from MS and matched healthy control blood for IFNγ‐mediated T‐bet induction under Tfh‐like culture conditions. After 11 days of stimulation with 3T3‐CD40L cells, IL‐21, and IFNγ, T‐bet was predominantly upregulated in B cells of MS patients (p = 0.021), whereas conditions without IFNγ did not show this. This in vitro–induced T‐bet was coexpressed with surface CXCR3 (r = 0.766, p = 0.0001), in line with our ex vivo results. These findings reveal that Th‐cell–derived IFNγ is a major trigger of peripheral CXCR3(T‐bet)+ B cells in MS.
IFNγ Stimulates Plasmablast Formation and Synergizes with CpG‐DNA to Establish IgG1 Switching during Human Tfh‐like B‐Cell Cultures
Besides IFNγ, TLR9 ligand CpG‐DNA has also been reported to induce T‐bet in murine B cells14, 18 and promote proinflammatory cytokine responses of B cells from MS patients.7 To assess how TLR9 signals integrate with IFNγ to regulate human T‐bet+ B‐cell development, we first determined whether naive or memory B cells are more prone to this type of coactivation. Naive (CD27−IgG−) and memory (CD27+IgG+) B cells were sorted from healthy donor blood and stimulated with 3T3‐CD40L cells, IL‐21, IFNγ, and/or CpG‐DNA. After 11 days of naive B‐cell cultures, both T‐bet and CXCR3 expression was induced by IFNγ, and further enhanced after addition of both IFNγ and CpG‐DNA (p = 0.001 and p = 0.021; Fig 5A). This additional effect of CpG‐DNA was not found when using sorted memory B cells (see Fig 5B). Both IFNγ‐induced and CpG‐DNA–induced T‐bet(CXCR3)+ B cells also showed strongly reduced CD21 expression (data not shown), a typical feature seen for T‐bet–expressing B cells.31
Figure 5.
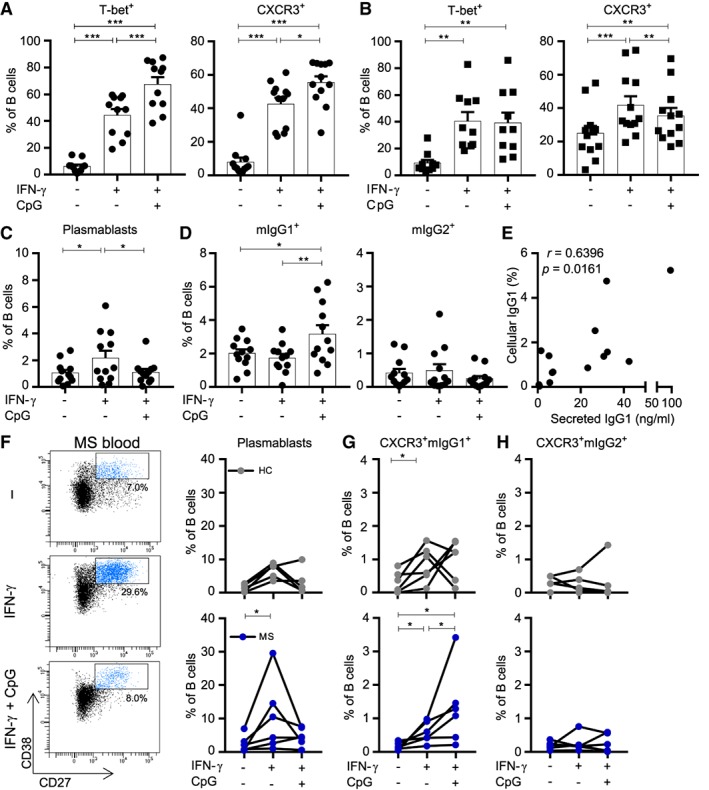
Interferon‐γ (IFNγ) induces plasmablast differentiation, whereas both IFNγ and CpG‐DNA further upregulate T‐bet and trigger IgG1 switching in B cells of multiple sclerosis (MS) patients. (A–D) Naive (IgG−CD27−; dots) and memory (IgG+CD27+; squares) B cells were sorted from peripheral blood of healthy donors and were cultured under T follicular helper (Tfh)‐like conditions with IL‐21 and 3T3‐CD40L cells, with or without IFNγ and/or CpG‐DNA. Frequencies of T‐bet+ and CXCR3+ B cells after 11 days of culture using (A) naive B cells (n = 12) and 6 days of culture using (B) memory B cells (n = 10–12) are shown. The frequencies of (C) plasmablasts (CD38highCD27+; n = 12) and (D) membrane‐bound (m) mIgG1+ and mIgG2+ B cells were analyzed after culturing naive populations for 11 days (n = 12). (E) Correlation between cellular expression and secretion of IgG1 was determined by FACS and enzyme‐linked immunosorbent assay (pooled stimulation conditions for 5 donors). (F–H) Naive B cells from the blood of MS patients (n = 6; dark blue dots) and healthy controls (HC; n = 6; gray dots) were cultured under the same Tfh‐like conditions and analyzed for (F) plasmablast (CD38highCD27+) and (G, H) CXCR3+mIgG1+ and CXCR3+mIgG2+ B‐cell differentiation after 11 days of culture. Data are presented as the mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001. The p values for A–D and F–H were calculated by the Wilcoxon matched‐pairs signed rank test. The correlation coefficient and p value for E were calculated by Spearman rank correlation.
During a germinal center response, naive B cells can either differentiate into plasmablast or memory populations, depending on the local inflammatory environment.10, 32 We investigated the effects of IFNγ and CpG‐DNA on plasmablast formation and IgG subclass switching during IL‐21–/CD40L‐induced naive B‐cell differentiation. After 11 days of culture, sorted naive B cells from healthy donors developed into plasmablasts under IFNγ stimulatory conditions only (p = 0.034; Fig 5). IFNγ and CpG‐DNA together did not induce plasmablast formation, but instead triggered IgG1 and not IgG2 expression on differentiating B cells (IFNγ only vs IFNγ + CpG‐DNA: p = 0.002). Interestingly, this in vitro‐induced IgG1 switching was subjected to differentiation of sorted naive and not memory (data not shown) B cells. CpG‐DNA alone did not upregulate CXCR3, T‐bet, and IgG1 in differentiating naive B cells (data not shown), indicating that both IFNγ and TLR9 signaling are required for enhanced expression of these markers. To verify that B‐cell intrinsic expression also corresponds with secretion of IgG1, we performed ELISAs on culture supernatants of these B cells. The percentage of IgG1+ B cells positively correlated to IgG1 secretion (r = 0.640, p = 0.016).
Finally, to address how this is regulated in MS, we isolated naive B cells from MS patients and performed similar culture experiments. IFNγ‐mediated plasmablast formation was more induced after 11 days of culture compared to matched controls (p = 0.031; see Fig 5F). This was not seen after stimulation with both IFNγ and CpG‐DNA. Instead, this type of triggering resulted in a robust induction of CXCR3+IgG1+, and not CXCR3+IgG2+ subsets in MS (see Fig 5G, H). Collectively, these data demonstrate that TLR9 signaling potentiates IFNγ‐induced T‐bet and CXCR3 expression during naive B‐cell differentiation in vitro, and that this is important for IFNγ‐mediated formation of IgG1+ memory B cells rather than plasmablasts under Tfh‐like circumstances in MS.
Discussion
Evidence has accumulated that at least in the periphery, antibody‐independent roles of B cells are driving the pathology of MS.7 However, local production of autoantibodies should not be ruled out as an underlying B‐cell mechanism in this disease.33 Although autoreactive naive B cells are highly active in MS blood,9 the vast majority of B cells identified in the MS brain have undergone further maturation into antibody‐producing cells.34, 35 It has also been demonstrated that memory B cells of MS patients are the most potent antigen‐presenting cells and likely have specific proinflammatory propensities, including the capacity to express enhanced levels of immune‐activating molecules.35 This is of special interest considering the presence of meningeal B‐cell–rich follicle‐like structures in MS and adjacent subpial cortical demyelinating injury,8 which probably contributes to progressive loss of neurological function in patients with MS. Thus, identification of the particular B‐cell subsets that can preferentially migrate into the CNS and clarification of how they may contribute to propagating local injury responses are of considerable interest in such an organ‐specific disease. In this study, we demonstrate that integrating IFNγ and pathogen‐associated TLR9 signals is critical for the development of human T‐bet+ memory B cells, probably underlying their selective recruitment to the CNS of MS patients.
Recent studies have shown that in MS identical B‐cell clones are present in both the periphery and CNS.2, 3, 4 That these B‐cell populations further undergo somatic hypermutation in the brain implies the presence of functional germinal centers within the CNS. Such structures have been identified in the meninges of MS patients and are obvious localizations that play a role here.8 The enrichment that we observe of CXCR3+ B cells in paired CSF, meninges, and brain tissue compartments compared to blood of MS patients is in line with studies that show higher levels of CXCR3 ligand CXCL10 in the CSF of MS patients.24 These results are also consistent with our previous findings that CXCR3+ T cells, including Th17.1, are abundant within the CNS,20, 24 suggesting a common CXCR3‐driven lymphocyte recruitment pathway in MS.24, 36, 37 Other studies have also put forward CXCR5 and its ligand CXCL13 as important contributors to B‐cell recruitment to the CNS.3, 38 We did not find differences in CXCR5‐expressing B cells between CNS tissues and paired blood. Hence, the CXCR5/CXCL13 axis is probably related to local organization rather than recruitment of pathogenic B (and T) cells,39, 40 in a process similar to that in secondary lymphoid organs.41 Although these studies indicate a role for germinal center B cells within the CNS, little is known about which peripheral mechanisms underlie their development and local recruitment. In mice, it has been shown that in an autoimmune setting, IFNγ, likely produced by activated Tfh cells, induces germinal centers,10 which can be found in meningeal follicle‐like structures.8 In these situations, IFNγ induced B‐cell–intrinsic expression of T‐bet, possibly resulting in enhanced Ig class switching and CXCR3 expression.13, 14 This points to a central role of IFNγ‐associated CXCR3+ B‐cell subsets in the meningeal process.10, 12
While the inducing effects of peripheral B cells on autoreactive Th1 cells are currently being elucidated,37 far less is known about the impact of Th1 cells on peripheral B‐cell differentiation and function in MS patients. Therefore, we were interested in the signals needed for B cells to differentiate into T‐bet+ cells and postulated that IFNγ‐ and IL‐21–producing Tfh1 cells in germinal centers can trigger development of such B cells. In MS patients, B cells were found to express higher T‐bet levels under Tfh1‐like culture conditions. Furthermore, IL‐21–based B‐ and T‐cell cocultures revealed that CXCR3‐expressing IgG+ memory B cells were less induced after blocking of IFNγ, which corresponds to studies that show IFNγ regulates CXCR3 expression in human B cells.29 Th‐cell proliferation, activation, and effector memory formation were also affected in these cultures. In line with our findings, a recent study demonstrated that memory B cells induce proliferation of CNS‐infiltrating Th1 cells in MS, which was inhibited after IFNγ abrogation.37 Therefore, in MS patients, peripheral interaction of CXCR3(T‐bet)+ B and IFNγ‐producing Th cells probably generates a feedforward loop, in which IFNγ enhances the potency of B cells as antigen‐presenting cells, resulting in the activation of (IFNγ‐producing) pathogenic Th cells.
Furthermore, we found that naive B cells from MS patients developed into plasmablasts rather than IgG1‐switched memory B cells under IFNγ‐only conditions. Because T‐bet mediates class‐switching in murine B cells,13 we expected that an additional signal would be required for triggering such a mechanism in human B cells. Besides Tfh1 cells, innate TLR signaling is also critical for naive B‐cell differentiation.42 Especially pathogen‐associated TLR9 and its ligand CpG‐DNA have been shown to promote the development of T‐bet+ B cells in mice.19, 31, 42 Correspondingly, we found that the induction of MS‐blood–derived naive B cells with both IFNγ and CpG‐DNA resulted in the development of IgG1‐switched, T‐bethigh B cells during Tfh‐like cultures. Likewise, CXCR3 surface expression was more enhanced under these conditions, reflecting the high CXCR3 levels on ex vivo IgG1+ B cells. This additional effect of TLR9 signaling on human T‐bet+ B cells in MS links to the role of TLR9 in driving neuroinflammatory responses, including increased production of chemokines in the CNS.43 Moreover, CXCR3+IgG1+ B cells showed an enhanced transmigration potential over brain endothelial layers, and selectively accumulated in MS blood after natalizumab therapy. The importance of pathogenic immune cells in contributing to MS disease progression, such as CXCR3+ memory B cells, has been put forward by the recurrence of often‐fatal clinical relapses in MS patients when discontinuing the use of natalizumab.27, 44, 45 During these rebounds, Epstein–Barr virus–infected memory B cells that have accumulated in the blood show massive influx into brain tissues of MS patients.45 Furthermore, persistent viral infections are suggested to sustain the development of T‐bet–expressing B cells,15 which further supports the enhanced differentiation and local recruitment of CXCR3(T‐bet)high memory B cells in an organ‐specific autoimmune disease such as MS (Fig 6).
Figure 6.
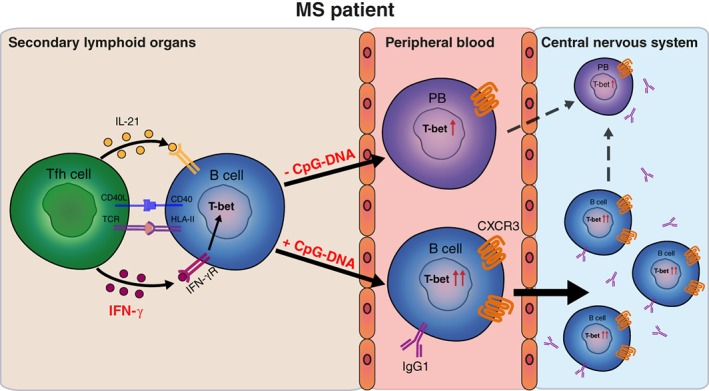
Interferon‐γ (IFNγ) and Toll‐like receptor 9 (TLR9) signaling upregulate T‐bet in peripheral B cells, likely driving CXCR3‐mediated recruitment and IgG1 production in the central nervous system of multiple sclerosis (MS) patients. Our findings suggest that in the secondary lymphoid organs of MS patients, IFNγ triggers naive B cells to differentiate into T‐bet–expressing populations in a T follicular helper (Tfh)‐dependent manner. Human T‐bet+ B cells can either develop into plasmablasts or undergo further differentiation into IgG1+ memory B cells mediated by TLR9 ligation. The enhanced CXCR3 expression on both IFNγ‐ and TLR9‐induced IgG1+ B cells makes these subsets highly capable of transmigrating across the blood–brain barrier and mediate local pathology in MS. TCR = T‐cell receptor.
Although the exact role of (local) autoantibody production in MS is not clear, the question of whether and how T‐bet+ B cells are involved in this process deserves further attention. In SLE, T‐bet+ B cells have autoreactive BCRs and are prone to differentiate into IgG autoantibody–producing plasmablasts.46 Inappropriate T‐bet expression in B cells also impaired CXCR3‐mediated plasmablast differentiation within germinal centers14 and autoantibody production.13 In our study, IFNγ‐induced and CpG‐DNA–induced human CXCR3(T‐bet)high B cells showed increased IgG1 expression and secretion. This strongly suggests that after preferential recruitment and reactivation in the CNS, CXCR3(T‐bet)highIgG1+ B cells are responsible for local production of IgG1 in MS (see Fig 6).28 Although B cells within the CNS of MS patients show characteristics of an antigen‐driven response, the specific antigens driving this response remain unknown. MS disease heterogeneity is reflected by the identification of several candidate target antigens, including nonmyelin proteins such as neurofilament light and RAS guanyl releasing protein 2 (RASGRP2).33, 37 In addition to this, increased Epstein–Barr nuclear antigen 1 (EBNA1)‐specific IgG1 titers have been found in active MS, which may be explained by the interaction of B cells with pathogen‐associated TLR ligands and EBNA1‐specific, IFNγ‐producing T cells that cross‐recognize myelin antigens.47 Therefore, we propose that the relevant antigen specificity of B cells in MS can be found within this subset, which should be further explored in the near future.
Taken together, not only a disrupted blood–brain barrier, but also peripheral T‐bet–mediated differentiation and transmigration of IgG1+ memory populations could explain how B cells are eventually able to mediate CNS pathology in MS patients (see Fig 6). The relevance of T‐ and B‐cell interaction in tolerance breakthrough is stressed by the finding that antigen‐specific B cells are potentially 1,000 to 10,000 times better presenters of autologous peptides to T cells than nonspecific B cells.48 We here reveal that human CXCR3(T‐bet)+ B cells are a product of T‐ and B‐cell interaction. Similar to SLE, such populations probably serve as potent antigen‐presenting cells in CNS‐specific autoimmune diseases such as MS.31 Anti‐CD20 therapy exerts immediate effects and is assumed to predominantly affect this function of B cells in MS patients.35 The potential role of CXCR3(T‐bet)+ B cells as prime targets of this therapy is further supported by their abundant CD20 expression, as shown in the current study. The development of new targeted strategies to inhibit T‐bet function have the potential to become a double‐edged sword in MS by suppressing pathogenic, IFNγ‐producing T (Th17.1) cells together with their counterpart CXCR3(T‐bet)+ B cells. Small molecule inhibitors of IFNγ signaling (jakinibs)49 and the TLR/myD88 pathway50 are already used in clinics for other inflammatory diseases, and are promising candidates for combined suppression of IFNγ and TLR signals to control pathogenic T‐bet+ B cells in autoimmune diseases such as MS.
Author Contributions
J.v.L., L.R., M.M.v.L., R.Q.H., P.P.A.U., S.M.v.H., and H.E.d.V. contributed to the study concept and design. J.v.L., L.R., A.F.W.‐W., M.‐J.M., M.J., P.‐P.A.U., T.A.S., and M.M.v.L. acquired and analyzed data. J.v.L., M.M.v.L., L.R., and R.Q.H. drafted the manuscript and figures.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This work was supported by the Dutch MS Research Foundation (14‐875 MS) and Erasmus Medical Center (Mrace grant; both to M.M.v.L.). This research was performed within the framework of the Erasmus Postgraduate School Molecular Medicine.
We thank P. van Geel and H. de Wit for FACS sorting.
References
- 1. Farh KKH, Marson A, Zhu J, et al. Genetic and epigenetic fine‐mapping of causal autoimmune disease variants. Nature 2015;518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. von Büdingen HC, Kuo TC, Sirota M, et al. B cell exchange across the blood‐brain barrier in multiple sclerosis. J Clin Invest 2012;122:4533–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eggers EL, Michel BA, Wu H, et al. Clonal relationships of CSF B cells in treatment‐naive multiple sclerosis patients. JCI Insight 2017;2(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenfield AL, Dandekar R, Ramesh A, et al. Longitudinally persistent cerebrospinal fluid B cells can resist treatment in multiple sclerosis. JCI Insight 2019;4(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stern JNH, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 2014;6:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hauser SL, Waubant E, Arnold DL, et al. B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 7. Bar‐Or A, Fawaz L, Fan B, et al. Abnormal B‐cell cytokine responses: a trigger of T‐cell–mediated disease in MS? Ann Neurol 2010;67:452–461. [DOI] [PubMed] [Google Scholar]
- 8. Magliozzi R, Howell O, Vora A, et al. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089–1104. [DOI] [PubMed] [Google Scholar]
- 9. Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest 2013;123:2737–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rawlings DJ, Metzler G, Wray‐Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol 2017;17:421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michel L, Touil H, Pikor NB, et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS‐compartmentalized inflammation. Front Immunol 2015;6:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson SW, Jacobs HM, Arkatkar T, et al. B cell IFN‐gamma receptor signaling promotes autoimmune germinal centers via cell‐intrinsic induction of BCL‐6. J Exp Med 2016;213:733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peng SL, Szabo SJ, Glimcher LH. T‐bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A 2002;99:5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piovesan D, Tempany J, Di Pietro A, et al. c‐Myb regulates the T‐bet‐dependent differentiation program in B cells to coordinate antibody responses. Cell Rep 2017;19:461–470. [DOI] [PubMed] [Google Scholar]
- 15. Barnett BE, Staupe RP, Odorizzi PM, et al. Cutting edge: B cell‐intrinsic T‐bet expression is required to control chronic viral infection. J Immunol 2016;197:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knox JJ, Buggert M, Kardava L, et al. T‐bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2017;2(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buljevac D, Flach HZ, Hop WC, et al. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain 2002;125(pt 5):952–960. [DOI] [PubMed] [Google Scholar]
- 18. Sindhava VJ, Oropallo MA, Moody K, et al. A TLR9‐dependent checkpoint governs B cell responses to DNA‐containing antigens. J Clin Invest 2017;127:1651–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jegerlehner A, Maurer P, Bessa J, et al. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol 2007;178:2415–2420. [DOI] [PubMed] [Google Scholar]
- 20. van Langelaar J, van der Vuurst de Vries RM, Janssen M, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain 2018;141:1334–1349. [DOI] [PubMed] [Google Scholar]
- 21. van Nierop GP, van Luijn MM, Michels SS, et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol 2017;134:383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weksler BB, Subileau EA, Perriere N, et al. Blood‐brain barrier‐specific properties of a human adult brain endothelial cell line. FASEB J 2005;19:1872–1874. [DOI] [PubMed] [Google Scholar]
- 23. Souwer Y, Griekspoor A, Jorritsma T, et al. B cell receptor‐mediated internalization of Salmonella: a novel pathway for autonomous B cell activation and antibody production. J Immunol 2009;182:7473–7481. [DOI] [PubMed] [Google Scholar]
- 24. Sørensen TL, Trebst C, Kivisäkk P, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co‐localization in the inflamed central nervous system. J Neuroimmunol 2002;127:59–68. [DOI] [PubMed] [Google Scholar]
- 25. Kalinowska‐Łyszczarz A, Szczuciński A, Pawlak MA, Losy J. Clinical study on CXCL13, CCL17, CCL20 and IL‐17 as immune cell migration navigators in relapsing−remitting multiple sclerosis patients. J Neurol Sci 2011;300:81–85. [DOI] [PubMed] [Google Scholar]
- 26. Elgueta R, Marks E, Nowak E, et al. CCR6‐dependent positioning of memory B cells is essential for their ability to mount a recall response to antigen. J Immunol 2015;194:505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saraste M, Penttila TL, Airas L. Natalizumab treatment leads to an increase in circulating CXCR3‐expressing B cells. Neurol Neuroimmunol Neuroinflamm 2016;3:e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eickhoff K, Kaschka W, Skvaril F, et al. Determination of IgG subgroups in cerebrospinal fluid of multiple sclerosis patients and others. Acta Neurol Scand 1979;60:277–282. [DOI] [PubMed] [Google Scholar]
- 29. Muehlinghaus G, Cigliano L, Huehn S, et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood 2005;105:3965–3971. [DOI] [PubMed] [Google Scholar]
- 30. O'Donnell H, McSorley S. Salmonella as a model for non‐cognate Th1 cell stimulation. Front Immunol 2014;5:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rubtsova K, Rubtsov AV, Thurman JM, et al. B cells expressing the transcription factor T‐bet drive lupus‐like autoimmunity. J Clin Invest 2017;127:1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Garcia‐Ibanez L, Toellner K‐M. Regulation of germinal center B‐cell differentiation. Immunol Rev 2016;270:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willis SN, Stathopoulos P, Chastre A, et al. Investigating the antigen specificity of multiple sclerosis central nervous system‐derived immunoglobulins. Front Immunol 2015;6:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class‐switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med 2014;6:248ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker D, Pryce G, Amor S, et al. Learning from other autoimmunities to understand targeting of B cells to control multiple sclerosis. Brain 2018;141:2834–2847. [DOI] [PubMed] [Google Scholar]
- 36. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP‐1α and IP‐10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A 1999;96:6873–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jelcic I, Al Nimer F, Wang J, et al. Memory B cells activate brain‐homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 2018;175:85–100.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up‐regulation is differentially linked to CNS immune cell recruitment. Brain 2006;129:200–211. [DOI] [PubMed] [Google Scholar]
- 39. Magliozzi R, Columba‐Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle‐like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 2004;148:11–23. [DOI] [PubMed] [Google Scholar]
- 40. Rainey‐Barger EK, Rumble JM, Lalor SJ, et al. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav Immun 2011;25:922–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Allen CDC, Ansel KM, Low C, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol 2004;5:943. [DOI] [PubMed] [Google Scholar]
- 42. Ruprecht CR, Lanzavecchia A. Toll‐like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol 2006;36:810–816. [DOI] [PubMed] [Google Scholar]
- 43. Butchi NB, Woods T, Du M, et al. TLR7 and TLR9 trigger distinct neuroinflammatory responses in the CNS. Am J Pathol 2011;179:783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sorensen PS, Koch‐Henriksen N, Petersen T, et al. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy in highly active MS patients. J Neurol 2014;261:1170–1177. [DOI] [PubMed] [Google Scholar]
- 45. Serafini B, Scorsi E, Rosicarelli B, et al. Massive intracerebral Epstein‐Barr virus reactivation in lethal multiple sclerosis relapse after natalizumab withdrawal. J Neuroimmunol 2017;307:14–17. [DOI] [PubMed] [Google Scholar]
- 46. Wang S, Wang J, Kumar V, et al. IL‐21 drives expansion and plasma cell differentiation of autoreactive CD11chiT‐bet+ B cells in SLE. Nat Commun 2018;9:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lünemann JD, Jelcić I, Roberts S, et al. EBNA1‐specific T cells from patients with multiple sclerosis cross react with myelin antigens and co‐produce IFN‐gamma and IL‐2. J Exp Med 2008;205:1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lanzavecchia A. Antigen‐specific interaction between T and B cells. Nature 1985;314:537–539. [DOI] [PubMed] [Google Scholar]
- 49. Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017;16:843. [DOI] [PubMed] [Google Scholar]
- 50. Capolunghi F, Rosado MM, Cascioli S, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology 2010;49:2281–2289. [DOI] [PubMed] [Google Scholar]