

Abstract
Multiple myeloma (MM) is a common hematologic malignancy for which the underlying molecular mechanisms remain largely unclear. This study aimed to elucidate key candidate genes and pathways in MM by integrated bioinformatics analysis. Expression profiles GSE6477 and GSE47552 were obtained from the Gene Expression Omnibus database, and differentially expressed genes (DEGs) with p < .05 and [logFC] > 1 were identified. Functional enrichment, protein–protein interaction network construction and survival analyses were then performed. First, 51 upregulated and 78 downregulated DEGs shared between the two GSE datasets were identified. Second, functional enrichment analysis showed that these DEGs are mainly involved in the B cell receptor signaling pathway, hematopoietic cell lineage, and NF‐kappa B pathway. Moreover, interrelation analysis of immune system processes showed enrichment of the downregulated DEGs mainly in B cell differentiation, positive regulation of monocyte chemotaxis and positive regulation of T cell proliferation. Finally, the correlation between DEG expression and survival in MM was evaluated using the PrognoScan database. In conclusion, we identified key candidate genes that affect the outcomes of patients with MM, and these genes might serve as potential therapeutic targets.
Keywords: bioinformatics analysis, immune‐associated genes, multiple myeloma, prognosis
1. INTRODUCTION
Multiple myeloma (MM) is an incurable cancer of B cells caused by an aberrant accumulation of malignant plasma cells in the bone marrow (Palumbo & Anderson, 2011). MM comprises 10% of all hematological cancers and is characterized by hypercalcemia, renal failure, anemia, and bone lesions, leading to fatal outcomes(Pawlyn & Morgan, 2017). Although the development of new drugs has improved patient outcomes, the response to treatment and survival of newly diagnosed patients with MM varies, and the median survival time ranges from 2 to >10 years (Sonneveld et al., 2016). Thus, identifying molecular biomarkers is critically important for early diagnosis, prevention, and personalized therapy.
Gene chips have been widely applied as a gene detection technology, and the corresponding data have been deposited in public databases (Vogelstein et al., 2013). Integrating and reanalyzing these genomic data offer possibilities for identifying certain disease‐related biomarkers. Recently, many studies have been carried out according to microarray data profiles to elucidate the pathogenesis of MM (C. Liu, Gu, & Jiang, 2017; Sun et al., 2015). However, the results are based on a single cohort study, creating poor reproducibility and consistency. To overcome these disadvantages, integrated bioinformatics methods should be combined with expression profiling techniques.
In the current study, we downloaded two microarray datasets, GSE6477 (Chng et al., 2007) and GSE47552 (Lopez‐Corral et al., 2014), from the NCBI‐Gene Expression Omnibus database (NCBI‐GEO), including gene expression data for purified plasma cell samples from 110 patients with MM and 20 healthy controls. We identified differentially expressed genes (DEGs) using the interactive web tool GEO2R with standard data processing and analyzed these DEGs were performed Gene Ontology (GO) and pathway enrichment analysis with the Database for Annotation, Visualization and Integrated Discovery (DAVID), KEGG pathways and REACTOME.
Subsequently, we integrated the DEG protein–protein interaction (PPI) network with module screening to identify hub genes in MM. Identifying DEGs and enriching their functions and signaling pathways may help reveal potential biosignatures for the diagnosis and management of MM.
2. MATERIALS AND METHODS
2.1. Microarray data information and DEG identification
The gene expression profiles GSE6477 and GSE47552 were obtained from NCBI‐GEO. The microarray data of GSE6477 are based on GPL96 platforms (HG‐U133A Affymetrix Human Genome U133A Array) and include purified plasma cell samples from 69 newly diagnosed patients with MM and 15 healthy controls. The GSE47552 data are based on GPL6244 platforms (HuGene‐1_0‐st Affymetrix Human Gene 1.0 ST Array) and include purified plasma cell samples from 41 newly diagnosed MM patients and 5 healthy controls.
GEO2R was used to identify DEGs between patients with MM and healthy controls. In this study, genes with p < .05 and [logFC] > 1 were defined as DEGs. The DEG data were processed at the Morpheus website (available online: https://software.broadinstitute.org/morpheus) to draw a heatmap of the top 100 significantly changed genes.
2.2. GO and pathway enrichment analysis
DAVID (https://david‐d.ncifcrf.gov/) was used to analyze candidate DEG functions and KEGG pathway enrichment. GO term enrichment analysis includes biological process (BP), cellular component (CC), and molecular function (MF). Pathway analysis was also carried out using another online database, REACTOME (available online: http://www.reactome.org). Interrelation analysis between pathways was developed using the ClueGo plug‐in Cytoscape software. A p value <.05 was considered the cut‐off criterion.
2.3. PPI network establishment and modular analysis
We evaluated the DEG‐encoded proteins and PPI information using the STRING database (available online: http://string‐db.org). Cytoscape software version 3.7.1 was applied to establish the protein interaction network.
MCODE, a plug‐in in Cytoscape, was utilized to screen the modules from the PPI network and identify the most significant module based on the MCODE score and node number.
2.4. Survival analysis
Correlation between DEG expression and survival in MM was analyzed using the PrognoScan database (http://dna00.bio.kyutech.ac.jp/PrognoScan/). PrognoScan searches for relationships between gene expression and MM disease‐specific survival (DSS) were based on the GSE2658 data set (n = 559). A Cox p value <.05 was considered statistically significant.
3. RESULTS
3.1. Identification of DEGs in MM
We obtained gene expression profiles of purified plasma cell samples from newly diagnosed patients with MM and healthy controls from the GSE6477 and GSE47552 datasets and analyzed DEGs using GEO2R. Setting the cut‐off criterion as p < .05 and [logFC] > 1, we identified 1,266 and 871 DEGs from GSE6477 and GSE47552, respectively (Figure 1a). Figure 1b shows the top 100 genes depicted as a heatmap. These genes were well clustered between patients with MM and healthy controls. Employing integrated bioinformatics analysis, we identified 51 upregulated genes and 78 downregulated genes as common to the two datasets (Figure 1c).
Figure 1.
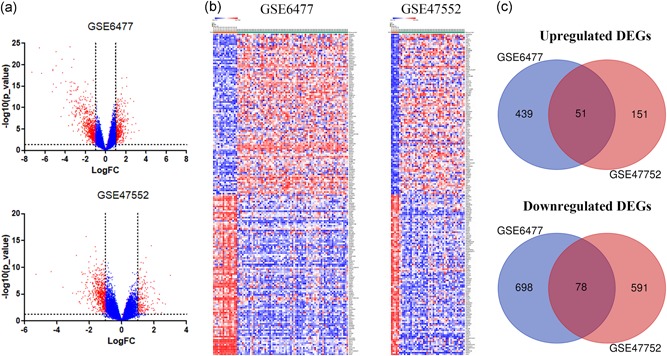
Identification of differentially expressed genes in two cohort profile datasets (GSE6477 and GSE47552). (a) Respective volcano plot of the two datasets. Red plots represent genes with [logFC] > 1 and p < .05. Blue plots represent the remaining genes with no significant difference. (b) Heatmap of the top 100 DEGs (100 up‐ and 100 downregulated genes). (c) Commonly changed DEGs in the two datasets (51 up‐ and 78 downregulated genes). DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
3.2. GO term enrichment analysis of DEGs
We performed DEG GO analysis using the DAVID gene annotation tool. As shown in Figure 2a, the DEGs were examined according to three subontologies: BP, CC, and MF. Figure 2b and Table 1 illustrate that for BP, upregulated genes were mainly enriched in translational elongation and translation; downregulated genes were largely enriched in positive regulation of response to the stimulus, positive regulation of immune system process, immune response, activation of the immune response, response to wounding and lymphocyte mediated immunity. For CC, enrichment of upregulated genes was primarily in the cytosolic ribosome, ribosomal subunit, and cytosolic part, and that of downregulated genes was mainly in the extracellular region part. Upregulated genes in MF are mainly involved in structural constituents of ribosome and structural molecule activity and the downregulated genes mainly in Ras guanyl‐nucleotide exchange factor activity.
Figure 2.
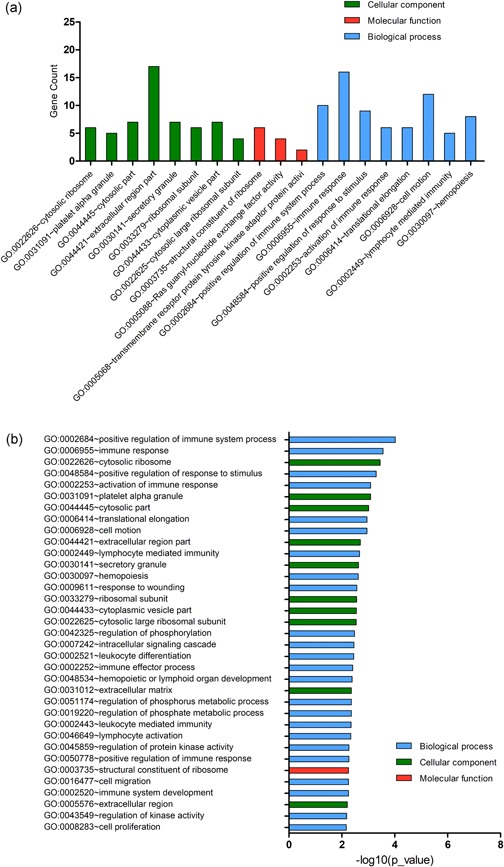
Gene Ontology (GO) term enrichment analysis of all DEGs. (a) GO analysis of DEGs consisting of three subontologies (biological process, molecular function and cellular component). (b) Significantly enriched GO terms for all DEGs. DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Gene Ontology term enrichment analysis of DEGs in multiple myeloma
| Term | Description | Count | p value |
|---|---|---|---|
| Upregulated | |||
| GO:0022626 | Cytosolic ribosome | 6 | 4.39 × 10−6 |
| GO:0006414 | Translational elongation | 6 | 1.27 × 10−5 |
| GO:0033279 | Ribosomal subunit | 6 | 4.08 × 10−5 |
| GO:0003735 | Structural constituent of ribosome | 6 | 5.19 × 10−5 |
| GO:0044445 | Cytosolic part | 6 | 9.25 × 10−5 |
| GO:0022625 | Cytosolic large ribosomal subunit | 4 | 2.06 × 10−4 |
| GO:0006412 | Translation | 7 | 4.48 × 10−4 |
| GO:0005840 | Ribosome | 6 | 4.64 × 10−4 |
| GO:0015934 | Large ribosomal subunit | 4 | 1.10 × 10−3 |
| GO:0005198 | Structural molecule activity | 7 | 4.04 × 10−3 |
| Downregulated | |||
| GO:0048584 | Positive regulation of response to stimulus | 9 | 1.29 × 10−5 |
| GO:0002684 | Positive regulation of immune system process | 9 | 1.37 × 10−5 |
| GO:0006955 | Immune response | 14 | 1.39 × 10−5 |
| GO:0002253 | Activation of immune response | 6 | 7.44 × 10−5 |
| GO:0009611 | Response to wounding | 11 | 1.54 × 10−4 |
| GO:0044421 | Extracellular region part | 14 | 2.02 × 10−4 |
| GO:0002449 | Lymphocyte mediated immunity | 5 | 3.11 × 10−4 |
| GO:0002252 | Immune effector process | 6 | 3.92 × 10−4 |
| GO:0006952 | Defense response | 11 | 5.07 × 10−4 |
| GO:0050778 | Positive regulation of immune response | 6 | 5.63 × 10−4 |
Abbreviations: DEGs, differentially expressed genes; GO, Gene Ontology.
3.3. Signaling pathway enrichment analysis
KEGG and REACTOME pathway enrichment analyses of the up‐ and downregulated DEGs were performed (Figure 3a; Table 2). Enrichment of upregulated DEGs was mostly in the formation of a pool of free 40S subunits, L13a‐mediated translational silencing of ceruloplasmin expression, and eukaryotic translation termination and SRP‐dependent cotranslational protein targeting to the membrane. Downregulated DEGs were mainly enriched in the transcriptional regulation of white adipocyte differentiation, the B cell receptor signaling pathway, platelet degranulation, chondroitin sulfate biosynthesis, and posttranslational protein phosphorylation. Subsequently, we performed an interrelation analysis of pathways by examining KEGG processes in ClueGO. All DEGs were primarily associated with the B cell receptor pathway, hematopoietic cell lineage, NF‐kappa B pathway, and the PPAR pathway (Figure 3b).
Figure 3.
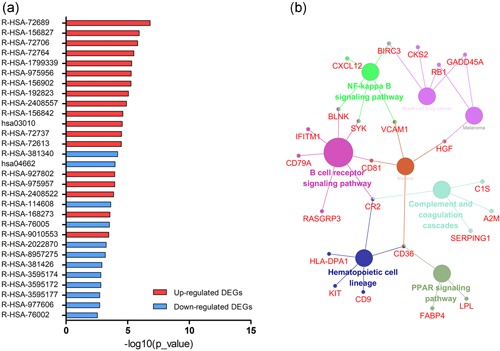
Signaling pathway enrichment analysis of DEGs. (a) KEGG and REACTOME pathway enrichment of up‐ and downregulated DEGs. (b) Interrelation analysis of pathways via assessment of KEGG processes in ClueGO. DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
Table 2.
Signaling pathway enrichment analysis of DEGs in multiple myeloma
| Pathway | Name | Gene count | p value | Genes |
|---|---|---|---|---|
| Upregulated DEG | ||||
| Reactome:R‐HSA‐72689 | Formation of a pool of free 40S subunits | 7 | 1.52 × 10−7 | RPS19;RPL32;EIF3K;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐156827 | L13a‐mediated translational silencing of Ceruloplasmin expression | 7 | 1.14 × 10−6 | RPS19;RPL32;EIF3K;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐72706 | GTP hydrolysis and joining of the 60S ribosomal subunit | 7 | 1.54 × 10−6 | RPS19;RPL32;EIF3K;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐72764 | Eukaryotic Translation Termination | 6 | 3.13 × 10−6 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐1799339 | SRP‐dependent cotranslational protein targeting to membrane | 6 | 4.64 × 10−6 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐975956 | Nonsense‐mediated decay (NMD) independent of the exon junction complex (EJC) | 6 | 5.10 × 10−6 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐156902 | Peptide chain elongation | 6 | 5.35 × 10−6 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐192823 | Viral mRNA Translation | 6 | 8.71 × 10−6 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐2408557 | Selenocysteine synthesis | 6 | 1.26 × 10−5 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| Reactome:R‐HSA‐156842 | Eukaryotic translation elongation | 6 | 2.45 × 10−5 | RPS19;RPL32;RPL24;RPLP2;RPL35;UBA52 |
| KEGG Pathway: hsa03010 | Ribosome | 6 | 2.73 × 10−5 | RPS19, RPL32, RPL35, RPLP2, RPL24, UBA52 |
| Downregulated DEG | ||||
| Reactome:R‐HSA‐381340 | Transcriptional regulation of white adipocyte differentiation | 6 | 6.50 × 10−5 | FABP4;LPL;CD36 |
| KEGG Pathway: hsa04662 | B cell receptor signaling pathway | 6 | 1.05 × 10−4 | CR2, RASGRP3, CD81, CD79A, BLNK, SYK |
| Reactome:R‐HSA‐114608 | Platelet degranulation | 6 | 2.34 × 10−4 | TF;SERPING1;CD9;CTSW;CD36;A2M |
| Reactome:R‐HSA‐76005 | Response to elevated platelet cytosolic Ca2+ | 6 | 3.05 × 10−4 | TF;SERPING1;CD9;CTSW;CD36;A2M |
| Reactome:R‐HSA‐2022870 | Chondroitin sulfate biosynthesis | 3 | 5.51 × 10−4 | CSGALNACT1;VCAN;DCN |
| Reactome:R‐HSA‐8957275 | Posttranslational protein phosphorylation | 5 | 6.44 × 10−4 | VCAN;TF;IGFBP5;CHRDL1;FBN1 |
| Reactome:R‐HSA‐381426 | Regulation of insulin‐like growth factor (IGF) transport and uptake by insulin‐like growth factor binding proteins (IGFBPs) | 5 | 1.3 × 10−3 | VCAN;TF;IGFBP5;CHRDL1;FBN1 |
| Reactome:R‐HSA‐3595174 | Defective CHST14 causes EDS, musculocontractural type | 2 | 1.51 × 10−3 | VCAN;DCN |
| Reactome:R‐HSA‐3595172 | Defective CHST3 causes SEDCJD | 2 | 1.51 × 10−3 | VCAN;DCN |
| Reactome:R‐HSA‐3595177 | Defective CHSY1 causes TPBS | 2 | 1.86 × 10−3 | VCAN;DCN |
| Reactome:R‐HSA‐977606 | Regulation of complement cascade | 5 | 1.88 × 10−3 | CR2;IGKC;C1S;CD81;SERPING1 |
Abbreviation: CR2, Component 3d receptor 2; DEGs, differentially expressed genes.
3.4. Immune analysis of downregulated DEGs
Based on the above results, we found the downregulated DEGs to be significantly associated with the immune system. Therefore, we performed the interrelation analysis by assessing the immune system process of downregulated DEGs in ClueGO. As depicted in Figure 4, downregulated DEGs were mainly enriched in B cell differentiation, positive regulation of monocyte chemotaxis, positive regulation of T cell proliferation and leukocyte tethering or rolling. In addition, we conducted an interrelation analysis between pathways in the BPs of downregulated DEGs (Figure 5). Consistent with the above results, we observed an enrichment of the downregulated DEGs largely in B cell differentiation, leukocyte tethering or rolling, platelet degranulation, positive regulation of T cell proliferation, positive regulation of monocyte chemotaxis and regulation of complement activation.
Figure 4.
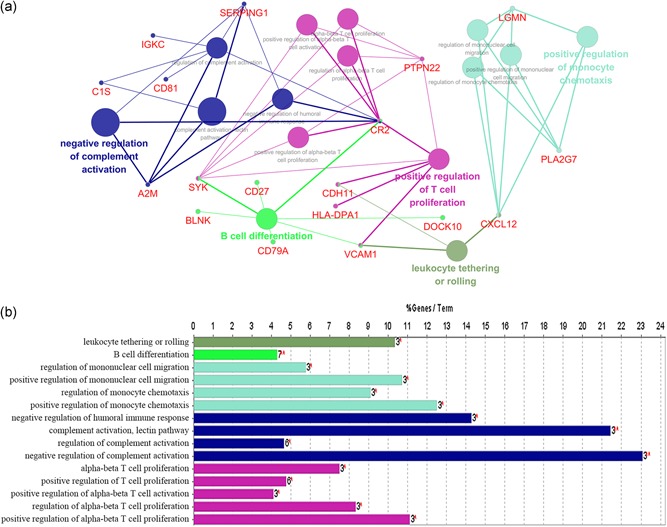
Interrelation analysis between pathways (immune system process) of downregulated DEGs. (a) The interrelation between immune system pathways. (b) Numbers of genes enriched in the identified pathways. DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
Figure 5.

Interrelation analysis between pathways (biological process) of downregulated DEGs. (a) Interrelation between biological process pathways. (b) Numbers of genes enriched in the identified pathways. DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
3.5. PPI construction and modular analysis
Based on the STRING online database and Cytoscape software, we constructed a DEG PPI network = containing 94 DEGs (41 upregulated and 53 downregulated), with 94 nodes and 224 edges identified, as shown in Figure 6a. By utilizing cluster analysis of the PPI network in Cytotype MCODE, we identified two significant modules based on the degree of importance. Module 1 contained 8 nodes and 27 edges (Figure 6b); Module 2 contained 5 nodes and 7 edges (Figure 6c). KEGG and REACTOME pathway enrichment analyses of the DEGs in these modules were then performed. DEGs in Module 1 were mainly enriched in the formation of a pool of free 40S subunits, L13a‐mediated translational silencing of ceruloplasmin expression and eukaryotic translation initiation (Table 3). DEGs in Module 2 were mainly enriched in the B cell receptor signaling pathway, the immune system, regulation of complement cascade and interferon alpha/beta signaling (Table 4).
Figure 6.
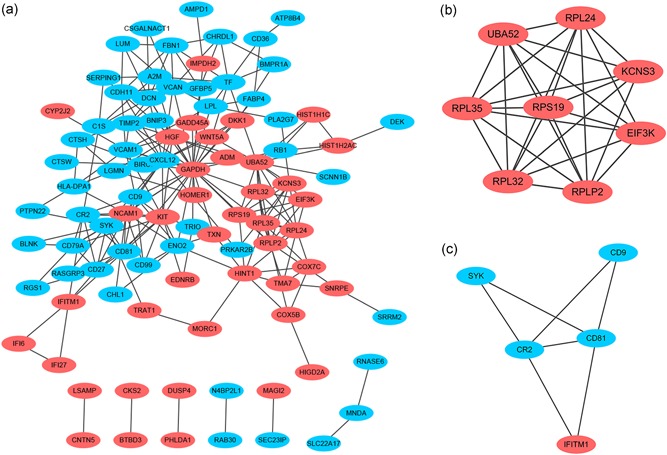
Protein–protein interaction (PPI) network of DEGs and module analysis. (a) Based on the STRING online database, a DEG PPI network was constructed containing 94 DEGs (41 upregulated DEGs labeled in red and 53 downregulated DEGs labeled in blue). (b) Identification of two significant modules based on the degree of importance. Module 1 contains 8 nodes and 27 edges. (c) Module 2 contains 5 nodes and 7 edges. DEGs, differentially expressed genes; PPI, protein–protein interaction [Color figure can be viewed at wileyonlinelibrary.com]
Table 3.
Pathway enrichment analysis of Module 1 genes function
| Term | Description | Count | p value |
|---|---|---|---|
| R‐HSA‐72689 | Formation of a pool of free 40 S subunits | 7 | 4.36 × 10–14 |
| R‐HSA‐156827 | L13a‐mediated translational silencing of Ceruloplasmin expression | 7 | 8.39 × 10–14 |
| R‐HSA‐72706 | GTP hydrolysis and joining of the 60 S ribosomal subunit | 7 | 8.93 × 10–14 |
| R‐HSA‐72613 | Eukaryotic translation initiation | 7 | 1.36 × 10–13 |
| R‐HSA‐72737 | Cap‐dependent translation initiation | 7 | 1.36 × 10–13 |
| R‐HSA‐156902 | Peptide chain elongation | 6 | 7.80 × 10–12 |
| R‐HSA‐72764 | Eukaryotic translation termination | 6 | 1.01 × 10–11 |
| R‐HSA‐2408557 | Selenocysteine synthesis | 6 | 1.01 × 10–11 |
| R‐HSA‐156842 | Eukaryotic translation elongation | 6 | 1.08 × 10–11 |
| R‐HSA‐975956 | Nonsense‐mediated decay (NMD) independent of the exon junction complex (EJC) | 6 | 1.15 × 10–11 |
| R‐HSA‐192823 | Viral mRNA translation | 6 | 1.56 × 10–11 |
| R‐HSA‐1799339 | SRP‐dependent cotranslational protein targeting to membrane | 6 | 3.05 × 10–11 |
| R‐HSA‐927802 | Nonsense‐mediated decay (NMD) | 6 | 3.75 × 10–11 |
| R‐HSA‐975957 | Nonsense‐mediated decay (NMD) enhanced by the exon junction complex (EJC) | 6 | 3.75 × 10–11 |
| R‐HSA‐2408522 | Selenoamino acid metabolism | 6 | 3.95 × 10–11 |
| R‐HSA‐72766 | Translation | 7 | 7.10 × 10–11 |
Table 4.
Pathway enrichment analysis of Module 2 genes function
| Term | Description | Count | p value |
|---|---|---|---|
| bta04662 | B cell receptor signaling pathway | 4 | 1.03 × 10−5 |
| R‐HSA‐168256 | Immune system | 5 | 1.23 × 10−3 |
| R‐HSA‐977606 | Regulation of complement cascade | 2 | 1.42 × 10−3 |
| R‐HSA‐166658 | Complement cascade | 2 | 1.79 × 10−3 |
| R‐HSA‐1300652 | Sperm: oocyte membrane binding | 1 | 2.13 × 10−3 |
| R‐HSA‐909733 | Interferon alpha/beta signaling | 2 | 2.48 × 10−3 |
| R‐HSA‐5336415 | Uptake and function of diphtheria toxin | 1 | 4.26 × 10−3 |
| R‐HSA‐9020558 | Interleukin‐2 signaling | 1 | 5.95 × 10−3 |
| R‐HSA‐1280218 | Adaptive immune system | 3 | 6.07 × 10−3 |
| R‐HSA‐76002 | Platelet activation, signaling and aggregation | 2 | 6.45 × 10−3 |
| R‐HSA‐1280215 | Cytokine signaling in immune system | 3 | 7.11 × 10−3 |
| R‐HSA‐198933 | Immunoregulatory interactions between a lymphoid and a non‐lymphoid cell | 2 | 7.14 × 10−3 |
| R‐HSA‐912631 | Regulation of signaling by CBL | 1 | 1.02 × 10−2 |
| R‐HSA‐913531 | Interferon signaling | 2 | 1.08 × 10−2 |
| R‐HSA‐1187000 | Fertilization | 1 | 1.32 × 10−2 |
| R‐HSA‐168249 | Innate immune system | 3 | 1.35 × 10−2 |
3.6. Survival analysis of DEGs
Finally, we explored the potential prognostic value of DEGs for patients with MM by evaluating the correlation between DEG expression and survival rates using PrognoScan. Based on the GSE2658 data set (n = 559), 31 DEGs (Table S1; 8 upregulated and 23 downregulated genes) were identified to be significantly associated with DSS in MM. Representative genes are shown in Figure 7. SNRPE, TXN, HIST1H1C, and CKS2 were upregulated, whereas DENND2D, CYP1B1, DCN, VCAN, HLA‐DPA1, TP73‐AS1, CHRDL1 and RCBTB2 were downregulated.
Figure 7.
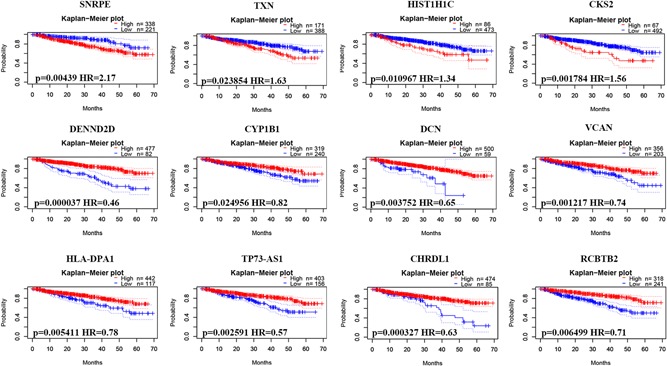
Correlation between individual DEG expression and MM disease‐specific survival. Kaplan–Meier survival curves comparing high and low expression of DEGs in MM in PrognoScan, as based on the GSE2658 data set (n = 559). DEGs, differentially expressed genes; MM, multiple myeloma [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
MM remains incurable for most patients despite dramatic improvements in new treatments. In this study, we analyzed the expression of genes in two microarray datasets based on purified plasma cells of MM patients and healthy controls. A total of 129 common DEGs were identified (51 upregulated and 78 downregulated), with p < .05 and [logFC] > 1. Subsequently, we utilized bioinformatics methods to deeply explore these DEGs, including GO term enrichment, signaling pathway enrichment, PPI network construction, and survival analysis.
GO term enrichment analysis consists of three groups: BP, CC, and MF. With regard to BPs, the DEGs identified are mainly involved in positive regulation of the immune system process, immune response and positive regulation of response to the stimulus. Evading immune destruction is one of the hallmarks of cancer (Hanahan & Weinberg, 2011). Indeed, cancer cells are involved in extensive and dynamic crosstalk with immune cells, and many molecular events mediate this interrelationship (Grivennikov, Greten, & Karin, 2010). Patients with MM have increased susceptibility to infections, and the immune system and immune responses play crucial roles in the pathogenesis of MM (Rossi, Botta, Correale, Tassone, & Tagliaferri, 2013). Myeloma cells primarily reside in the bone marrow immune microenvironment, which also contains innate immune cells (such as macrophages, myeloid‐derived suppressor cells, and natural killer cells) and adaptive immune cells (T and B lymphocytes; Guillerey, Nakamura, Vuckovic, Hill, & Smyth, 2016). Some of the genes identified in the present study have been shown to participate in the crosstalk between MM cells with immune cells, such as vascular cell adhesion molecule 1 (VCAM1) and CD9 (Belloni et al., 2018; De Bruyne et al., 2008), leading to progression of MM.
DEGs in CC are mainly enriched in cytosolic ribosomes. Consistent with this, DEGs in MF are mainly associated with the structural constituents of ribosomes. Ribosome biogenesis is a process by which ribosomes are generated for protein synthesis, and this process is critical for cells to survive, grow and proliferate (Iadevaia, Liu, & Proud, 2014; Lessard, Brakier‐Gingras, & Ferbeyre, 2019). In cancer, ribosome biogenesis is commonly enhanced to meet the increased anabolic requirements related to malignant transformation and tumor development. In addition, the ribosome serves as a central information hub, and numerous oncogenic signaling pathways modulate its function[12 13]. For instance, RPS9, identified as an upregulated DEG in this study, contributes to cancer cell metastasis through c‐Myc (K. C. Chen et al., 2018). Several studies on MM have revealed that ribosome biogenesis is associated with the MM risk allele and response to bortezomib treatment (Ali, Ajore, Wihlborg, & Niroula, 2018; Hofman et al., 2017).
Next, interrelation analysis of pathways was conducted by utilizing KEGG processes in ClueGO. The results showed all DEGs to mainly be associated with the B cell receptor signaling pathway, hematopoietic cell lineage, the NF‐kappa B signaling pathway, and the PPAR signaling pathway. The B cell receptor signaling pathway has been reported to mediate PD‐L1 signaling and metabolism in hematologic malignancies (Li et al., 2018; Vangapandu et al., 2017). Consistent with our results, this pathway was found to be the most significant pathway in another MM polygenic interaction study (Chattopadhyay et al., 2018). The NF‐kappa B signaling pathway plays a crucial role in the proliferation, survival and treatment resistance of MM, and dysregulation of NF‐kB may result from genetic and microenvironment‐driven mechanisms (Demchenko et al., 2010; Keats et al., 2007; McMillin et al., 2010). Furthermore, important therapeutic advances have emerged through alteration of the activity of NF‐kB with pharmacological interventions (Adams, 2004; Moreau et al., 2012). The three subtypes of PPAR (PPARalpha, beta/delta, and gamma) exhibit differential expression patterns in vertebrates. PPARγ agonists significantly suppress the adhesive interaction between MM and bone marrow stromal cells (Wang, Yang, Zhang, & Farrar, 2007) and increase the cytotoxic effect of other drugs in MM cells (Aouali et al., 2009).
Subsequently, interrelation analysis was performed. Immune system process analysis of downregulated DEGs indicated that B cell differentiation, positive regulation of T cell proliferation, and leukocyte tethering or rolling interact with each other through key genes, such as spleen‐associated tyrosine kinase (SYK; related to MM cell survival and migration; Lorenz et al., 2016), complement Component 3d receptor 2 (linked to the innate complement‐mediated immune response; Sorman, Zhang, Ding, & Heyman, 2014), VCAM1 (contributing to drug resistance in MM; Belloni et al., 2018) and cadherin 11 (CDH11; involved in MM progression and bone destruction; Christopoulos et al., 2017). Interrelation analysis via assessment of the BPs of downregulated DEGs was consistent with the above results. A variety of mechanisms to evade immune surveillance have been reported in MM, including impaired antibody production (Rawstron et al., 1998), deregulation of the T cell compartment (Koike et al., 2002), upregulation of inhibitory ligands (Paiva et al., 2015), disruption of antigen presentation (Brown, Pope, Yuen, Gibson, & Joshua, 1998) and recruitment of immunosuppressive cells (Franssen et al., 2015; Malek et al., 2016). Thus, various immunotherapy strategies have been developed, such as immunomodulatory drugs, checkpoint inhibitors, monoclonal antibodies, chimeric antigen receptor T‐cells, and vaccines (Kumar & Anderson, 2016; Rodriguez‐Otero, Paiva, Engelhardt, Prosper, & San Miguel, 2017). These strategies have shown encouraging results in patients with relapsed refractory MM and also in improving patient outcomes (Neri, Bahlis, & Lonial, 2016).
A PPI network of DEGs was constructed, containing 94 nodes and 224 edges. In this network, screening revealed two significant modules based on the degree of importance. Many of the core genes within the subnetworks have been reported to play a crucial role in the development of MM, such as SYK, CD81, and CD9. SYK, a protein kinase, is a critical element in B cell receptor signaling (Knoll et al., 2017). Moreover, SYK has been demonstrated to be involved in ectoenzyme CD38‐, chemokine receptor CXCR4‐, and integrin ligand VCAM1 signaling (Buchner et al., 2010). CD38 is characteristically expressed on myeloma cells, and CXCR4 and VCAM1 play a crucial role in MM‐microenvironmental crosstalk (Schuler, Ewerth, Waldschmidt, Wasch, & Engelhardt, 2013). Targeting SYK has shown promising therapeutic effects in B cell malignancies, including chronic lymphoid leukemia (D. Liu & Mamorska‐Dyga, 2017), and in MM, inhibition of SYK resulted in significant decreases in cell survival and migration (Lorenz et al., 2016). Previous studies have demonstrated that CD81 is an independent prognostic factor in MM and may be associated with MM pathogenesis (Arana et al., 2018; F. Chen et al., 2018; Paiva et al., 2017). Expression of CD9 in MM cells is related to the disease status and survival of patients with MM, and CD9 was found to be involved in transendothelial invasion in an MM mouse model (De Bruyne et al., 2006). Moreover, CD9 expression was found to be downregulated during disease progression, which led to decreased susceptibility of MM cells to NK cell–mediated cytolysis (De Bruyne et al., 2008). Our survival analysis revealed 31 DEGs to be significantly associated with survival in MM patients, and further investigation of these genes in clinical research is warranted.
In conclusion, integrated bioinformatics analysis of multiple datasets of newly diagnosed MM patients and healthy controls was performed. Common DEGs were identified that are significantly enriched in various pathways, especially immune responses, followed by evaluation of the prognostic value for patients with MM. Notably, most of the prognostic genes are immune‐related genes. The results of this study increase our understanding of the molecular drivers that underlie MM initiation and progression, and the critical genes and pathways identified constitute potential therapeutic targets.
CONFLICT OF INTERESTS
The authors declare that there is no conflict of interests.
AUTHOR CONTRIBUTIONS
H.Y. and G.Z. conceived of and designed the study. J.Q., Y.L., and X.H. performed the data analysis. H.Y., E.Z., and Z.C. wrote the paper.
DATA AVAILABILITY
The data that support the findings of this study are available in GEO at https://www.ncbi.nlm.nih.gov/geo/. These data were derived from the following resources available in the public domain: GSE6477 and GSE47552.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors thank the GEO databases for the availability of the data. This study was supported by grants from the National Natural Science Foundation of China (Grant number 91742110) and Zhejiang Provincial Natural Science Foundation (Grant number LY19H080004).
Yan H, Zheng G, Qu J, et al. Identification of key candidate genes and pathways in multiple myeloma by integrated bioinformatics analysis. J Cell Physiol. 2019;234:23785–23797. 10.1002/jcp.28947
Contributor Information
Enfan Zhang, Email: 54380234@qq.com.
Zhen Cai, Email: caiz@zju.edu.cn.
References
REFERENCES
- Adams, J. (2004). The development of proteasome inhibitors as anticancer drugs. Cancer Cell, 5(5), 417–421. [DOI] [PubMed] [Google Scholar]
- Ali, M. , Ajore, R. , Wihlborg, A. K. , & Niroula, A. (2018). The multiple myeloma risk allele at 5q15 lowers ELL2 expression and increases ribosomal gene expression. Nature Communications, 9(1), 1649 10.1038/s41467-018-04082-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouali, N. , Palissot, V. , El‐Khoury, V. , Moussay, E. , Janji, B. , Pierson, S. , … Berchem, G. (2009). Peroxisome proliferator‐activated receptor gamma agonists potentiate the cytotoxic effect of valproic acid in multiple myeloma cells. British Journal of Haematology, 147(5), 662–671. 10.1111/j.1365-2141.2009.07902.x [DOI] [PubMed] [Google Scholar]
- Arana, P. , Paiva, B. , Cedena, M. T. , Puig, N. , Cordon, L. , Vidriales, M. B. , … San Miguel, J. F. (2018). Prognostic value of antigen expression in multiple myeloma: A PETHEMA/GEM study on 1265 patients enrolled in four consecutive clinical trials. Leukemia, 32(4), 971–978. 10.1038/leu.2017.320 [DOI] [PubMed] [Google Scholar]
- Belloni, D. , Heltai, S. , Ponzoni, M. , Villa, A. , Vergani, B. , Pecciarini, L. , … Ferrero, E. (2018). Modeling multiple myeloma‐bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica, 103(4), 707–716. 10.3324/haematol.2017.167486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, R. D. , Pope, B. , Yuen, E. , Gibson, J. , & Joshua, D. E. (1998). The expression of T cell related costimulatory molecules in multiple myeloma. Leukemia and Lymphoma, 31(3‐4), 379–384. 10.3109/10428199809059231 [DOI] [PubMed] [Google Scholar]
- De Bruyne, E. , Andersen, T. L. , De Raeve, H. , Van Valckenborgh, E. , Caers, J. , Van Camp, B. , … Vanderkerken, K. (2006). Endothelial cell‐driven regulation of CD9 or motility‐related protein‐1 expression in multiple myeloma cells within the murine 5T33MM model and myeloma patients. Leukemia, 20(10), 1870–1879. 10.1038/sj.leu.2404343 [DOI] [PubMed] [Google Scholar]
- De Bruyne, E. , Bos, T. J. , Asosingh, K. , Vande Broek, I. , Menu, E. , Van Valckenborgh, E. , … Van Riet, I. (2008). Epigenetic silencing of the tetraspanin CD9 during disease progression in multiple myeloma cells and correlation with survival. Clinical Cancer Research, 14(10), 2918–2926. 10.1158/1078-0432.ccr-07-4489 [DOI] [PubMed] [Google Scholar]
- Buchner, M. , Baer, C. , Prinz, G. , Dierks, C. , Burger, M. , Zenz, T. , … Zirlik, K. (2010). Spleen tyrosine kinase inhibition prevents chemokine‐ and integrin‐mediated stromal protective effects in chronic lymphocytic leukemia. Blood, 115(22), 4497–4506. 10.1182/blood-2009-07-233692 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, S. , Thomsen, H. , da Silva Filho, M. I. , Weinhold, N. , Hoffmann, P. , Nothen, M. M. , … Forsti, A. (2018). Enrichment of B cell receptor signaling and epidermal growth factor receptor pathways in monoclonal gammopathy of undetermined significance: A genome‐wide genetic interaction study. Molecular Medicine, 24(1), 30 10.1186/s10020-018-0031-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Hu, Y. , Wang, X. , Fu, S. , Liu, Z. , & Zhang, J. (2018). Expression of CD81 and CD117 in plasma cell myeloma and the relationship to prognosis. Cancer Medicine, 7(12), 5920–5927. 10.1002/cam4.1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. C. , Hsu, W. H. , Ho, J. Y. , Lin, C. W. , Chu, C. Y. , Kandaswami, C. C. , … Cheng, C. H. (2018). Flavonoids luteolin and quercetin inhibit RPS19 and contributes to metastasis of cancer cells through c‐Myc reduction. Journal of Food and Drug Analysis, 26(3), 1180–1191. 10.1016/j.jfda.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng, W. J. , Kumar, S. , Vanwier, S. , Ahmann, G. , Price‐Troska, T. , Henderson, K. , … Fonseca, R. (2007). Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Research, 67(7), 2982–2989. 10.1158/0008-5472.can-06-4046 [DOI] [PubMed] [Google Scholar]
- Christopoulos, P. F. , Sfikakis, P. P. , Nikolaou, E. , Terpos, E. , Koutsilieris, M. , & Kyrtsonis, M. C. (2017). Cadherin‐11 (CDH11) expression in the peripheral blood of patients with active multiple myeloma. British Journal of Haematology, 177(5), 813–816. 10.1111/bjh.14105 [DOI] [PubMed] [Google Scholar]
- Demchenko, Y. N. , Glebov, O. K. , Zingone, A. , Keats, J. J. , Bergsagel, P. L. , & Kuehl, W. M. (2010). Classical and/or alternative NF‐kappaB pathway activation in multiple myeloma. Blood, 115(17), 3541–3552. 10.1182/blood-2009-09-243535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen, L. E. , van de Donk, N. W. , Emmelot, M. E. , Roeven, M. W. , Schaap, N. , Dolstra, H. , … Mutis, T. (2015). The impact of circulating suppressor cells in multiple myeloma patients on clinical outcome of DLIs. Bone Marrow Transplantation, 50(6), 822–828. 10.1038/bmt.2015.48 [DOI] [PubMed] [Google Scholar]
- Grivennikov, S. I. , Greten, F. R. , & Karin, M. (2010). Immunity, inflammation, and cancer. Cell, 140(6), 883–899. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillerey, C. , Nakamura, K. , Vuckovic, S. , Hill, G. R. , & Smyth, M. J. (2016). Immune responses in multiple myeloma: Role of the natural immune surveillance and potential of immunotherapies. Cellular and Molecular Life Science, 73(8), 1569–1589. 10.1007/s00018-016-2135-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. [DOI] [PubMed] [Google Scholar]
- Hofman, I. J. F. , van Duin, M. , De Bruyne, E. , Fancello, L. , Mulligan, G. , Geerdens, E. , … Vandenberghe, P. (2017). RPL5 on 1p22.1 is recurrently deleted in multiple myeloma and its expression is linked to bortezomib response. Leukemia, 31(8), 1706–1714. 10.1038/leu.2016.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadevaia, V. , Liu, R. , & Proud, C. G. (2014). mTORC1 signaling controls multiple steps in ribosome biogenesis. Seminars in Cell and Developmental Biology, 36, 113–120. 10.1016/j.semcdb.2014.08.004 [DOI] [PubMed] [Google Scholar]
- Keats, J. J. , Fonseca, R. , Chesi, M. , Schop, R. , Baker, A. , Chng, W. J. , … Bergsagel, P. L. (2007). Promiscuous mutations activate the noncanonical NF‐kappaB pathway in multiple myeloma. Cancer Cell, 12(2), 131–144. 10.1016/j.ccr.2007.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll, M. , Winther, S. , Natarajan, A. , Yang, H. , Jiang, M. , Thiru, P. , … Lamming, D. W. (2017). SYK kinase mediates brown fat differentiation and activation. Nature Communications, 8(1), 2115 10.1038/s41467-017-02162-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike, M. , Sekigawa, I. , Okada, M. , Matsumoto, M. , Iida, N. , Hashimoto, H. , & Oshimi, K. (2002). Relationship between CD4(+)/CD8(+) T cell ratio and T cell activation in multiple myeloma: Reference to IL‐16. Leukemia Research, 26(8), 705–711 [DOI] [PubMed] [Google Scholar]
- Kumar, S. K. , & Anderson, K. C. (2016). Immune therapies in multiple myeloma. Clinical Cancer Research, 22(22), 5453–5460. 10.1158/1078-0432.ccr-16-0868 [DOI] [PubMed] [Google Scholar]
- Lessard, F. , Brakier‐Gingras, L. , & Ferbeyre, G. (2019). Ribosomal proteins control tumor suppressor pathways in response to nucleolar stress. BioEssays, 41(3), e1800183 10.1002/bies.201800183 [DOI] [PubMed] [Google Scholar]
- Li, L. , Zhang, J. , Chen, J. , Xu‐Monette, Z. Y. , Miao, Y. , Xiao, M. , … Pham, L. V. (2018). B‐cell receptor‐mediated NFATc1 activation induces IL‐10/STAT3/PD‐L1 signaling in diffuse large B‐cell lymphoma. Blood, 132(17), 1805–1817. 10.1182/blood-2018-03-841015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Gu, X. , & Jiang, Z. (2017). Identification of novel targets for multiple myeloma through integrative approach with Monte Carlo cross‐validation analysis. Journal of Bone Oncology, 8, 8–12. 10.1016/j.jbo.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. , & Mamorska‐Dyga, A. (2017). Syk inhibitors in clinical development for hematological malignancies. Journal of Hematology & Oncology, 10(1), 145 10.1186/s13045-017-0512-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Corral, L. , Corchete, L. A. , Sarasquete, M. E. , Mateos, M. V. , Garcia‐Sanz, R. , Ferminan, E. , … Gutierrez, N. C. (2014). Transcriptome analysis reveals molecular profiles associated with evolving steps of monoclonal gammopathies. Haematologica, 99(8), 1365–1372. 10.3324/haematol.2013.087809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, J. , Waldschmidt, J. , Wider, D. , Follo, M. , Ihorst, G. , Chatterjee, M. , … Engelhardt, M. (2016). From CLL to multiple myeloma ‐ spleen tyrosine kinase (SYK) influences multiple myeloma cell survival and migration. British Journal of Haematology, 174(6), 985–989. 10.1111/bjh.13825 [DOI] [PubMed] [Google Scholar]
- Malek, E. , de Lima, M. , Letterio, J. J. , Kim, B. G. , Finke, J. H. , Driscoll, J. J. , & Giralt, S. A. (2016). Myeloid‐derived suppressor cells: The green light for myeloma immune escape. Blood Reviews, 30(5), 341–348. 10.1016/j.blre.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin, D. W. , Delmore, J. , Weisberg, E. , Negri, J. M. , Geer, D. C. , Klippel, S. , … Mitsiades, C. S. (2010). Tumor cell‐specific bioluminescence platform to identify stroma‐induced changes to anticancer drug activity. Nature Medicine (New York, NY, United States), 16(4), 483–489. 10.1038/nm.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, P. , Richardson, P. G. , Cavo, M. , Orlowski, R. Z. , San Miguel, J. F. , Palumbo, A. , & Harousseau, J. L. (2012). Proteasome inhibitors in multiple myeloma: 10 years later. Blood, 120(5), 947–959. 10.1182/blood-2012-04-403733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri, P. , Bahlis, N. J. , & Lonial, S. (2016). New strategies in multiple myeloma: Immunotherapy as a novel approach to treat patients with multiple myeloma. Clinical Cancer Research, 22(24), 5959–5965. 10.1158/1078-0432.ccr-16-0184 [DOI] [PubMed] [Google Scholar]
- Paiva, B. , Azpilikueta, A. , Puig, N. , Ocio, E. M. , Sharma, R. , Oyajobi, B. O. , … Melero, I. (2015). PD‐L1/PD‐1 presence in the tumor microenvironment and activity of PD‐1 blockade in multiple myeloma. Leukemia, 29(10), 2110–2113. 10.1038/leu.2015.79 [DOI] [PubMed] [Google Scholar]
- Paiva, B. , Puig, N. , Cedena, M. T. , de Jong, B. G. , Ruiz, Y. , Rapado, I. , … San‐Miguel, J. F. (2017). Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia, 31(2), 382–392. 10.1038/leu.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo, A. , & Anderson, K. (2011). Multiple myeloma. New England Journal of Medicine, 364(11), 1046–1060. 10.1056/NEJMra1011442 [DOI] [PubMed] [Google Scholar]
- Pawlyn, C. , & Morgan, G. J. (2017). Evolutionary biology of high‐risk multiple myeloma. Nature Reviews Cancer, 17(9), 543–556. 10.1038/nrc.2017.63 [DOI] [PubMed] [Google Scholar]
- Rawstron, A. C. , Davies, F. E. , Owen, R. G. , English, A. , Pratt, G. , Child, J. A. , … Morgan, G. J. (1998). B‐lymphocyte suppression in multiple myeloma is a reversible phenomenon specific to normal B‐cell progenitors and plasma cell precursors. British Journal of Haematology, 100(1), 176–183 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Otero, P. , Paiva, B. , Engelhardt, M. , Prosper, F. , & San Miguel, J. F. (2017). Is immunotherapy here to stay in multiple myeloma? Haematologica, 102(3), 423–432. 10.3324/haematol.2016.152504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, M. , Botta, C. , Correale, P. , Tassone, P. , & Tagliaferri, P. (2013). Immunologic microenvironment and personalized treatment in multiple myeloma. Expert Opinion on Biological Therapy, 13(Suppl 1), S83–93. 10.1517/14712598.2013.799130 [DOI] [PubMed] [Google Scholar]
- Schuler, J. , Ewerth, D. , Waldschmidt, J. , Wasch, R. , & Engelhardt, M. (2013). Preclinical models of multiple myeloma: A critical appraisal. Expert Opinion on Biological Therapy, 13(Suppl 1), S111–123. 10.1517/14712598.2013.799131 [DOI] [PubMed] [Google Scholar]
- Sonneveld, P. , Avet‐Loiseau, H. , Lonial, S. , Usmani, S. , Siegel, D. , Anderson, K. C. , … Orlowski, R. (2016). Treatment of multiple myeloma with high‐risk cytogenetics: A consensus of the International Myeloma Working Group. Blood, 127(24), 2955–2962. 10.1182/blood-2016-01-631200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorman, A. , Zhang, L. , Ding, Z. , & Heyman, B. (2014). How antibodies use complement to regulate antibody responses. Molecular Immunology, 61(2), 79–88. 10.1016/j.molimm.2014.06.010 [DOI] [PubMed] [Google Scholar]
- Sun, J. , Wen, X. , Jin, F. , Li, Y. , Hu, J. , & Sun, Y. (2015). Bioinformatics analyses of differentially expressed genes associated with bisphosphonate‐related osteonecrosis of the jaw in patients with multiple myeloma. OncoTargets and Therapy, 8, 2681–2688. 10.2147/ott.s88463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangapandu, H. V. , Havranek, O. , Ayres, M. L. , Kaipparettu, B. A. , Balakrishnan, K. , Wierda, W. G. , … Gandhi, V. (2017). B‐cell receptor signaling regulates metabolism in chronic lymphocytic leukemia. Molecular Cancer Research, 15(12), 1692–1703. 10.1158/1541-7786.mcr-17-0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , Papadopoulos, N. , Velculescu, V. E. , Zhou, S. , Diaz, L. A., Jr. , & Kinzler, K. W. (2013). Cancer genome landscapes. Science, 339(6127), 1546–1558. 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. H. , Yang, X. Y. , Zhang, X. , & Farrar, W. L. (2007). Inhibition of adhesive interaction between multiple myeloma and bone marrow stromal cells by PPARgamma cross talk with NF‐kappaB and C/EBP. Blood, 110(13), 4373–4384. 10.1182/blood-2006-07-038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are available in GEO at https://www.ncbi.nlm.nih.gov/geo/. These data were derived from the following resources available in the public domain: GSE6477 and GSE47552.