

Abstract
The B12 cofactors instill a natural curiosity regarding the primordial selection and evolution of their corrin ligand. Surprisingly, this important natural macrocycle has evaded molecular scrutiny, and its specific role in predisposing the incarcerated cobalt ion for organometallic catalysis has remained obscure. Herein, we report the biosynthesis of the cobalt‐free B12 corrin moiety, hydrogenobyric acid (Hby), a compound crafted through pathway redesign. Detailed insights from single‐crystal X‐ray and solution structures of Hby have revealed a distorted helical cavity, redefining the pattern for binding cobalt ions. Consequently, the corrin ligand coordinates cobalt ions in desymmetrized “entatic” states, thereby promoting the activation of B12‐cofactors for their challenging chemical transitions. The availability of Hby also provides a route to the synthesis of transition metal analogues of B12.
Keywords: cobalamins, cobalt, synthetic biology, vitamins, X-ray structures
The unique structural1 and biosynthetic features2 of coenzyme B12 and its biological homologues raise fundamental questions concerning the evolution and selection of the corrin ligand,3 as well as the adoption of B12 cofactors into key metabolic roles across the three domains of life. The combined selection of the corrin macrocycle and of cobalt as the specific transition metal center for bio‐organometallic catalysis is an intriguing aspect of the B12 cofactors.4 The resistance of cobalt corrins against the removal of cobalt without concomitant destruction of the corrin ligand5 has made a study of cobalt‐free natural corrins a major scientific challenge.6 Consequently, despite the 40 years since vitamin B12 was prepared by total synthesis,7 the special partnership of the ligand and the cobalt ion of the natural B12 cofactors remains largely unexplored.4a
Two pathways for B12 biosynthesis have highlighted intriguing “ring contraction” steps2 that tailor the “coordination hole” of the tetrapyrrolic macrocycle to the effective size of cobalt ions.4a, 8 Surprisingly, B12’s own ligand, hydrogenobyric acid (Hby) (Figure 1), is not a biosynthetic intermediate in either of them.2 However, metabolic engineering of the B12 biosynthetic pathway has allowed the development of strategies to access metal‐free corrins by design.2b, 9 We recently reported recombinant E. coli strains that generated metal‐free corrins, such as hydrogenobyrinic acid a,c‐diamide (HBAD).9, 10 Normally, in the aerobic B12 biosynthetic pathway, HBAD is next chelated with cobalt.2 However, when grown in the absence of cobalt, some purple sulfur bacteria produce cobalt‐free corrinoids,11 including a compound tentatively identified as Hby,11b, 11c providing hope for the biological synthesis of Hby.2b Herein, we describe an engineered B12 biosynthesis pathway variant containing the enzyme CobQ for the effective preparation of Hby, and present a thorough analysis of the structure of this metal‐free corrin, which is critical for binding cobalt ions and for bestowing B12 biocatalysts with their exceptional reactivity.4a, 12
Figure 1.
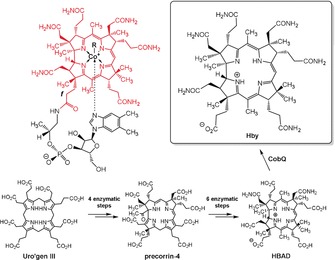
Structural formulae of hydrogenobyric acid (Hby) and of the cobalamins (Cbls) coenzyme B12 (R=5′‐adenosyl, AdoCbl), methyl‐cobalamin (R=CH3, MeCbl), vitamin B12 (R=CN, CNCbl) and cob(II)‐alamin (R=e−, CblII), and key steps of the designed de novo biosynthesis of Hby. A complete outline of the engineered biosynthesis of Hby is included in Figure S1 in the Supporting Information.
A pathway variant was explored for the biosynthesis of Hby by integrating cobQ from a purple sulfur bacterium11a into the existing repertoire of HBAD biosynthetic genes to generate a Hby‐operon in an E. coli strain called ED661. With the Hby‐operon integrated in the genome under the control of a T7 promoter, Hby was found to be excreted into the culture medium. A 4 L fermentation of this strain furnished 11.8 mg (12.8 μmol) of crystalline Hby (Figure 1 and Supporting Information, SI), providing an unprecedented opportunity to study a metal‐free natural corrin. When buffered to pH 5–7, and kept in the dark, aqueous solutions of Hby were found to be relatively stable at room temperature (at higher pH Hby was converted into “yellow corrinoids”).11a, 11b
In aqueous solution, Hby exhibited UV/Vis absorption11b with maxima at 270 nm, 330 nm, 499 nm and 524 nm, and emitted fluorescence with maxima at 552 and 609 nm (Figure 2), comparable to a natural “metal‐free red corrin”.13 The absorption and emission maxima (at 524 and 552 nm, respectively) position the lowest singlet excited state of Hby at 223 kJ mol−1 (for additional data see SI, Figure S2). NMR‐ and mass spectra (Figure 2 and see SI) established the structure of Hby. The signals of all H, C and N atoms of Hby were assigned via (1H,1H)‐homonuclear and (1H,13C)‐ and (1H,15N)‐heteronuclear single and multiple bond correlations. Two lowfield signals gave evidence for two “inner” H‐atoms at N2 and N4, specifying the structure of the cationic corrin ligand core in metal‐free Hby. Other NH tautomers, such as 1,3 Hby with “inner” H atoms at N1 and N3, were not detected (Figure 2). However, the HN2 and HN4 protons undergo unsymmetrical transannular H‐bonding with N1 and N3, detected with 15N‐labelled Hby, clarifying the question6 of the location and H‐bonding pattern of the “inner” H atoms in a natural metal‐free corrin. The H atoms H(N2) and H(N4) of Hby were also observed to interact mutually by NOE correlations and by an additional nonbonding through‐space interaction, diagnosed through substitution of either one of these H(N)s by D (see SI, Figure S4).
Figure 2.
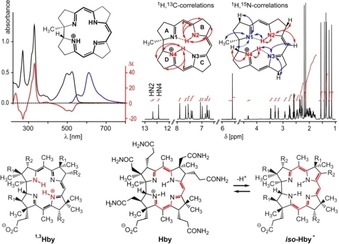
Spectra and structure of Hby. Top left: UV/Vis‐absorption (black trace), fluorescence emission (blue trace) and CD spectra (red trace) of Hby, recorded at 25 °C. Top right: 700 MHz 1H NMR spectrum of Hby in H2O/D2O (49:1) at pH 5 and correlations locating two “inner” HN protons at N2 and N4 and establishing their H‐bonds to N1 and N3. Bottom: The structure of Hby in water is represented best by the formula shown, while the tautomer 1,3 Hby (left) was not detected. Deprotonation of Hby generates an iso‐corrin anion, presumably iso ‐Hby− (right), but not a “neutral” corrin (see SI for formulae); R1=CH2CONH2, R2=CH2CH2CONH2 in the formulae of 1,3 Hby and iso ‐Hby−.
Both of the two “inner” H atoms are tightly bound by the corrin ligand, despite their fast exchange with water with rates of 21.9 s−1 (HN2) and 6.3 s−1 (HN4) at 308 K (SI, Figure S5). Indeed, the corrin moiety of Hby, a weak acid with pK a(Hby)=11.2,11b is deprotonated at the corrin periphery, presumably at C8 (Figure 2), as was first deduced by Eschenmoser and Fischli for the model corrin HCor+ (formula and crystal structure in SI, Figure S6).6, 14, 15 Poignantly, a monoprotonated “neutral” corrin ligand6 remains elusive. These features of Hby are supported by DFT analyses, which are consistent with the experimentally found stable zwitterionic form of Hby with two unsymmetrical H‐bonds N1–HN2 and N3–HN4, support peripheral C8 as the most acidic position of Hby and indicate protomers of Hby with a single “inner” H atom, either at N4 or at N2, to be significantly less stable (see SI, Figures S7 and S8; Table S5).
Hby generated single crystals from H2O/MeCN at 5 °C, with space group P21. X‐ray analysis revealed a pseudo‐C2‐symmetric helical arrangement of the core part of Hby, with similar structural features observed in the crystal as in solution (Figure 3 and SI, Figures S6 and S9). Electron density for two “inner” H atoms was located at N2 and N4, which were at a distance of only 2.27 Å from each other. The two H atoms are also close to N1 and N3 with distances of 1.91 Å and 2.06 Å, respectively, consistent with the NMR‐derived unsymmetrical H‐bonding. The distance between N2 and N4 of Hby is 3.97 Å, that is, about 0.3 Å longer than that between N1 and N3 (3.67 Å). By contrast, in HCor+, the “inner” H atoms are located at N1 and N3.6, 14, 15 However, H(N1) of HCor+ undergoes H‐bonding interactions to an EtOH molecule, giving the C4–C5 bond of HCor+ a 24.8° twist.6, 15 In both, Hby and HCor+, the “inner” H atoms break the inherent C2 symmetry of the corrin core, contrasting with the situation in the more regularly structured cobalt corrins and in the “expanded”, symmetrical porphyrins.16
Figure 3.
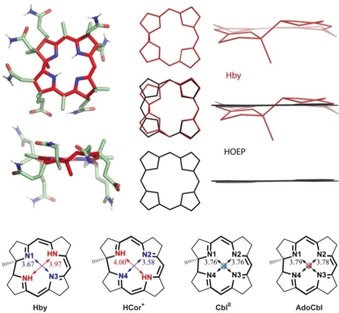
The ring‐contracted corrin ligand is a uniquely skewed helix. Top left: Two projections of the crystal structure of Hby (color coding: carbons of corrin core: red; of substituents: green; nitrogens: blue; oxygens: red; hydrogens: white). Top center and right: Projections of core structures of Hby (red), of octaethyl‐porphyrin (HOEP, black) and their superposition (middle). Bottom: Core structures of the metal‐free corrins Hby and HCor+,14, 15 and of the Cbls CblII and AdoCbl, in which effects of “inner” H atoms or of Co ions on the lengths of diagonals are highlighted.
The corrin Hby features a coordination hole with an average diameter of 3.83 Å, indicating an effective ring contraction of roughly 0.3 Å, compared to octaethylporphyrin (HOEP).16 Hence, the effective coordination radius in Hby (1.916 Å) is close to the average equatorial (Co–N) bond in AdoCbl (1.897 Å),17 MeCbl (1.898 Å)18 and in CblII (1.88 Å).19 At first sight, the corrin ligand appears to be well adapted for coordination of CoIII and CoII ions.17, 18, 19 However, the corrin‐specific trans junction between rings A and D imposes a distinctly helical structure.1 Consequently, the four chelating N atoms of the corrin macrocycle of Hby represent a screw‐like coordination hole, leading to a coordinative misfit for cobalt ions that is particularly strong for CoIII.
The mutual conformational adaptation of the corrin ligand and the coordinated cobalt ions was evaluated by two structure parameters: i) The corrin helicity h of the innermost coordination space of the corrin ligand provided by the four corrin nitrogen atoms, defined by the dihedral angle N1‐N2‐N3‐N4 (see Figure 4). In the metal‐free corrin Hby it amounts to h(Hby)=12.9°. CoIII corrins feature strongly reduced h values, e.g., h(AdoCbl)=3.5° and h(MeCbl)=4.6°. Hence, the ligand is strongly flattened by CoIII binding in AdoCbl and MeCbl. On the other hand, the four‐coordinate CoII center (CblIIACA) of the human adenosyl‐transferase ACA fits the corrin ligand better, displaying h(CblIIACA)=8°.20 Five‐coordinate CoII corrins display lower intermediate levels (see Figure 4). ii) The interplanar angle φ, which concerns the equatorial coordination sphere at the cobalt center, indicating coordinative strain in cobalt corrins when deviating from 0° (see Figure 4 and SI for details). The reference value of Hby is φ=13.5°. In CblIIACA φ=17°, in the two CoII corrins, CblII and CbinII 21 φ is 12.5°, respectively 7.6°. In CoIII corrins, like AdoCbl and MeCbl, φ is only 4–5°. Hence, h and φ decrease in a roughly correlated fashion from Hby to CoII and to CoIII corrins, indicating significant directional coordinative misfit in CoIII corrins.
Figure 4.
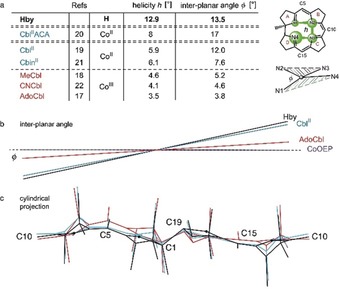
Structural characteristics of the coordinative interaction between cobalt ion and corrin ligand in B12 derivatives. a) Table with data describing the mutual adaptation of cobalt ions and the natural corrin ligand, expressed by the corrin helicity h (the calculated dihedral angle N1‐N2‐N3‐N4) and by the angle φ between the planes [N1‐cobalt‐N4] and [N2‐cobalt‐N3] (see drawings at right). In the order Hby, CoII corrins and CoIII corrins, h and φ decrease both in a roughly correlated fashion. b) The interplanar angle φ is large in helical Hby and in CblII, but strongly reduced in AdoCbl. In four‐coordinate CoII and six‐coordinate CoIII porphyrins (CoOEP) φ=ca. 0°.16 c) Cylinder projections of the structures of Hby, AdoCbl and CbinII, highlighting conformational differences in the corrin ligand. The conformation of Hby (black trace) is largely retained in the CoII corrin CbinII (blue trace), contrasting with its stronger adaptation to CoIII binding in AdoCbl (red trace).
The structural analysis of the helical corrin ligand Hby of B12 derivatives has revealed key elements helping to “demystify vitamin B12”.3, 4 It has confirmed the postulated “fit”3, 4, 8 of the “ring‐contracted” corrin ligand Hby to the size of CoIII and CoII ions (in AdoCbl and CblII). However, the corrin ligand Hby is distinctly helical, dissatisfying the octahedral coordination preference of CoIII centers, while better meeting the requirements of CoII and CoI ions (Figures 4 and 5). The inferior accommodation of CoIII over CoII centers implies a previously overlooked coordinative strain for CoIII corrins that promotes homolytic (Co–C) bond cleavage. This effect is crucial for the homolysis of AdoCbl to CblII in the B12‐dependent radical isomerization reactions.4c, 23 The same type of strain also activates the cobalt‐bound methyl group of MeCbl for abstraction by radicals24 in B12‐dependent radical SAM enzymes.25 A similar strain decrease may also accompany the heterolytic abstraction of the cobalt‐bound methyl of MeCbl by nucleophiles in B12‐dependent enzymatic methyl group transfer, producing CoI cobalamin.26 In the critical adenosyl‐transferase ACA, an unstable four‐coordinate form of CblII (CblIIACA)20 undergoes the reduction to the four‐coordinate CoI species. Such essential four‐coordinate CoII and CoI forms, which are hard to generate metabolically,25b, 27 appear to be well accommodated by the helical coordination hole of the corrin ligand. Since CoI corrins are not structurally characterized, model DFT calculations were used. They indicate a reduction of coordinative strain, by about 7 kJ mol−1, for the transition from six‐coordinate CoIII to four‐coordinate CoI ions, when bound by four N atoms in a nonplanar arrangement, as in Hby. The analogous CoIII‐to‐CoII transition experiences a strain decrease of about 10 kJ mol−1 (SI, Figure S10). Hence, the inherently helical corrin ligand acts as a “Procrustean bed” that destabilizes CoIII centers towards loss of axial ligands and formation of CoII or CoI forms, enhancing catalysis by the B12 cofactors.
Figure 5.
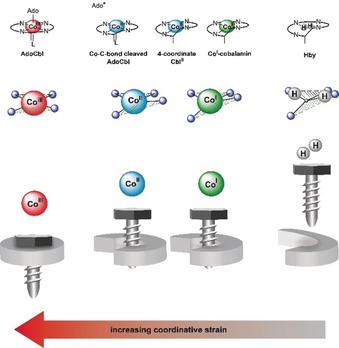
The helical corrin ligand binds cobalt centers in a strained state, promoting the cleavage of axial bonds and formation of reduced corrinoids. This is symbolized at the top for the CoIII corrin AdoCbl (before and after Co–C bond cleavage), for four‐coordinate CoII and CoI cobalamin, and for Hby. Middle and bottom: The corrin ligand is flattened and interplanar angle φ decreased most strongly at CoIII centers, less at CoII and CoI ions. Both parameters indicate an increasing misfit and strain in the series CoI/CoII and CoIII corrins; numerical data for h and φ are collected in Figure 4.
The previously unrecognized role of the flexible helical corrin ligand in activating organometallic CoIII corrins for catalysis classifies the B12 cofactors AdoCbl and MeCbl as “entatic state” molecules. The term “entatic” state was initially applied to proteins with metal centers bound in a strained coordination sphere to lower activation barriers for enzyme catalysis.28 Herein, we infer that cobalt corrins have been selected4a since they represent “entatic state” complexes in which ligand‐imposed strain activates CoIII centers for catalysis. A related situation exists in coenzyme F430, a Ni corphinoid, in which radial strain results from a misfit between the size of the coordinated Ni ions and the porphyrinoid macrocycle.4a, 29
The availability of the metal‐free Hby has also opened the door to the direct preparation of transition metal analogues of the cobalamins, the “metbalamins” (Metbls), a “Holy Grail” of bioinorganic chemistry.6, 9, 11b, 11c, 30 Hence, Hby has served as an effective starting material for the synthesis of transition metal B12 analogues, to be reported shortly. As described with AdoRhbl, the RhIII analogue of AdoCbl,9 suitably structured Metbls hold a significant potential as “antivitamins B12”, in biological imaging or as novel antibiotics.31 The exciting prospect of investigations with transition metal complexes of the skewed corrins will interest experimental scientists and theoretical chemists alike.
Experimental Section
CCDC 1881269 (Hby, see SI) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by grants from the Biotechnology and Biological Sciences Research Council (BBSRC; BB/L010208/1, BB/K009249/1 and BB/S002197/1) to M.J.W., and from the Austria Science Fund (FWF, P‐28892) to B.K. and Lise Meitner Fellowship (M‐2005) to M.P., who is also grateful to the Tyrolean Science Fund (TWF, UNI‐0404/1980), the Vienna Scientific Cluster (VSC3) and the University of Innsbruck HPC infrastructure for support of the computational work.
C. Kieninger, E. Deery, A. D. Lawrence, M. Podewitz, K. Wurst, E. Nemoto-Smith, F. J. Widner, J. A. Baker, S. Jockusch, C. R. Kreutz, K. R. Liedl, K. Gruber, M. J. Warren, B. Kräutler, Angew. Chem. Int. Ed. 2019, 58, 10756.
Dedicated to Professor Albert Eschenmoser on the occasion of his 94th birthday
Contributor Information
Prof. Dr. Martin J. Warren, Email: M.J.Warren@kent.ac.uk.
Prof. Dr. Bernhard Kräutler, Email: bernhard.kraeutler@uibk.ac.at.
References
- 1. Hodgkin D. C., Science 1965, 150, 979–988. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Battersby A. R., Science 1994, 264, 1551–1557; [DOI] [PubMed] [Google Scholar]
- 2b. Moore S. J., Lawrence A. D., Biedendieck R., Deery E., Frank S., Howard M. J., Rigby S. E. J., Warren M. J., Proc. Natl. Acad. Sci. USA 2013, 110, 14906–14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eschenmoser A., Angew. Chem. Int. Ed. 2011, 50, 12412–12472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12618–12681. [Google Scholar]
- 4.
- 4a. Eschenmoser A., Angew. Chem. Int. Ed. Engl. 1988, 27, 5–39; [Google Scholar]; Angew. Chem. 1988, 100, 5–40; [Google Scholar]
- 4b. da Silva J. J. R. F., Williams R. J. P., The Biological Chemistry of the Elements, Clarendon Press, Oxford, 1991; [Google Scholar]
- 4c. Halpern J., Science 1985, 227, 869–875. [DOI] [PubMed] [Google Scholar]
- 5. Lewis N. J., Nussberger R., Kräutler B., Eschenmoser A., Angew. Chem. Int. Ed. Engl. 1983, 22, 736–737; [Google Scholar]; Angew. Chem. 1983, 95, 744–746. [Google Scholar]
- 6. Blaser H. U., Winnacker E. L., Fischli A., Hardegger B., Bormann D., Hashimoto N., Schossig J., Keese R., Eschenmoser A., Helv. Chim. Acta 2015, 98, 1845–1920. [Google Scholar]
- 7.
- 7a. Eschenmoser A., Wintner C. E., Science 1977, 196, 1410–1426; [DOI] [PubMed] [Google Scholar]
- 7b. Woodward R. B. in Vitamin B12, Proceedings of the Third European Symposium on Vitamin B12 and Intrinsic Factor (Eds.: B. Zagalak, W. Friedrich), Walter de Gruyter, Berlin, 1979, p. 37. [Google Scholar]
- 8. Pratt J. M. in Chemistry and Biochemistry of B12 (Ed.: R. Banerjee), Wiley, New York, 1999, pp. 73–112. [Google Scholar]
- 9. Widner F. J., Lawrence A. D., Deery E., Heldt D., Frank S., Gruber K., Wurst K., Warren M. J., Kräutler B., Angew. Chem. Int. Ed. 2016, 55, 11281–11286; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11451–11456. [Google Scholar]
- 10. Deery E., Schroeder S., Lawrence A. D., Taylor S. L., Seyedarabi A., Waterman J., Wilson K. S., Brown D., Geeves M. A., Howard M. J., Pickersgill R. W., Warren M. J., Nat. Chem. Biol. 2012, 8, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Toohey J. I., Proc. Natl. Acad. Sci. USA 1965, 54, 934–942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Koppenhagen V. B., in B12, Vol. 2 (Ed.: D. Dolphin), Wiley, New York, 1982, pp. 105–150; [Google Scholar]
- 11c. Koppenhagen V. B., Pfiffner J. J., J. Biol. Chem. 1970, 245, 5865–5867. [PubMed] [Google Scholar]
- 12. Gruber K., Puffer B., Kräutler B., Chem. Soc. Rev. 2011, 40, 4346–4363. [DOI] [PubMed] [Google Scholar]
- 13. Thomson A. J., J. Am. Chem. Soc. 1969, 91, 2780–2785. [Google Scholar]
- 14. Fischli A., Eschenmoser A., Angew. Chem. Int. Ed. Engl. 1967, 6, 866–868; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1967, 79, 865–867. [Google Scholar]
- 15. Edmond E. D., Hodgkin D. C., Helv. Chim. Acta 1975, 58, 641–654. [Google Scholar]
- 16. Scheidt W. R. in Handbook of Porphyrin Science Vol. 24 (Ed.: K. M. Kadish, K. M. Smith, R. Guilard), World Scientific, Singapore, 2012, pp. 1–179. [Google Scholar]
- 17. Ouyang L., Rulis P., Ching W. Y., Nardin G., Randaccio L., Inorg. Chem. 2004, 43, 1235–1241. [DOI] [PubMed] [Google Scholar]
- 18. Randaccio L., Furlan M., Geremia S., Slouf M., Srnova I., Toffoli D., Inorg. Chem. 2000, 39, 3403–3413. [DOI] [PubMed] [Google Scholar]
- 19. Kräutler B., Keller W., Kratky C., J. Am. Chem. Soc. 1989, 111, 8936–8938. [Google Scholar]
- 20. Maurice M. S. St., Mera P., Park K., Brunold T. C., Escalante-Semerena J. C., Rayment I., Biochemistry 2008, 47, 5755–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kräutler B., Keller W., Hughes M., Caderas C., Kratky C., J. Chem. Soc. Chem. Commun. 1987, 1678–1680. [Google Scholar]
- 22. Kräutler B., Konrat R., Stupperich E., Färber G., Gruber K., Kratky C., Inorg. Chem. 1994, 33, 4128–4139. [Google Scholar]
- 23. Buckel W., Golding B. T., Annu. Rev. Microbiol. 2006, 60, 27–49. [DOI] [PubMed] [Google Scholar]
- 24. Mosimann H., Kräutler B., Angew. Chem. Int. Ed. 2000, 39, 393–395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 417–419. [Google Scholar]
- 25.
- 25a. Zhang Q., van der Donk W. A., Liu W., Acc. Chem. Res. 2012, 45, 555; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Kräutler B., Puffer B. in Handbook of Porphyrin Science, Vol. 25 (Ed.: K. M. Kadish, K. M. Smith, R. Guilard), World Scientific, Singapore, 2012, pp. 133–265; [Google Scholar]
- 25c. Fujimori D. G., Curr. Opin. Chem. Biol. 2013, 17, 597–604; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25d. McLaughlin M. I., van der Donk W. A., Biochemistry 2018, 57, 4967–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matthews R. G., Acc. Chem. Res. 2001, 34, 681–689. [DOI] [PubMed] [Google Scholar]
- 27. Pallares I. G., Moore T. C., Escalante-Semerena J. C., Brunold T. C., Biochemistry 2014, 53, 7969–7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vallee B. L., Williams R. J., Proc. Natl. Acad. Sci. USA 1968, 59, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kratky C., Waditschatka R., Angst C., Johansen J. E., Plaquevent J. C., Schreiber J., Eschenmoser A., Helv. Chim. Acta 1985, 68, 1312–1337. [Google Scholar]
- 30. Brenig C., Prieto L., Oetterli R., Zelder F., Angew. Chem. Int. Ed. 2018, 57, 16308–16312; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16546–16550. [Google Scholar]
- 31.
- 31a. Kräutler B., Chem. Eur. J. 2015, 21, 11280–11287; [DOI] [PubMed] [Google Scholar]
- 31b. Zelder F., Sonnay M., Prieto L., ChemBioChem 2015, 16, 1264–1278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary