

Abstract
Aspartimide (Asi) formation is a notorious side reaction in peptide synthesis that is well characterized and described in literature. In this context, we observed significant amounts of chain termination in Fmoc‐SPPS while synthesizing the N‐terminal Xaa‐Asp‐Yaa motif. This termination was caused by the formation of piperazine‐2,5‐diones. We investigated this side reaction using a linear model peptide and independently synthesizing its piperazine‐2,5‐dione derivative. Nuclear magnetic resonance (NMR) data of the side product present in the crude linear peptide proves that exclusively the six‐membered ring is formed whereas the theoretically conceivable seven‐membered 1,4‐diazepine‐2,5‐dione is not found. We propose a mechanism where nucleophilic attack of the N‐terminal amino function takes place at the α‐carbon of the carbonyl group of the corresponding Asi intermediate.
In addition, we systematically investigated the impact of (a) different adjacent amino acid residues, (b) backbone protection, and (c) side chain protection of flanking amino acids. The side reaction is directly related to the Asi intermediate. Hence, hindering or avoiding Asi formation reduces or completely suppresses this side reaction.
Keywords: 1,4‐diazepine‐2,5‐diones; Asp‐Gly motif; Asp β‐carboxy protection; aspartimides; backbone protection; chain termination; diketodiazepines; diketopiperazines; Fmoc‐SPPS; piperazine‐2,5‐diones; truncated sequences; Xaa‐Asp‐Yaa motif
Abbreviations
- As recommended in J. Peptide Sci. 1999; 5: 465‐471, with the following additions and variations: Asi (according to Albericio [1])
Aspartimide
- Dmb
2,4‐dimethoxybenzyl
- DKD
1,4‐diazepine‐2,5‐diones (diketodiazepines)
- DKP
piperazine‐2,5‐diones (diketopiperazines)
- EDT
1,2‐ethane‐dithiol
- Hmb
2‐hydroxy‐4‐methoxy benzyl
- iPrOH
isopropanol
- LC‐MS
liquid chromatography coupled with mass spectrometry
- OMpe
3‐methylpent‐3‐yl ester
- PG
protecting group
- PS
polystyrene
- RT
retention time
- RRT
relative retention time
- J
coupling constant
1. INTRODUCTION
Aspartimide (Asi) formation is a notorious side reaction in peptide synthesis that is well characterized and described in literature.1, 2, 3, 4, 5, 6 In addition, nucleophilic ring opening of the Asi intermediate is extensively described in literature (eg, Subirós‐Funosas et al and Wade et al1, 7). The formation of cyclic by‐products in liquid phase peptide synthesis was previously observed and described. In this context, cyclo(‐Xaa‐Asp)‐Yaa peptides that can either be N‐terminal six‐membered piperazine‐2,5‐diones (DKP) or seven‐membered diazepine‐2,5‐diones (DKD) via Asi intermediates (Figure 1) were identified.8 Deamidation and/or Asi‐related synthetic pathways for Asp‐ and Asn containing peptides have been described.9, 10, 11
Figure 1.

N‐terminal six‐membered piperazine‐2,5‐dione (DKP) vs seven‐membered diazepine‐2,5‐dione (DKD)
We observed Fmoc‐SPPS chain termination at the N‐terminal Xaa‐Asp‐Yaa motif apparently caused by formation of cyclo(‐Xaa‐Asp)‐Yaa peptides. These peptides were detected and quantified by LC‐MS as minor components in crude material from SPPS after Trifluoroacetic acid (TFA) cleavage. In the context of SPPS such Asi‐related cyclizations, possibly leading to either DKDs and/or DKPs, are so far exclusively known for Asp β‐benzyl esters. To the best of our knowledge, there is no published study describing whether a six‐ or seven‐membered species or a mixture is formed and to what extent this side‐reaction occurs.12, 13
Although Electrospray ionization‐Mass spectrometry (ESI‐MS) studies of cyclic dipeptides are described in literature,14 we were not able to discriminate between the two possible cyclic structures (ie, six‐ vs seven‐membered ring) by MS. Therefore, the nature of this side reaction was not known when we initiated our investigations.4, 15 Obviously, cyclo(‐Xaa‐Asp)‐Yaa peptides are not amenable for further acylation. Hence, they are present as truncated sequences in the crude material of the SPPS. This is in contrast to the Asi formation at the Asp‐Yaa site, where chain elongation is readily possible.
We chose the model peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 for further investigation. Its DKP analog was independently synthesized, and extensive NMR studies were performed for structural elucidation.
In addition, we investigated the impact of flanking amino acid residues on the side reaction. Also, the impact of backbone and side chain protection is discussed.
2. RESULTS AND DISCUSSION
2.1. Chain termination at Xaa‐Asp‐Yaa motif
During Fmoc‐SPPS cyclo(‐Xaa‐Asp)‐Yaa, peptides may be formed by cyclization via nucleophilic attack of the free amino group of the Xaa residue at either the β‐carboxy group of Asp and/or the α‐ and β‐carbonyl groups of an Asi intermediate (if present) after deprotection of Fmoc‐Xaa‐peptide‐resin. Aspartimides are ubiquitous by‐products in peptides with the Asp‐Yaa motif synthesized via Fmoc‐SPPS. It is thus conceivable that both a seven‐membered DKD following pathways A or B in Scheme 1 and/or a six‐membered DKP following pathway C can be formed.13 However, the formation of a seven‐membered DKD was not observed in this study.
Scheme 1.
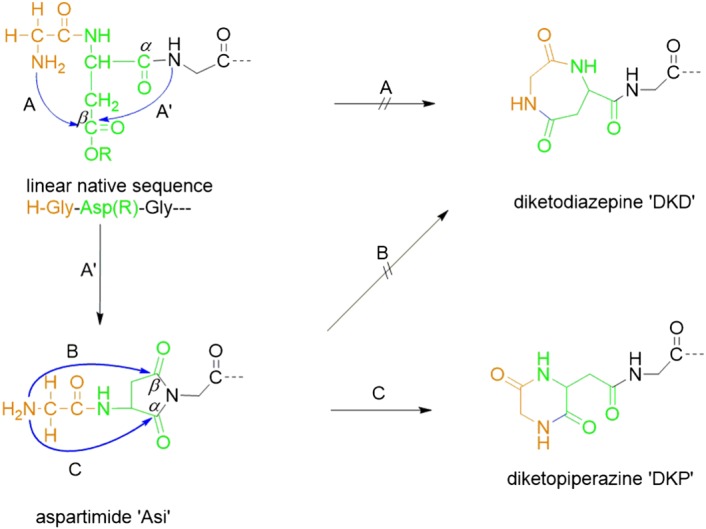
Xaa‐Asp‐Yaa motif: Xaa, Yaa = Gly for simplicity. R = Asp carboxylic acid protecting group. Alternative possible chain termination pathways A, B, and C for nucleophilic attack of N‐terminal amine at Asp‐carboxy or at carbonyl groups of Asi intermediate. A' for nucleophilic attack of secondary amine at Asp‐carboxy group. No formation of diazepine‐2,5‐diones (DKD) was observed in this study. Stereochemistry omitted for simplification.
2.2. Structure elucidation of the synthesized DKP reference compound
The DKP reference compound of the model peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 was synthesized. Therefore, the linear precursor H‐Gly‐β‐Asp (OAll)‐(Dmb)Gly‐Ala‐Lys (Boc)‐Phe‐NH‐ (Scheme 2) with a β‐Asp was synthesized and cyclized on the resin. The β‐Asp‐OAll protection in conjunction with Gly backbone blockage excludes Asi formation and thus allows for direct and straightforward formation of the six‐membered DKP. Allyl alkoxide is obviously an excellent leaving group. Hence, cyclization occurs without additional activation under mild basic conditions. Please note that the potential competing formation of the respective seven‐membered DKD is excluded by using this approach.
Scheme 2.

On‐resin formation of cyclo(‐Gly‐Asp)‐(Dmb)Gly‐Ala‐Lys (Boc)‐Phe‐NH‐. Subsequent TFA cleavage from the resin/deprotection of side chain and backbone protecting groups yielding the crude piperazine‐2,5‐diones (DKP) reference compound is not shown
The chemical structure of the DKP is shown in Figure 2. All 1H, 13C, and 15N NMR chemical shifts for the independently synthesized reference compound were unambiguously assigned via the corresponding 1D and 2D NMR experiments (a detailed NMR resonance assignment with all correlations observed is summarized in the Supporting Information). On the basis of the observed spin systems, the assignment of resonances to the individual amino acids Phe,6 Lys,5 Ala,4 and Gly3 towards the C‐terminus was straightforward. With the aid of crucial 1H‐13C HMBC correlations to the amide carbons and of cross signals of neighboring amide protons in the ROESY NMR spectra, the linkages between the four amino acid residues (Gly‐Ala‐Lys‐Phe) were clearly identified. In the NMR spectra of the linear peptide sequence, all chemical shifts identified for these four amino acid residues are almost identical to the chemical shifts evaluated for the DKP product (see data in the Supporting Information). For the linear peptide, the NMR data of the N‐terminal acetylated Gly1 and the Asp2 residue were unambiguously assigned as well. In the case, of the purified DKP reference material NMR resonances assignable to three amide, two methylene, and one methine groups were identified, all groups belonging to the originally present Gly1 and Asp2 residues. Apparently, both signals of the acetyl protecting group originally identified for the linear peptide were absent. In addition, it was clear from the DQF‐COSY NMR spectrum that the detected methine group was directly bound to one of the methylene groups, whereas the remaining methylene group was present solitary.
Figure 2.
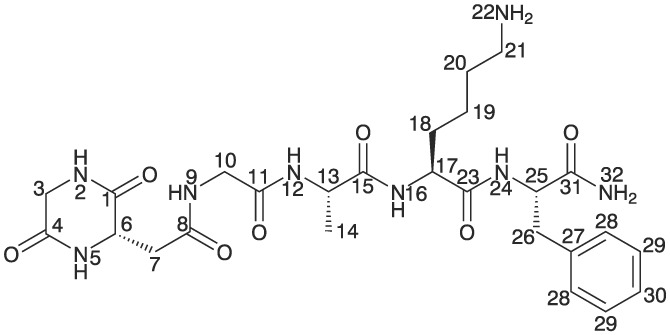
Chemical structure of the piperazine‐2,5‐diones (DKP) with assignments of positions used for 1H, 13C, and 15N chemical shifts
From the 2D NMR experiments, the chemical structure including the six‐membered C‐terminal ring shown in Figure 2 was unambiguously identified. The main characteristics of a six‐membered cycle is obtained from the 1H‐13C long range correlations of amide protons (Figure 3, where the correlations of H‐2 and H‐5 to carbons C‐1 and C‐4 confirm the presence of this cycle. For a seven‐membered ring (such as described in Süli‐Vargha et al13), the correlation of H‐5 to C‐1 would not be observed. Although the long range correlation of H‐9 to C‐7 is rather weak, it is a further proof that the methylene group C7 must be exocyclic (for a full list of long range correlations, see the Supporting Information). All long range correlations of NH protons to the directly bound amide carbons are usually strong (eg, prominent cross signals of H‐12 → C‐11, Figure 3), but only weak correlations were observed for H‐2 → C‐1 or H‐5 → C‐4, respectively. This could be associated with the six‐membered ring structure (ring strain). Consistently with the identification of a similar DKP ring system, no (or only a weak) HMBC correlation from an endocyclic amide proton onto the directly bound amide carbon was reported16 (see Supporting Information). All additional 1H‐13C heteronuclear multiple bond correlation (HMBC) correlations of protons of interest to amide carbons further confirm the formation of a cyclic product of residues Gly1 and Asp2 (H‐3 → C‐1, C‐4; H‐6 → C‐1, C‐4, C‐8; H‐7 → C‐1, C‐8; H‐10 → C‐8, C‐11, see the Supporting Information).
Figure 3.

Region showing correlations of amide protons to amide carbons of the piperazine‐2,5‐diones (DKP) reference with assignment of crucial 1H‐13C HMBC NMR correlations from H‐2 and H‐5 to C‐1,4 (spectrum recorded with selection of 4 Hz H‐C coupling constant)
The formation of the six‐membered ring of the DKP was further confirmed by correlations observed in the 1H‐15N HMBC NMR experiments. From the 1H and 13C NMR assignments, the resonances attributed to Gly,3 Ala,4 Lys,5 and Phe6 were already known. Subsequently, the amide nitrogen atoms were assigned (positions 9, 12, 16, 22, 24, and 32, see Figure 2), and, therefore, the two remaining nitrogen atoms must belong to positions 2 and 5. The correlations found for methylene protons H‐3 to nitrogens N‐2 and N‐5 indicate the presence of a cycle. As highlighted by circles in Figure 4, the exocyclic protons H‐7 correlate to N‐5 of the cycle and to N‐9 of residue Gly.3 In aqueous solution, additionally, the correlations of the methine proton H‐6 to exactly the same nitrogens N‐2 and N‐5 as for H‐3 of the solitaire methylene group were observed (see the Supporting Information). This is a further proof for the presence of the six‐membered cycle.
Figure 4.

Regions of interest with assignment of crucial 1H‐15N HMBC NMR correlations for the piperazine‐2,5‐diones (DKP) reference. The spectra were recorded in DMF‐d7 with selection of 4 Hz (left) and 2 Hz (right) long‐range couplings
The most prominent by‐product in the crude product mixture of the model peptide has been identified by LC‐MS (M‐60, m/z = 575.29 u, approximately 4 % peak area in HPLC, see Figure 5). Minor cleavage related impurities (<1% area by HPLC) were identified but are disregarded in this context.
Figure 5.

HPLC UV trace overlay of crude Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 (blue trace, linear target retention time [RT] 7.85 min) and same crude spiked with piperazine‐2,5‐diones (DKP) reference (black trace, RT 7.30 min). Peak assignments by liquid chromatography coupled with mass spectrometry (LC‐MS) (mass relative to target M)
HPLC coelution with the independently synthesized and purified DKP reference compound with the corresponding truncated sequence (see black HPLC trace overlay in Figure 5) indicates that chain termination occurs via pathway C (Scheme 1). Unfortunately, the attempts to synthesize the seven‐membered DKD were not successful due to dominating intermolecular reactions instead of the intended intramolecular cyclization (see, eg, Moure et al17). We synthesized the structure described in the literature13 as a seven‐membered DKD and could show by NMR experiments that the six‐membered DKP structure is formed (data shown in the supporting information).
Most of the NMR resonances identified for the linear peptide sequence and for the synthesized DKP product are almost identical (in the 2D NMR spectra as well), of course with exception of the N‐terminal regions of the sequences. Figure 6 shows an overlay of 1H‐NMR spectra of the crude native peptide and the synthesized six‐ring DKP reference compound. The signals of the reference compound are also observed in the spectra of the crude native peptide.
Figure 6.

1H‐NMR of (a) crude native peptide and (b) six‐ring peptide in DMF with signals of the six‐membered ring peptide by‐product indicated in the spectrum of the crude native peptide
In Figure 7, enlarged regions of the HSQC NMR spectra of the crude native peptide and of the purified DKP reference material are shown. It can be clearly observed (Figure 7A), that cross peaks assignable to positions 3, 6, and 7 of the DKP are also present in the spectrum of the crude native peptide. Additionally, slightly less split cross peaks are observed for positions 10, 13, and 17 assigned to residues Gly,3 Ala,4 and Lys,5 respectively. Further evidence for the presence of the DKP by‐product in the crude native peptide are the correlation signals somewhat outstanding of the main resonances detectable in the HMBC and DQF‐COSY NMR spectra appearing at exactly the same positions as in the spectra recorded for the independently synthesized DKP reference (see Figure 8). Signal integration with respect to the 1H NMR resonance of H‐25 at 4.58 ppm resulted in ≈4 to 5 mol‐% of the DKP by‐product in the crude peptide mixture, which is well aligned with the analytical HPLC peak area relative to main peak of 4%. A similar amount was determined from the 1H NMR data recorded in aqueous solution (integrated 1H NMR spectra shown in Supporting Information).
Figure 7.
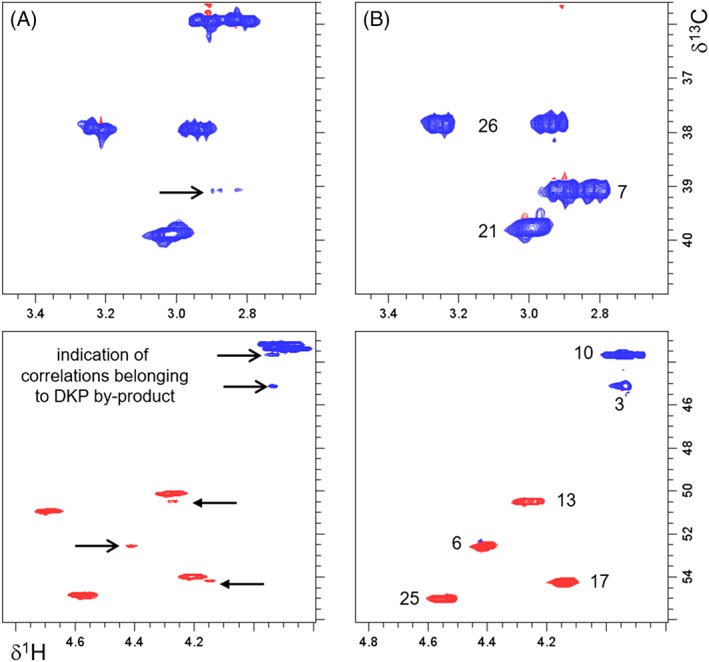
Enlarged regions of 1H‐13C HSQC NMR spectra recorded in DMF‐d7 of (a) crude native peptide with indication of residual resonances of the piperazine‐2,5‐diones (DKP) by‐product and (b) synthesized purified DKP product with assignment of correlations (chemical structure with atom numbering shown in Figure 2)
Figure 8.

Enlarged regions of 1H‐13C HMBC and 1H‐1H DQF‐COSY NMR spectra recorded in DMF‐d7 of (a) crude native peptide with indication of residual resonances assignable to the piperazine‐2,5‐diones (DKP) by‐product and (b) synthesized purified DKP reference
It is noteworthy that we did not observe any additional sets of resonances in the NMR spectra of the crude peptide mixture showing noticeable signal intensities (Limit of quantitation (LOQ) at approximately 1%), which could be assigned to a species with a terminal seven‐membered ring.
Following the identification of the six‐membered ring by‐product in the crude mixture of the linear peptide, we made efforts to verify the presence of this species already on‐resin prior to cleavage from the resin. In Figure 9B, a section of the HSQC HR‐MAS NMR spectrum of the resin bound peptide is shown. It is evident that not all observed correlations are detected at exactly the same 1H and 13C chemical shifts as in the spectra of the crude linear peptide (Figure 9C). The DMF swollen on‐resin species (a) are still protected by t‐butoxy groups at residues Asp2 and Lys5 (effects mainly on positions 7 and 21), (b) the peptide is still linked to the tricyclic amide linker resin (influence mainly on position 25), and (c) the presence of the polystyrene back‐bone additionally can slightly modify chemical shifts. All resonances unambiguously assignable to specific positions of the linear peptide (see Supporting Information) are denoted in Figure 9, and the correlations of the six‐membered ring by‐product are marked by arrows. In the spectral regions shown in Figure 9, an additional correlation in this region of interest (marked by circles, Figure 9A,B) could be attributed to the tricyclic amide linker resin. In summary, it is clear from our HR‐MAS NMR investigation, that the formation of the six‐ring by‐product already takes place on‐resin and that the by‐product formation is not a consequence of the reaction conditions during the cleavage of the peptide from the solid support or of the deprotection step.
Figure 9.
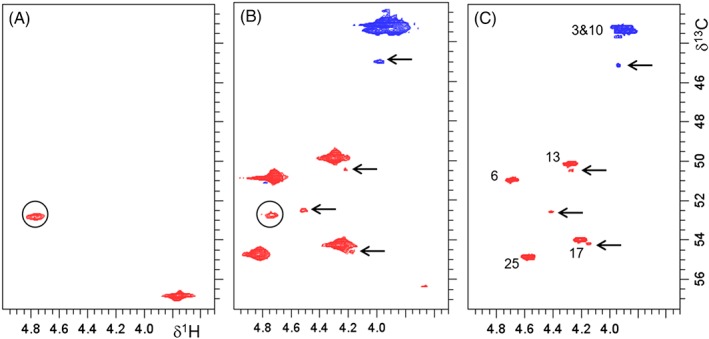
Enlarged regions of interest of1H‐13C HR‐MAS HSQC NMR spectra recorded in DMF‐d7 of (a) tricyclic amide linker, (b) on‐resin peptide, and (c) crude peptide with assignment of resonances of the main product. Resonances assigned to the six‐ring product in (b) and (c) are labeled by arrows, resonances assignable to the tricyclic amide linker resin in the HR‐MAS NMR spectra are marked with circles. The additional correlation at 3.76/56.8 ppm in (a) is assigned to the α amide position of phenylalanine
Although we have not proven that solely the DKP is formed in peptides other than our model peptide, the fact that both NMR data and coelution of the reference compound only show the DKP in case of our model peptide, we conclude that this Asi‐related side reaction leads to N‐terminal DKP.
After 5 years of storage of the peptide resin from this experiment, the same amount of DKP was found by analytical HPLC. Hence, it can be excluded that the modification occurs on the peptide‐resin after the SPPS is completed (data not shown).
2.3. Influence of the flanking residues Xaa and Yaa in the Ac‐Xaa‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 sequence
It is well‐known that side reactions such as Asi formation are highly dependent on the peptide sequence3, 6, 18 and on the Asp β ‐carboxy protecting group.1, 2, 4, 6 The peptide sequence Ac‐Xaa‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 has been defined to further investigate the influence of different parameters such as the flanking amino acid residues Xaa and Yaa, the Fmoc cleavage conditions, and the Asp carboxy protecting group on the level of side reaction.
To investigate the impact of the neighboring amino acids in the peptide chain, two sets of peptides each consisting of 20 peptides (general sequence Ac‐Xaa‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2) were synthesized. One set used fixed Xaa = Gly and Yaa = 20 common amino acids (Figure 10), and the second set used Xaa = 20 common amino acids and fixed Yaa = Gly (Figure 11). In all cases, we found a similar impurity profile with the DKP by‐product eluting at relative retention time (RRT) 0.9 and the Asi by‐product eluting at RRT 1.1 in the analytical HPLC system. The detected amounts of DKP (blue bars) and the corresponding Asi (red bars) are depicted relative to linear target. The results indicate that the DKP by‐products are formed via Asi intermediates. The detected sum of these two may be an indicator of the sequence‐specific susceptibility to this side reaction whereas the individual levels of Asi and DKP could also depend on process variables such as, eg, basic and acidic conditions,7, 8 temperatures, and water content in organic media. In some crude products, we found substantial amounts of the according β‐Asp peptides. It is noteworthy that nucleophilic ring opening of Asi also occurs by hydrolysis leading to linear α‐/β‐Asp peptides.4, 7 The latter is a competing reaction to the herein discussed DKP formation and diminishes the observable Asi level to a certain extent.
Figure 10.
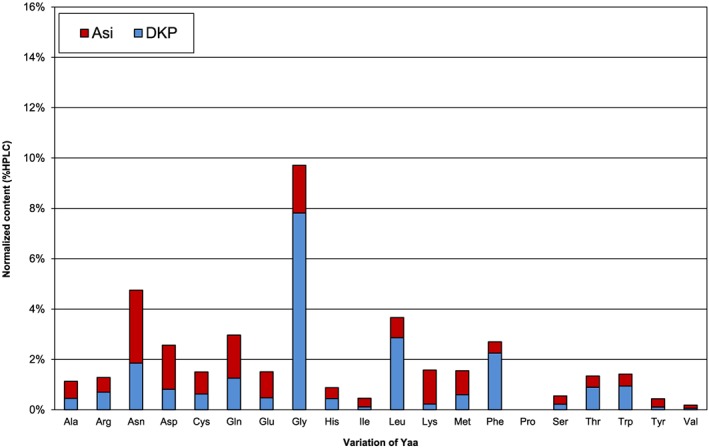
Amount of by‐products with variation of Yaa (sequence = Ac‐Gly‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2) relative to linear target
Figure 11.

Amount of by‐products with variation of Xaa (sequence = Ac‐Xaa‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2) relative to linear target
In the Xaa = Gly series, chain termination is most prominent with Yaa = Gly (8% of cyclo(‐Gly‐Asp) peptide detected, see Figure 10). Yaa = Gly is especially prone to Asi formation.4, 7 As expected for N‐alkyl amino acids bearing no acidic backbone amide proton, no Asi by‐products were found for Yaa = Pro or when Yaa = Gly was incorporated as backbone‐protected Fmoc‐(Dmb)Gly‐OH (data not shown4, 19). Consequently, no cyclo(‐Gly‐Asp) peptides were observed in these cases, and it is obvious that chain termination via pathway A in Scheme 1 can be excluded. When incorporating pseudo proline dipeptides in Yaa postion, the same effect as for Pro is achieved, and Asi‐related chain termination is avoided.20, 21, 22
The set of experiments with Yaa = Gly is the “worst case motif” for Asi formation. Highest levels of DKP were detected for Xaa = Gly, Asp, Ser, and Ala (blue bars in Figure 11). Additionally, the corresponding Asi (red bars in Figure 11) was detected in amounts up to 13% depending on Xaa.
Hence, not only adjacent Yaa but also the residue Xaa has a certain contribution to the observed level of Asi at the Xaa‐Asp‐Yaa motif and consequently of N‐terminal DKP. A similar observation was previously described for DKD formation from Asp β‐benzyl esters.13
2.4. Influence of Asp‐carboxy protecting groups and Fmoc deprotection conditions
The peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 was synthesized using different Asp protecting groups (β‐PG: [OtBu] vs [OMpe]; α‐PG: ‐OtBu) and different reaction times for Fmoc removal.
It is noticeable that the formation of DKP and Asi is increasing linearly with increasing Fmoc deprotection time. The effect is shown in Table 1.
Table 1.
Asp protecting group (PG) and Fmoc cleavage duration variations
| % Area HPLC | |||||
|---|---|---|---|---|---|
| Fmoc cleavage duration/min | PG | DKP | Linear target sequence | Asi | Sum of linear and cyclic products |
| 1 × 10' | α‐OtBu | 28.0 | 31.8 | 23.9 | 83.7 |
| 1 × 10' | β‐(OtBu) | 2.3 | 81.6 | 1.4 | 85.3 |
| 1 × 10' | β‐(OMpe) | 0.8 | 83.9 | 0.7 | 85.4 |
| 2 × 10' | α‐OtBu | 51.0 | 14.8 | 17.4 | 83.2 |
| 2 × 10' | β‐(OtBu) | 5.4 | 77.4 | 1.9 | 84.7 |
| 2 × 10' | β‐(OMpe) | 1.6 | 82.4 | 0.7 | 84.7 |
| 3 × 10' | α‐OtBu | 69.3 | 3.8 | 9.4 | 82.5 |
| 3 × 10' | β‐(OtBu) | 9.0 | 74.6 | 2.3 | 85.9 |
| 3 × 10' | β‐(OMpe) | 2.7 | 81.8 | 0.8 | 85.3 |
| 4 × 10' | α‐OtBu | 77.1 | 1.0 | 3.0 | 81.1 |
| 4 × 10' | β‐(OtBu) | 14.3 | 67.8 | 2.6 | 84.7 |
| 4 × 10' | β‐(OMpe) | 4.1 | 80.9 | 0.9 | 85.9 |
Compared with OtBu, OMpe23 considerably reduces formation of Asi2 and consequently of DKP. In the series, the extent of cyclization is β‐PG = (OAll) > (OtBu) > (OMpe) (ie, coupling of α‐carboxy of Asp) and even remarkably higher with α‐OtBu or α‐OAll protection (ie, coupling of β‐carboxy of Asp). A similar trend is also known for Asi formation.2, 4, 24 Recently, the OMpe protecting group was surpassed by β‐tri‐alkylmethyl esters (alkyl = ethyl/propyl/butyl) for Asi suppression.25 Longer Fmoc cleavage reaction times lead to increasing levels of Asi or of DKP, respectively. Approximately constant levels of the sums of cyclic side products and linear target are observed, which is aligned with the proposal that DKP is formed via ring opening of the Asi intermediates (see Table 1).
To investigate the role of water content in 20% piperidine/DMF mixtures, two control experiments were performed. Two samples of the peptide resin Fmoc‐Asp (OtBu)‐Gly‐Ala‐Lys (Boc)‐Phe‐NH‐ were treated separately with 20% piperidine/DMF solutions containing 0.7% and 5.3% water, respectively. After Fmoc‐Gly‐OH coupling a second, Fmoc removal with the same respective water contents in 20% piperidine/DMF followed. The % area of DKP and Asi by‐products in both crude samples after final acetylation and cleavage from resin is given in Table 2. It is noticeable that higher water content leads to formation of more cyclic by‐products.
Table 2.
Influence of water content during Fmoc cleavage (2 × 15 min). (Experiments were performed independently of results shown in Figure 9 and Figure 10 resulting in slightly differing results for piperazine‐2,5‐diones [DKP] and Asi content)
| % Area HPLC | ||||
|---|---|---|---|---|
| Water content in pip/DMF (%) | DKP | Linear target sequence | Asi | Sum of linear and cyclic products |
| 0.7 ± 0.2 | 5.9 | 89.3 | 1.2 | 96.4 |
| 5.3 ± 0.4 | 13.2 | 80.0 | 1.8 | 95.0 |
A control experiment was performed applying varying conditions for N‐terminal Ac‐Gly incorporation and with different Asp protecting groups. Ac‐Gly‐OH instead of Fmoc‐Gly‐OH followed by Fmoc removal and final acetylation was coupled onto H‐Asp (OtBu)‐Gly‐Ala‐Lys (Boc)‐Phe‐NH‐ with and without final 20% piperidine/DMF treatment in order to simulate high pH stress due to final Fmoc deprotection.
As expected, almost no DKP is formed in case of coupling Ac‐Gly‐OH. However, Asi is found, especially when treating the peptide after coupling of Ac‐Gly‐OH with 20% piperidine/DMF in order to simulate proceeding SPPS (Table 3). This finding supports the formation pathway of DKP via the Asi intermediates as the N‐terminal nucleophilic attack at Asi is impossible by using the Ac‐Gly building block. It also shows that DKP is formed after Fmoc deprotecting following the Xaa coupling step, whereas Asi is formed throughout all SPPS cycles following Asp coupling.
Table 3.
Coupling of Ac‐Gly‐OH building block and Asp protecting group (PG) variations
| % area HPLC | |||||
|---|---|---|---|---|---|
| Conditions | PG | DKP | Target | Asi | Sum of linear and cyclic products |
| Fmoc‐Gly‐OH coupling + piperidine treatment + acetylation | α‐OtBu | 58.9 | 5.4 | 14.4 | 78.7 |
| β‐(OtBu) | 7.1 | 71.6 | 7.7 | 86.4 | |
| β‐(OMpe) | 1.9 | 79.1 | 7.2 | 88.2 | |
| Ac‐Gly‐OH coupling | α‐OtBu | 0.6 | 55.8 | 23.2 | 79.6 |
| β‐(OtBu) | 0.6 | 80.4 | 8.8 | 89.8 | |
| β‐(OMpe) | 0.6 | 82.6 | 7.9 | 91.1 | |
| Ac‐Gly coupling + piperidine treatment | α‐OtBu | 0.3 | 2.0 | 42.2 | 44.5 |
| β‐(OtBu) | 0.4 | 47.4 | 26.3 | 74.1 | |
| β‐(OMpe) | 0.4 | 69.1 | 10.1 | 79.6 | |
3. CONCLUSION
Chain termination during Fmoc‐SPPS at the Asi‐prone Xaa‐Asp‐Yaa motif is described. NMR data of the model peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 peptide and its DKP analog clearly indicate that the by‐product is an N‐terminal DKP. Hence, we conclude that the truncated DKP is formed via nucleophilic attack of the N‐terminal Xaa amino function at the α‐carbonyl of the Asi intermediate. The extent of this side reaction depends on (a) the peptide sequence (with Glycine as adjacent residues as a worst case, see Figure 10 and Figure 11), (b) the Asp side chain protecting group, and (c) Fmoc deprotection duration (Table 1). Hold times under basic conditions (Fmoc cleavage), temperature, and water content in organic media used (data not shown) have been found to promote Asi and consequently N‐terminal DKP formation.
In conclusion, we found that chain termination can be overcome by reducing Asi formation eg, by shortened Fmoc deprotection duration, acidic modifiers,26 and/or modifications of the Asp building block. Three strategies to reduce Asi formation during SPPS are the usage of the OMpe protecting group for Asp, the usage of a backbone amide protecting group27 (Figure 12), and the usage of pseudoprolines.
Figure 12.

Xaa‐1‐Xaa‐Asp‐Yaa‐Zaa motif (Xaa, Yaa = Gly for simplicity). Usage of protecting groups (R2 and R3) as well as pseudoprolines during SPPS can reduce or avoid aspartimide (Asi)‐related chain termination
4. MATERIALS AND METHODS
4.1. General automated SPPS
All peptides, unless described otherwise, were synthesized on tricyclic amide linker resin (150 μmol scale, substitution B0 = 0.65 mmol/g) using an automated peptide synthesizer (Symphony from Protein Technology Inc.). All reactions were carried out at room temperature. Couplings were performed with Fmoc protected amino acid derivatives or AcOH (2 eq., 300 μmol) and TBTU/DIPEA (1.8 eq., 275 μmol/3 eq., 450 μmol) in DMF (5 mL). All derivatives and reagents were obtained from Bachem AG, Bubendorf, Switzerland. Fmoc was removed using 20 % (v/v) piperidine in DMF (2 × 15 min, 2 × 3.75 mL), and unless stated differently, Asp was introduced as Fmoc‐Asp (OtBu)‐OH. Crude peptides were obtained after cleavage with TFA/water/EDT 92:5:3 (v/v) and analyzed by Ultra High Performance Liquid Chromatography (UHPLC) (Waters Acquity C18, 1.7 μm; linear gradient of Acetonitrile (ACN) in 0.1% TFA; flow 0.4 mL/min, λ = 220 nm). All LC‐MS spectra were acquired in positive ion mode with a Bruker microTOF ESI‐MS mass spectrometer coupled to a Dionex Ultimate 3000 UHPLC system. Peak assignment was performed using LC‐MS analysis of the crude peptides (see Supporting Information). Aspartimide side product peaks were assigned by their mass −18 μ relative to the linear target sequence (less intense isobaric peaks, if present, and piperidides were disregarded for simplicity).
4.2. Manual SPPS
The Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 peptides were synthesized at a 150 μmol scale using 20 % (v/v) piperidine in DMF (2 × 15 min) with a varying water content of 0.7% and 5.3% for Fmoc removal starting from Fmoc‐Asp (OtBu)‐Gly‐Ala‐Lys (Boc)‐Phe‐NH‐. Fmoc‐Gly‐OH (2 eq., 300 μmol) coupling was performed with Oxyma/DIC (3.1 eq., 465 μmol/2.7 eq., 405 μmol). Remaining conditions matched the general automated SPPS.
4.3. Synthesis of Cyclo(‐Gly‐Asp)‐Gly‐Ala‐Lys‐Phe‐NH2 trifluoroacetate salt (DKP) reference compound
Fmoc‐Gly‐β‐Asp (OAll)‐(Dmb)Gly‐Ala‐Lys (Boc)‐Phe‐NH‐ was synthesized on Xanthenyl linker resin (7.5 mmol scale, substitution B0 = 0.40 mmol/g) using an automated peptide synthesizer (PTI Sonata). Couplings were performed with amino acid derivative (except Fmoc‐Asp‐OAll) (2 eq., 15 mmol) and TBTU/DIPEA (1.9 eq., 14 mmol/3 eq., 23 mmol) in DMF. For Fmoc‐Asp‐OAll, HATU was used instead of TBTU; 24 g of peptide resin were obtained.
An aliquot of Fmoc‐Gly‐Asp‐OAll‐(Dmb)Gly‐Ala‐Lys (Boc)‐Phe‐NH‐ (10.0 g, approximately 3 mmol) was swollen in DMF (3 × 100 mL). Fmoc was cleaved with 20% piperidine in DMF for a total of 30 minutes. After washing with DMF and iPrOH (100 mL each), DIPEA was added dropwise until a basic pH value was obtained (estimated using wetted pH indicator paper). The mixture was gently agitated for 60 hours. The resin was washed with DMF and iPrOH (100 mL each) and dried under vacuum to obtain 9.0 g TFA cleavage and precipitation yielded the crude DKP with 87% purity (UHPLC, Waters Acquity C18, 1.7 μm; linear gradient of ACN in 0.1% TFA; flow 0.4 mL/min, λ = 220 nm), which was subsequently purified using preparative RP‐HPLC on C18 and a gradient of ACN in 0.1% (v/v) TFA. The purified TFA salt was obtained after lyophilization with 99% HPLC purity (0.53 g overall gross yield, TFA content by HPLC 16.4%, net peptide content estimated by elemental analysis 77.4% [N theory 19.50%; N found 15.09%], 0.7 mmol, approximately 24% yield). ESI‐MS: [M+H]+ found 575.29 μ, theory 575.29 μ.
4.4. Varying flanking residues Xaa and Yaa
The peptide sequence Ac‐Xaa‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 has been used to investigate the impact of the neighboring amino acids in the peptide chain, two sets of peptides each consisting of 20 peptides (general sequence Ac‐Xaa‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2) were synthesized. One set used fixed Xaa = Gly and Yaa = 20 common amino acids, and the second set used Xaa = 20 common amino acids and fixed Yaa = Gly (see Figure 10 and Figure 11 in the Results and Discussion section). DKP and Asi side products in peptide crudes were identified via LC‐MS and corresponding content determined with HPLC (see data in the Supporting Information).
4.5. NMR experiments
1H, 13C, and 15N NMR spectra were recorded at 400.1, 100.6, and 40.5 MHz on a Bruker Avance 400 NMR spectrometer (Bruker Biospin AG, Fällanden, Switzerland). The 1D 1H and 13C NMR spectra, and the 1H‐13C HSQC, 1H‐13C HMBC, 1H‐15N HSQC, 1H‐15N HMBC, 1H‐1H DQF‐COSY, TOCSY, and ROESY 2D correlation NMR experiments were performed at 298 K on a 5‐mm BBO CryoProbe Prodigy equipped with z‐gradient applying 90° pulse lengths of 11.4 (1H), 10.0 (13C), and 13.5 μs (15N). The NMR experiments of the crude peptide and of the purified DKP reference compound were recorded in DMF‐d7, D2O, or H2O/D2O (9/1) solutions using Bruker standard pulse programs and parameter sets selecting coupling constants of 145 Hz (HSQC), 8 or 4 Hz (1H‐13C HMBC), and 95, 4, or 2 Hz, respectively (1H‐15N HSQC and HMBC). Mixing times of 120 and 250 ms were applied in the TOCSY and ROESY NMR experiments, respectively. On‐resin material was investigated using a HR‐MAS NMR probe applying spinning rates of 4000 Hz. To remove excess solvents resins were washed five times with CDCl3 and one time with DMF‐d7. The slightly dried resins (approximately 20 mg) were filled into 4 mm zirconia rotors (active volume of 50 μL) and swollen in DMF‐d7. The 1H and 13C chemical shifts were referenced to the remaining resonances of the solvent DMF at 2.75 and 29.76 ppm or externally referenced relative to the signals of 3‐trimethylsilyl 2,2,3,3‐tetradeutero sodium propionate dissolved in D2O at −0.09 and −1.7 ppm, respectively. The 15N NMR spectra were referenced to an external sample of nitromethane at 0.0 ppm. Coupling constants J are reported, in Hz and for 1H NMR data, coupling patterns are described as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. For 13C NMR data s = quaternary carbon, d = CH, t = CH2, q = CH3 and w = weak HMBC correlation.
Supporting information
Table S1.1 Identities of relevant peaks identified by LC‐MS of Ac‐Xaa‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 and Ac‐Gly‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 flanking residue variations.
Figure S2.1 HMBC correlation used for the identification of a diketopiperazine sub‐structure [Ref. 16, main text].
Figure S3.1 Chemical structure of the purified DKP by‐product with assignments of positions used for 1H, 13C and 15N chemical shifts.
Figure S3.2 Chemical structure of the native peptide with assignments of positions used for 1H and 13C chemical shifts.
Figure S3.3 Chemical structure of the resin bound linear protected peptide with assignments of positions used for 1H and 13C chemical shifts.
Figure S3.4 Chemical structure of resin bound phenylalanine with assignments of positions used for 1H and 13C chemical shifts. This compound was synthesized for the identification of 1H/13C chemical shifts of the solid support.
Figure S4.1 Enlarged regions of 1H NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.2 Enlarged regions of 13C{1H} NMR spectra with peak picking of the DKP reference recorded in DMF‐d7.
Figure S4.3 1H‐13C HSQC NMR spectrum of DKP reference with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in DMF‐d7.
Figure S4.4 1H‐13C HMBC NMR spectra selecting 8 Hz (top) or 4 Hz (bottom) H‐C couplings of the DKP reference recorded in DMF‐d7.
Figure S4.5 1H‐1H DQF‐COSY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.6 1H‐1H TOCSY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S 4.7 1H‐1H ROESY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.8 1H‐15N HSQC NMR spectrum with assignment of positions for the DKP reference (DMF‐d7). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.9 1H‐15N HMBC NMR spectra selecting 4 Hz (top) or 2 Hz (bottom) H‐N couplings of the DKP reference (DMF‐d7).
Figure S4.10 1H NMR spectra with integrals of the DKP reference recorded in D2O (top) and in H2O/D2O 9/1 (bottom, spectrum recorded with suppression of water resonance).
Figure S4.11 Enlarged regions of 13C{1H} NMR spectra with peak picking of the DKP reference recorded in D2O.
Figure S4.12 1H‐13C HSQC NMR spectrum of the DKP reference with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in D2O.
Figure S4.13 1H‐13C HMBC NMR spectrum of the DKP reference recorded in D2O (top) and in H2O/D2O 9/1 (bottom).
Figure S4.14 1H‐1H DQF‐COSY NMR spectrum of the DKP reference recorded in D2O.
Figure S4.15 1H‐15N HMQC NMR spectrum with assignment of positions for the DKP reference (H2O/D2O 9/1). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.16 1H‐15N HMBC NMR spectra selecting 4 Hz (top) or 2 Hz (bottom) H‐N couplings of the DKP reference (D2O).
Figure S4.17 Region of interest with assignment of crucial 1H‐15N HMBC NMR correlations for the DKP reference (recorded in D2O, selection of 2 Hz coupling constant).
Figure S4.18 1H NMR spectra with integrals of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7. Below the integration of selected resonances for the quantification of the DPK by‐product is shown.
Figure S4.19 Enlarged regions of 13C{1H} NMR spectra with peak picking of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.20 1H‐13C HSQC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in DMF‐d7.
Figure S4.21 1H‐13C HMBC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.22 1H‐1H DQF‐COSY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.23 1H‐1H TOCSY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.24 1H‐1H ROESY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.25 1H‐15N HSQC NMR spectrum with assignment of positions of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 (DMF‐d7). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.26 1H NMR spectra with integrals of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in D2O. Below the integration of selected resonances for the quantitative determination of the DPK by‐product is shown.
Figure S4.27 Enlarged regions of 13C{1H} NMR spectra with peak picking of the native peptide Ac‐GDGAKF‐NH2 recorded in D2O.
Figure S4.28 1H‐13C HSQC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in D2O.
Figure S4.29 1H‐13C HMBC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in D2O.
Figure S4.30 1H HR‐MAS NMR spectrum of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.31 13C{1H} HR‐MAS NMR spectrum of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.32 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.33 1H‐13C HMBC HR‐MAS NMR spectrum and enlarged regions of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.34 1H HR‐MAS NMR spectrum of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.35 13C{1H} HR‐MAS NMR spectrum of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.36 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.36 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S5.1 Chemical structure of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 with assignments of positions used for 1 H and 13 C chemical shifts
Figure S5.2 Enlarged regions of 1 H NMR spectrum with integrals and resonance assignments of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.3 Enlarged regions of 13 C{1H} NMR spectrum with peak picking and resonance assignments of cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.4 1 H‐ 13 C HSQC NMR spectrum with resonance assignments of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 with spectral editing (CH and CH 3 groups in red, CH 2 in blue) recorded in DMSO‐d 6 .
Figure S5.5 1 H‐ 13 C HMBC NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.6 1 H‐ 1 H DQF‐COSY NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.7 Enlarged regions with crucial correlations of a) 1 H‐ 1 H DQF‐COSY and b‐d) 1 H‐ 13 C HMBC NMR spectra for the assignments of the aspartimide part of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.8 Chemical structure of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 with assignments of positions used for 1 H and 13 C chemical shifts
Figure S5.9 Enlarged regions of 1 H NMR spectrum with integrals and resonance assignments of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.10 Enlarged regions of 13 C{1H} NMR spectrum with peak picking and resonance assignments of cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.11 1 H‐ 13 C HSQC NMR spectrum with resonance assignments of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 with spectral editing (CH and CH 3 groups in red, CH 2 in blue) recorded in DMSO‐d 6 .
Figure S5.12 1 H‐ 13 C HMBC NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.13 1 H‐ 1 H DQF‐COSY NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.14 Enlarged regions with crucial correlations of a) 1 H‐ 1 H DQF‐COSY and b‐d) 1 H‐ 13 C HMBC NMR spectra for the assignments of the aspartimide part of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Table S6.1 Product compositions determined by HPLC of experiments with Ac‐Xaa‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 and Ac‐Gly‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 flanking residue variations.
Samson D, Rentsch D, Minuth M, Meier T, Loidl G. The aspartimide problem persists: Fluorenylmethyloxycarbonyl‐solid‐phase peptide synthesis (Fmoc‐SPPS) chain termination due to formation of N‐terminal piperazine‐2,5‐diones. J Pep Sci. 2019;25:e3193 10.1002/psc.3193
A preliminary account of this work was presented. D. Samson and G. Loidl, A novel side reaction in Fmoc‐SPPS: Formation of cyclo(‐Xaa‐Asp)‐Yaa peptides. Proceedings of the 22nd American Peptide Symposium, San Diego 2011 58‐59 (2012).
Footnotes
Our attempts to synthesize the DKD analog were not successful and mainly unreacted linear peptide or intermolecular oligomers were obtained, data not shown.
No potential m/z signal for the analogous DKD was found.
REFERENCES
- 1. Subirós‐Funosas R, El‐Faham A, Albericio F. Aspartimide formation in peptide chemistry: occurrence, prevention strategies and the role of N‐hydroxylamines . Tetrahedron. 2011;67(45):8595‐8606. 10.1016/j.tet.2011.08.046 [DOI] [Google Scholar]
- 2. Mergler M, Dick F. The aspartimide problem in Fmoc‐based SPPS . Part III J Pept Sci. 2005;11(10):650‐657. 10.1002/psc.668 [DOI] [PubMed] [Google Scholar]
- 3. Mergler M, Dick F, Sax B, Stähelin C, Vorherr T. The aspartimide problem in Fmoc‐based SPPS . Part II J Pept Sci. 2003;9(8):518‐526. 10.1002/psc.473 [DOI] [PubMed] [Google Scholar]
- 4. Mergler M, Dick F, Sax B, Weiler P, Vorherr T. The aspartimide problem in Fmoc‐based SPPS . Part I J Pept Sci. 2003;9(1):36‐46. 10.1002/psc.430 [DOI] [PubMed] [Google Scholar]
- 5. Bodanszky M, Kwei JZ, SIDE REACTIONS IN PEPTIDE SYNTHESIS: VII* . Sequence dependence in the formation of aminosuccinyl derivatives from β‐benzyl‐aspartyl peptides . J Pept Protein Res. 1978;12(2):69‐74. [PubMed] [Google Scholar]
- 6. Lauer JL, Fields CG, Fields GB. Sequence dependence of aspartimide formation during 9‐fluorenylmethoxycarbonyl solid‐phase peptide synthesis . Lett Pept Sci. 1995;1(4):197‐205. 10.1007/bf00117955 [DOI] [Google Scholar]
- 7. Wade JD, Mathieu MN, Macris M, Tregear GW. Base‐induced side reactions in Fmoc‐solid phase peptide synthesis: minimization by use of piperazine as Nα‐deprotection reagent . Lett Pept Sci. 2000;7(2):107‐112. 10.1007/bf02443569 [DOI] [Google Scholar]
- 8. Schön I, Kisfaludy L. Formation of aminosuccinyl peptides during acidolytic deprotection followed by their transformation to piperazine‐2,5‐dione derivatives in neutral media . Int J Pept Protein Res. 1979;14(5):485‐494. 10.1111/j.1399-3011.1979.tb01960.x [DOI] [PubMed] [Google Scholar]
- 9. Curnis F, Cattaneo A, Longhi R, et al. Critical role of flanking residues in NGR‐to‐isoDGR transition and CD13/integrin receptor switching . J Biol Chem. 2010;285(12):9114‐9123. 10.1074/jbc.M109.044297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeHart MP, Anderson BD. The role of the cyclic imide in alternate degradation pathways for asparagine‐containing peptides and proteins . J Pharm Sci. 2007;96(10):2667‐2685. 10.1002/jps.20905 [DOI] [PubMed] [Google Scholar]
- 11. Zhang X, Højrup P. Cyclization of the N‐terminal X‐Asn‐Gly motif during sample preparation for bottom‐up proteomics . Anal Chem. 2010;82(20):8680‐8685. 10.1021/ac1019478 [DOI] [PubMed] [Google Scholar]
- 12. Akaji K, Teruya K, Akaji M, Aimoto S. Synthesis of cyclic RGD derivatives via solid phase macrocyclization using the Heck reaction . Tetrahedron. 2001;57(12):2293‐2303. 10.1016/S0040-4020(01)00113-2 [DOI] [Google Scholar]
- 13. Süli‐Vargha H, Schlosser G, Ilaš J. 1,4‐diazepine‐2,5‐dione ring formation during solid phase synthesis of peptides containing aspartic acid β‐benzyl ester . J Pept Sci. 2007;13(11):742‐748. 10.1002/psc.885 [DOI] [PubMed] [Google Scholar]
- 14. Guo YC, Cao SX, Zong XK, Liao XC, Zhao YF. ESI‐MSn study on the fragmentation of protonated cyclic‐dipeptides . J Spectrosc. 2009;23(3‐4):131‐139. 10.3233/spe-2009-0388 [DOI] [Google Scholar]
- 15. Samson D , Loidl G . A novel side reaction in Fmoc‐SPPS: formation of cyclo(‐Xaa‐Asp)‐Yaa peptides . Proceedings of the 22nd American Peptide Symposium, San Diego 2011.; 58‐59.2012
- 16. Wang MH, Li XM, Li CS, Ji NY, Wang BG. Secondary metabolites from Penicillium pinophilum SD‐272, a marine sediment‐derived fungus . Mar Drugs. 2013;11(6):2230‐2238. 10.3390/md11062230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moure A, Sanclimens G, Bujons J, et al. Chemical modulation of peptoids: synthesis and conformational studies on partially constrained derivatives . Chem Eur J. 2011;17(28):7927‐7939. 10.1002/chem.201100216 [DOI] [PubMed] [Google Scholar]
- 18. Capasso S, Balboni G, Di Cerbo P. Effect of lysine residues on the deamidation reaction of asparagine side chains . Biopolymers. 2000;53(2):213‐219. 10.1002/(SICI)1097-0282(200002)53:2<213::AID-BIP11>3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- 19. Conroy T, Jolliffe KA, Payne RJ. Synthesis of N‐linked glycopeptides via solid‐phase aspartylation . Org Biomol Chem. 2010;8(16):3723‐3733. 10.1039/c003673k [DOI] [PubMed] [Google Scholar]
- 20. Haack T, Mutter M. Serine derived oxazolidines as secondary structure disrupting, solubilizing building blocks in peptide synthesis . Tetrahedron Lett. 1992;33(12):1589‐1592. 10.1016/S0040-4039(00)91681-2 [DOI] [Google Scholar]
- 21. Mutter M, Nefzi A, Sato T, Sun X, Wahl F, Wohr T. Pseudo‐prolines (psi Pro) for accessing "inaccessible" peptides . Pept Res. 1995;8(3):145‐153. [PubMed] [Google Scholar]
- 22. Wöhr T, Wahl F, Nefzi A, et al. Pseudo‐prolines as a solubilizing, structure‐disrupting protection technique in peptide synthesis . J Am Chem Soc. 1996;118(39):9218‐9227. 10.1021/ja961509q [DOI] [Google Scholar]
- 23. Karlström A, Undén A. A new protecting group for aspartic acid that minimizes piperidine‐catalyzed aspartimide formation in Fmoc solid phase peptide synthesis . Tetrahedron Lett. 1996;37(24):4243‐4246. 10.1016/0040-4039(96)00807-6 [DOI] [Google Scholar]
- 24. Vigil‐Cruz SC, Aldrich JV. Unexpected aspartimide formation during coupling reactions using Asp (OAl) in solid‐phase peptide synthesis . Lett Pept Sci. 1999;6(1):71‐75. 10.1023/a:1008871528559 [DOI] [Google Scholar]
- 25. Behrendt R, Huber S, White P. Preventing aspartimide formation in Fmoc SPPS of Asp‐Gly containing peptides—practical aspects of new trialkylcarbinol based protecting groups . J Pept Sci. 2016;22(2):92‐97. 10.1002/psc.2844 [DOI] [PubMed] [Google Scholar]
- 26. Subirós‐Funosas R, El‐Faham A, Albericio F. Use of Oxyma as pH modulatory agent to be used in the prevention of base‐driven side reactions and its effect on 2‐chlorotrityl chloride resin . Pept Sci. 2012;98(2):89‐97. 10.1002/bip.21713 [DOI] [PubMed] [Google Scholar]
- 27. Packman LC. N‐2‐hydroxy‐4‐methoxybenzyl (Hmb) backbone protection strategy prevents double aspartimide formation in a ‘difficult’ peptide sequence . Tetrahedron Lett. 1995;36(41):7523‐7526. 10.1016/0040-4039(95)01522-1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.1 Identities of relevant peaks identified by LC‐MS of Ac‐Xaa‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 and Ac‐Gly‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 flanking residue variations.
Figure S2.1 HMBC correlation used for the identification of a diketopiperazine sub‐structure [Ref. 16, main text].
Figure S3.1 Chemical structure of the purified DKP by‐product with assignments of positions used for 1H, 13C and 15N chemical shifts.
Figure S3.2 Chemical structure of the native peptide with assignments of positions used for 1H and 13C chemical shifts.
Figure S3.3 Chemical structure of the resin bound linear protected peptide with assignments of positions used for 1H and 13C chemical shifts.
Figure S3.4 Chemical structure of resin bound phenylalanine with assignments of positions used for 1H and 13C chemical shifts. This compound was synthesized for the identification of 1H/13C chemical shifts of the solid support.
Figure S4.1 Enlarged regions of 1H NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.2 Enlarged regions of 13C{1H} NMR spectra with peak picking of the DKP reference recorded in DMF‐d7.
Figure S4.3 1H‐13C HSQC NMR spectrum of DKP reference with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in DMF‐d7.
Figure S4.4 1H‐13C HMBC NMR spectra selecting 8 Hz (top) or 4 Hz (bottom) H‐C couplings of the DKP reference recorded in DMF‐d7.
Figure S4.5 1H‐1H DQF‐COSY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.6 1H‐1H TOCSY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S 4.7 1H‐1H ROESY NMR spectrum of the DKP reference recorded in DMF‐d7.
Figure S4.8 1H‐15N HSQC NMR spectrum with assignment of positions for the DKP reference (DMF‐d7). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.9 1H‐15N HMBC NMR spectra selecting 4 Hz (top) or 2 Hz (bottom) H‐N couplings of the DKP reference (DMF‐d7).
Figure S4.10 1H NMR spectra with integrals of the DKP reference recorded in D2O (top) and in H2O/D2O 9/1 (bottom, spectrum recorded with suppression of water resonance).
Figure S4.11 Enlarged regions of 13C{1H} NMR spectra with peak picking of the DKP reference recorded in D2O.
Figure S4.12 1H‐13C HSQC NMR spectrum of the DKP reference with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in D2O.
Figure S4.13 1H‐13C HMBC NMR spectrum of the DKP reference recorded in D2O (top) and in H2O/D2O 9/1 (bottom).
Figure S4.14 1H‐1H DQF‐COSY NMR spectrum of the DKP reference recorded in D2O.
Figure S4.15 1H‐15N HMQC NMR spectrum with assignment of positions for the DKP reference (H2O/D2O 9/1). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.16 1H‐15N HMBC NMR spectra selecting 4 Hz (top) or 2 Hz (bottom) H‐N couplings of the DKP reference (D2O).
Figure S4.17 Region of interest with assignment of crucial 1H‐15N HMBC NMR correlations for the DKP reference (recorded in D2O, selection of 2 Hz coupling constant).
Figure S4.18 1H NMR spectra with integrals of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7. Below the integration of selected resonances for the quantification of the DPK by‐product is shown.
Figure S4.19 Enlarged regions of 13C{1H} NMR spectra with peak picking of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.20 1H‐13C HSQC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in DMF‐d7.
Figure S4.21 1H‐13C HMBC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.22 1H‐1H DQF‐COSY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.23 1H‐1H TOCSY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.24 1H‐1H ROESY NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in DMF‐d7.
Figure S4.25 1H‐15N HSQC NMR spectrum with assignment of positions of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 (DMF‐d7). Note the different chemical shift region for the ε‐NH2 of residue Lys5.
Figure S4.26 1H NMR spectra with integrals of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in D2O. Below the integration of selected resonances for the quantitative determination of the DPK by‐product is shown.
Figure S4.27 Enlarged regions of 13C{1H} NMR spectra with peak picking of the native peptide Ac‐GDGAKF‐NH2 recorded in D2O.
Figure S4.28 1H‐13C HSQC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 with spectral editing (CH and CH3 groups in red, CH2 in blue) recorded in D2O.
Figure S4.29 1H‐13C HMBC NMR spectrum of the native peptide Ac‐Gly‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 recorded in D2O.
Figure S4.30 1H HR‐MAS NMR spectrum of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.31 13C{1H} HR‐MAS NMR spectrum of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.32 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.33 1H‐13C HMBC HR‐MAS NMR spectrum and enlarged regions of the linear protected peptide Ac‐Gly‐Asp(O‐tBu)‐Gly‐ALa‐Lys (Boc)‐Phe‐tricyclic amide linker‐resin recorded in DMF‐d7.
Figure S4.34 1H HR‐MAS NMR spectrum of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.35 13C{1H} HR‐MAS NMR spectrum of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.36 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S4.36 1H‐13C HSQC HR‐MAS NMR spectrum and enlarged regions of Phe‐tricyclic amide linker resin recorded in DMF‐d7.
Figure S5.1 Chemical structure of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 with assignments of positions used for 1 H and 13 C chemical shifts
Figure S5.2 Enlarged regions of 1 H NMR spectrum with integrals and resonance assignments of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.3 Enlarged regions of 13 C{1H} NMR spectrum with peak picking and resonance assignments of cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.4 1 H‐ 13 C HSQC NMR spectrum with resonance assignments of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 with spectral editing (CH and CH 3 groups in red, CH 2 in blue) recorded in DMSO‐d 6 .
Figure S5.5 1 H‐ 13 C HMBC NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.6 1 H‐ 1 H DQF‐COSY NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.7 Enlarged regions with crucial correlations of a) 1 H‐ 1 H DQF‐COSY and b‐d) 1 H‐ 13 C HMBC NMR spectra for the assignments of the aspartimide part of reference peptide cyclo[Gly‐Asp]‐Cys (Acm)‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.8 Chemical structure of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 with assignments of positions used for 1 H and 13 C chemical shifts
Figure S5.9 Enlarged regions of 1 H NMR spectrum with integrals and resonance assignments of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.10 Enlarged regions of 13 C{1H} NMR spectrum with peak picking and resonance assignments of cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.11 1 H‐ 13 C HSQC NMR spectrum with resonance assignments of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 with spectral editing (CH and CH 3 groups in red, CH 2 in blue) recorded in DMSO‐d 6 .
Figure S5.12 1 H‐ 13 C HMBC NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.13 1 H‐ 1 H DQF‐COSY NMR spectrum of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Figure S5.14 Enlarged regions with crucial correlations of a) 1 H‐ 1 H DQF‐COSY and b‐d) 1 H‐ 13 C HMBC NMR spectra for the assignments of the aspartimide part of reference peptide cyclo[Gly‐Asp]‐Ala‐Gly‐NH 2 recorded in DMSO‐d 6 .
Table S6.1 Product compositions determined by HPLC of experiments with Ac‐Xaa‐Asp‐Gly‐Ala‐Lys‐Phe‐NH2 and Ac‐Gly‐Asp‐Yaa‐Ala‐Lys‐Phe‐NH2 flanking residue variations.