

Abstract
Conductivity, carrier mobility, and a suitable Gibbs free energy are important criteria that determine the performance of catalysts for a hydrogen evolution reaction (HER). However, it is a challenge to combine these factors into a single compound. Herein, we discover a superior electrocatalyst for a HER in the recently identified Dirac nodal arc semimetal PtSn4. The determined turnover frequency (TOF) for each active site of PtSn4 is 1.54 H2 s−1 at 100 mV. This sets a benchmark for HER catalysis on Pt‐based noble metals and earth‐abundant metal catalysts. We make use of the robust surface states of PtSn4 as their electrons can be transferred to the adsorbed hydrogen atoms in the catalytic process more efficiently. In addition, PtSn4 displays excellent chemical and electrochemical stabilities after long‐term exposure in air and long‐time HER stability tests.
Keywords: Dirac semimetal, electrocatalysis, hydrogen evolution reaction, PtSn4, surface states
Introduction
The hydrogen evolution reaction (2 H+ + 2 e− → H2, HER) that occurs at the solid–liquid interface plays a vital role in clean energy conversion and for the understanding of other complicated heterogeneous reactions. The use of suitable catalysts that have high efficiency, selectivity, and stability is crucial for practical applications.1 Many advanced electrocatalysts have been designed and developed by employing a variety of strategies.2 However, a suitable lower cost replacement of the benchmark Pt catalyst has not yet been developed. The dominance of Pt as the best catalyst is an inevitable result of several key characteristics. 1) The excellent chemical stability of Pt provides active sites for obtaining a high turnover frequency (TOF). 2) Its ultra‐high electrical conductivity guarantees fast electron transport and reduces the Schottky barrier at the interface between the Pt and adsorbates. 3) A suitable d‐band center favors the metal‐adsorbate interaction and hydrogen release process.3
Recent observations of unoccupied metallic topological surface states (TSSs) suggest that certain noble metals, such as Pt are in fact Z 2 topological metals.4 The metallic surface states of these metals are protected by time reversal symmetry and are robust against surface modification and defects under ambient conditions.5 These are precisely the conditions that are beneficial for the various surface reactions that occur on the crystal surface.6 Our previous study on a range of topological materials from bismuth chalcogenide topological insulators7 to Weyl semimetals,8 demonstrated that each of these could be promising candidates for use as catalysts.
However, both topological insulators and Dirac/Weyl semimetals exhibit very low carrier densities around the Fermi level owing to weak electrostatic screening. We approach this problem by considering a topological nodal arc. In the case of Dirac node arc semimetals, the band crossing points around the Fermi level form a Dirac node arc in momentum space. Many exotic properties, such as ultrahigh magnetoresistivity, carrier mobility are related to the Dirac fermions and topological surface states.9 This suggests the following question: can the Pt atomic layer be combined with the exotic physical properties related to the Dirac node arc to speed up the HER process?
To pursue this question, we examined the bulk single crystal Dirac semimetal PtSn4. Recently, a high density of conduction electrons and Dirac node arcs were observed in single‐crystalline PtSn4 and subsequently confirmed using de Haas‐van Alphen quantum oscillations.9c, 10 The extremely weak bonding between the Sn‐Pt‐Sn layers of the compound enables the exposure of the Pt skin without the presence of metastable dangling bond surface states. We observe an ultra‐low resistivity of only 42 μΩ cm and a high mobility of 200 cm2 V−1 s−1 at room temperature in bulk single‐crystalline PtSn4, where the robust surface states are mainly derived from the p orbitals of Pt skin atoms. These lend a considerable contribution to the local density of states (DOS) near the Fermi Level and could act as electron donors for adsorbed molecules. With the bulk single crystal acting as both the cathode and catalyst for the HER, Pt‐like activity is exhibited together with a decrease in Pt usage of 71 % in mass. The TOF value for the PtSn4 bulk single crystals reached 1.54 H2 s−1 at 100 mV, surpassing those of state‐of‐the‐art Pt/C, MoS2, and other noble‐metal based catalysts. Of greater importance is the superior electrocatalytic stability of the single crystal after a long‐time and high current density test. These observations may be used to develop a new design principle for high intrinsic electrochemical catalysts with defined active sites.
Results and Discussion
Starting with Pt, we checked all of the Ptx My (M is a main group element) binary compounds, and selectively short‐listed specific representative cases that has the lowest Pt content with a certain M element. As shown in Figure 1 a, PtSn4 is chosen for this study because of its facile large‐scale synthesis and high electrical conductivity. As a widely accepted descriptor in prediction HER activity, the Gibbs free‐energy of the intermediate state, |ΔG H*|, was calculated for different models (Figure S1 in the Supporting Information).11 A Pt55 metal cluster was also included for comparison.12 As shown in the Volcano plot (Figure 1 b), Pt exhibits the optimal level for H adsorption with a near zero value of ΔG H*=−0.11 eV, suggesting the best electrocatalytic activity from a thermodynamic viewpoint. In stark contrast, the exposure of the Pt layer in the PtSn4 (010) plane resulted in a ΔG H* value of −0.28 eV, which is better than that of the Pt55 clusters, with a value of −0.33 eV. These results indicate that the Pt atomic layer of the PtSn4 single crystal could be an ideal surface for HERs.
Figure 1.
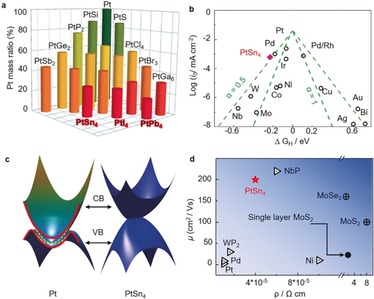
Motivations for searching for advanced electrocatalysts beyond pure Pt: a) The Pt mass ratios in binary compounds that include Pt. b) HER volcano plot for PtSn4 and selected metal catalysts. The Gibbs free energy of PtSn4 is very close to that of pure Pt. c) Band structure of Pt. The metallic topological surface states are shown in yellow. The bulk states (upper and lower parts) and surface states (middle part) are separated by the red line. The band structure of PtSn4 with non‐closed Dirac node arc is also shown. d) The room temperature mobility and conductivity of PtSn4 with the selected advanced electrocatalysts.
Figure 1 c shows the band structure of Pt, and the topological surface states above the Fermi energy on the (111) surfaces owing to the bulk Z 2 topological order can be observed. Although the Z 2 topological order does not exist in PtSn4, it hosts a Dirac nodal arc in the bulk band structure (Figure 1 c).9c This results in a large density of states around the Fermi level. Dirac electrons exist in the sheet, which generally correspond to a large carrier mobility. Figure 1 d shows a comparison of the conductivity and mobility of a selection of advanced HER catalysts, which indicates the advantages of the PtSn4 single crystal. The mobilities of the electrons and holes are very similar between 5 and 300 K and reach 200 cm2 V−1 s−1 at room temperature, which is even higher than that of back‐gated MoS2, MoSe2, WS2, WSe2, and black phosphorus on SiO2 substrates.13
With the above theoretical investigation and experimental facts in mind, high quality PtSn4 single crystals were grown out of an Sn‐rich binary melt. The HER performance evaluation of this single crystal was carried out in a conventional three‐electrode electrochemical cell in 1 m KOH electrolyte (the details are provided in the Methods section, Figure S2). The obtained polarization curves are shown in Figure 2 a. A sharp increase in the cathode current with an onset overpotential of approximately 0 mV was observed on the PtSn4 single crystal, with a burst of gas bubbles that appeared on the crystal surface (Figure S3 and Movie S1). To achieve a current density of 10 mA cm−2, a low overpotential of 37 mV is required, which is close to that of the Pt/C nanostructure (28 mV) and much less than that of Pt foil (71 mV). It should be noted that the PtSn4 single crystal demonstrates a higher activity than that of even pure Pt foils and Pt/C nanostructures (at higher current densities) in alkaline media, although Pt has a much smaller ΔG H* value. This may be caused by the sluggish water dissociation kinetics of Pt in alkaline media, which need not be considered under acidic conditions. Thus, we calculated the water dissociation energy barrier for the Pt (111) and PtSn4 (010) surfaces, respectively. Indeed, as illustrated in Figure 4 e, PtSn4 has a much lower energy barrier of 0.41 eV than that for Pt (0.91 eV).
Figure 2.
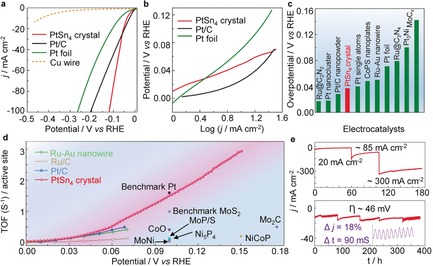
Electrochemical performance of PtSn4 single‐crystal catalyst. a) HER polarization curves for the Cu wire, Pt foil, 20 % Pt/C, and PtSn4 single crystal. b) Corresponding Tafel plots for the Pt foil, 20 % Pt/C, and PtSn4 single crystal in 1 m KOH. c) Overpotential of the PtSn4 single‐crystal catalyst at 10 mA cm−2 and some recently reported results for HER electrocatalysts. d) TOF values for the PtSn4 single‐crystal catalyst and other recently reported results for HER electrocatalysts. e) Current–time (i–t) chronoamperometric response of the PtSn4 electrocatalyst for increasing current densities from 20–300 mA cm−2, at an overpotential of 46 mV. The inset shows the changes in the current density during stirring of the electrolyte with a magnetic bar.
Figure 4.
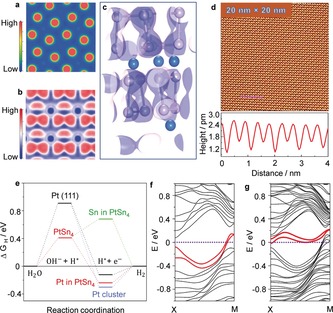
Bonding information in the crystal and the electron transfer mechanism. a) Contour plots of the total charge distribution of PtSn4 in the (010) plane. The electronic charges are almost all distributed in the vicinity of the Pt and Sn atoms. b) The 3D‐electron localization function mapping of the (010) surface shows that there is a weak bonding between the in‐plane Sn atoms. c) The 3D‐electron localization function (ELF), with an iso‐surface value of 0.72. d) Atomically resolved STM topography of the PtSn4 (010) surface. Inset: the corresponding fast Fourier transform (FFT). The line profile measured along the pink line in (d) indicates a lattice parameter of 0.451 nm; the bias voltage was V b=100 mV with a tunnel current of I set=200 pA. e) The Gibbs free energy diagram of the PtSn4 (010) surface with an exposed Sn layer, Pt layer, Pt metal (111) face, and Pt nanocluster (55 atoms) calculated at the equilibrium potential of different models, and the water dissociation energy berries on the Pt (111) and PtSn4 (010) surfaces. f) Band structure of the PtSn4 (010) surface with an exposed Pt layer, but without H adsorption. The robust surface states are along the direction Χ → Μ in the Brillouin zone, located just below the Fermi energy. g) Evolution of the Pt derived surfaces states after hydrogen adsorption when exposing the Pt layer. The surface states donate electrons to the adsorbed H 1s orbital, and shift above the Fermi level.
A Tafel analysis yields Tafel slopes of 39, 34, and 74 mV dec−1. for the PtSn4 single crystal, 20 % Pt/C, and Pt foil, respectively, implying a rapid HER rate with a Volmer‐Heyrovsky pathway as the rate‐determining step (Figure 2 b). The calculated HER exchange current density (j 0) for the PtSn4 single crystal is 0.585 mA cm−2, which is greater than that of the nanostructure Pt/C catalyst (0.518 mA cm−2). This indicates that PtSn4 is a superior electrocatalyst in alkaline media, with a better performance compared with those of nanostructures such as Pt3Ni nanowires (51 mV),14 Pt‐Ni excavated nano‐multipods (71 mV),15 and PtSe2 nanofilms (327 mV)16 (Figure 2 c and Table S5).17 TOF for each active site of PtSn4 is calculated to determine the intrinsic activity (Figure S4, see the Supporting Information). The TOF values of PtSn4 at 50 and 100 mV are 0.375 and 1.54 H2 s−1, respectively, which are greater than those of Pt/C (0.27 H2 s−1 @ 50 mV) and pure Au‐Ru nanowires (0.31 H2 s−1 @ 50 mV),18 and much greater than those of other nanostructured electrocatalysts (Figure 2 d).19 The multiple‐step chrono potentiometric process employed for the PtSn4 electrode is displayed in Figure S5, where the current density is increased from 10 to 110 mA cm−2, without an iR correction. Simultaneous changes in the current densities and applied voltage demonstrated excellent mass transport, conductivity, and mechanical robustness of the single crystalline electrode.20
We further investigated the long‐term electrochemical and chemical stability of the single‐crystal electrode. The PtSn4 bulk single crystal shows excellent stability for an increasing current of 20 to 300 mA cm−2, with a total measurement time of 175 h. At a constant overpotential of 46 mV, the current density is maintained for approximately 400 h without significant activity loss (Figure 2 e). Moreover, the HER activity can be retained even after long‐time air exposure. As shown in Figure S6, the polarization curves were recorded after three‐months of air exposure, and 10 000 cyclic voltammetry sweeps, showing a similar performance to that of the initial tests.
To better understand the origin of the HER activity, the structure of the single crystal was determined by single‐crystal X‐ray diffraction (Table S1–S3). The structure was solved in the centrosymmetric space group Ccca (No. 68), with lattice parameters a=6.4208(7), b=11.3592(1), and c=6.3766(7) Å at 300 K. Figures 3 a and S7 demonstrate the layered structure of PtSn4, which is composed of stacked Sn‐Pt‐Sn layers along the b axis with a Pt–Sn distance of 2.7905(9) Å (selected crystallographic data are provided in the Supporting Information, CCDC 1841795 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.). A homogeneous composition and orientation can be observed from the uniform contrast of the backscatter electron images (Figure 3 b and Figure S8). A thin lamella sample for high‐resolution transmission electron microscopy (TEM) was fabricated by focused ion beam (FIB). Clear lattice fringes with an interplane distance of 0.32 nm, which correspond to the (200) plane of PtSn4 can be seen from the lattice‐resolved TEM image (Figure S9a,b). The high quality of the single crystal was further demonstrated by an excellent match between the experimental and simulated electron‐diffraction patterns along the [010] direction (see the inset in Figure 3 c and Figure S10). The atomic structure of the PtSn4 single crystal was further investigated with noise corrected high‐resolution scanning TEM (HR‐STEM) (Figure 3 c). The Sn‐Pt‐Sn stacking sequence was retained, with a 100 % occupancy of the Pt and Sn sites.
Figure 3.
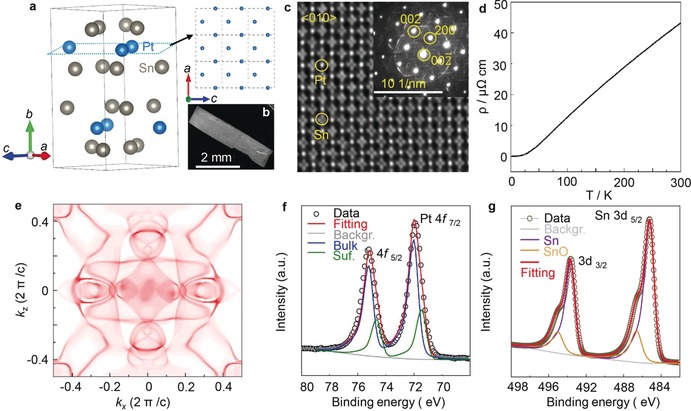
Structure information of bulk single‐crystal PtSn4. a) Crystal structure of the PtSn4 derived from the single‐crystal XRD at 300 K. The exposed Pt atomic layer was constructed as shown by the upper left panel. b) A typical SEM image of the obtained PtSn4 single crystal. c) HRTEM image of the PtSn4 sample prepared using the focused ion beam technique (FIB). Inset: the selected area diffraction (SAED) pattern recorded along the [010] crystal orientation. d) Temperature‐dependent resistivity along the [010] direction. An ultralow ρ of 42 μΩ cm at room temperature was observed. e) Calculated surface Fermi surface based on the semi‐infinite model. The XPS results for the f) Pt 4f and g) Sn 3d.
The in‐plane resistivity (ρ) of the PtSn4 crystal was measured in the temperature range 2–300 K, which displays a linear temperature dependence above 50 K (Figure 3 d), reflecting the dominance of the acoustic phonon scattering over the electron transport. The resistivity exhibits an ultralow value of ρ≈42 μΩ cm at room temperature, which is as low as the values of pure Pt and Sn. A very large residual resistivity ratio RRR=ρ (300 K)/ ρ (2 K)=1000 was obtained, together with a non‐saturating quadratic magneto‐resistivity up to 9 T (Figure S11a), suggesting the high quality of the single crystal. The positive slope of the Hall resistivity as a function of applied magnetic field, as well as the positive thermopower (Figure S11b), indicate that the holes were the dominant charge carriers. This ultrahigh conductivity and mobility may be derived from the unique Fermi surface and band dispersion of the PtSn4 single‐crystal, which is a general characteristic of nodal arc semimetals. Indeed, our band structure calculation showed a band crossing just below the Fermi level (Figure S12). The (010) face Fermi surfaces were constructed as shown in Figure 3 e, which consist of large electron and hole pockets.9c
The electronic state of the elements on the PtSn4 single‐crystal surface was determined by X‐ray photoelectron spectroscopy (XPS). The XPS survey spectrum confirmed the presence of Pt and Sn (Figure S13). As shown in Figure 3 f, the main peak at a binding energy of 72.0 eV can be assigned to zero‐valence platinum. It is interesting to note that no Pt‐O oxidation states were observed (72.8 eV for Pt2+ and 74 eV for Pt4+).21 The peak at lower binding energy originates from the surface Pt in PtSn4 single crystal. This confirms the ultra‐high stability of the Pt sites in the crystal. The main peak with a binding energy of 485.2 eV can be attributed to Sn0 3d5/2.22 All of these results are consistent with the previous studies on Pt single crystal surface and Pt alloys, where it was claimed that the formation of Pt−O bonding is very difficult.23
To understand the bonding character, the electron density distribution and electron localization function (ELF) were calculated. The total charge density distribution map is shown in Figure 4 a,b, demonstrating that the electronic charge is almost completely distributed in the vicinity of the Pt and Sn atoms. This indicates the presence of a metallic bond character in the compound.24 The ELF iso‐surface value of 0.72 further highlights the metallic nature of the bonding between Pt and Sn, and suggests a certain amount of electron localization between the intralayer Sn atoms (Figure 4 c). The relatively high ELF value between the Sn atoms indicates the weak covalent nature of the Sn‐Sn bonds. Finally, to confirm the exposure of the Pt layer, scanning tunneling microscopy (STM) was used to examine the in situ surface structure of the PtSn4 (010) surface (Figure S14a). The atomically resolved image in Figure 4 d visualizes a square Pt (010) net with an interatomic distance of 0.45 nm at various points in the spirals, which is identical to the bulk orientation and atomic model of the PtSn4 (010) face, and in perfect agreement with the value obtained by single‐crystal X‐ray diffraction. This is further demonstrated by the significant difference in the ΔG H* values of the Sn and Pt layers (Figure 4 e).
The ultrahigh chemical stability and defined crystal surface of PtSn4 provides a good opportunity to investigate the related activity mechanism. DFT calculations were performed on the exposed (010) surface of the PtSn4 single crystal. First, the exposure of Sn atoms was considered (the computation details are provided in Supporting Information). Figure S15 displays the calculated surface band structure for the (010) surface with an exposed Sn layer. The abrupt breaking of the weak Sn–Sn bonding creates a Sn‐ derived occupied s‐type surface state that typically covers a broad energy range of the surface Brillouin zone. After hydrogen adsorption, the surface states corresponding to the Sn–H occupied bonding states are submerged into the bulk valence bands (Figure S16), with the H s orbital observed in the deep bulk band (Figure S17). However, when the Pt layer is exposed, the surfaces could not be passivated or eliminated by hydrogen as there is no Fermi level pinning at the crystal surface. Indeed, as shown in Figure S18, we observe surface states along the Χ–Μ axis of the surface Brillouin zone (Figures 4 f and Figure S18). The orbital analysis showed that the surface states are mainly formed of p‐like electronic states from the topmost Pt layer (Figures S19a and S19b). To investigate their interaction with hydrogen, the band structures of the hydrogen‐covered surface were also determined. The hydrogen adsorption shifts the surface states above the Fermi level (Figure 4 g and Figure S20). This indicates the transfer of electrons from the surface state to the hydrogen s‐like orbitals. The downshift of hydrogen bands to a position far below the Fermi energy provided further proof of this electron transfer (Figure S21).
Conclusion
The bulk single‐crystal PtSn4 exhibits Pt‐like electrocatalytic activity as a catalyst for HERs. The ultra‐high chemical and electrochemical stability presented by this compound, together with a significant decrease in Pt usage, makes it an ideal candidate to replace Pt catalysts. The linear band crossing in this semimetal provides large room‐temperature carrier mobility and conductivity, which can be attributed to the high Fermi velocity of massless Dirac states.25 This facilitates the rapid charger transfer in the catalysis process and thereby increases the HER kinetics. Intrinsic and stable surface states were found in the Pt layer of the PtSn4 single crystal. This led to the direct donation of electrons from the occupied robust surface states to adsorbed hydrogen. Rather than adopting the traditional approaches of creating active sites by nanosizing, doping, straining, and edging, the present study indicates that the manipulation of robust surface states, as well as providing high conductivity and mobility, may be used to provide a new guide for obtaining high‐performance catalysts.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
We gratefully acknowledge Dr. Qiunan Xu and Dr. Yang Zhang for helpful discussions. This work was financially supported by the European Research Council (ERC Advanced Grant No. 291472 “Idea Heusler”) and ERC Advanced Grant (No. 742068). TOPMAT′. The calculations were carried out at the HPC Platform of Shanghaitech University.
G. Li, C. Fu, W. Shi, L. Jiao, J. Wu, Q. Yang, R. Saha, M. E. Kamminga, A. K. Srivastava, E. Liu, A. N. Yazdani, N. Kumar, J. Zhang, G. R. Blake, X. Liu, M. Fahlman, S. Wirth, G. Auffermann, J. Gooth, S. Parkin, V. Madhavan, X. Feng, Y. Sun, C. Felser, Angew. Chem. Int. Ed. 2019, 58, 13107.
Contributor Information
Dr. Guowei Li, Email: Guowei.li@cpfs.mpg.de.
Dr. Yan Sun, Email: Yan.Sun@cpfs.mpg.de.
Prof. Claudia Felser, Email: Claudia.Felser@cpfs.mpg.de.
References
- 1.
- 1a. Rao C. N. R., Dey S., Proc. Natl. Acad. Sci. USA 2017, 114, 13385–13393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Sivula K., Zboril R., Le Formal F., Robert R., Weidenkaff A., Tucek J., Frydrych J., Gratzel M., J. Am. Chem. Soc. 2010, 132, 7436–7444; [DOI] [PubMed] [Google Scholar]
- 1c. Miao J., Xiao F. X., Yang H. B., Khoo S. Y., Chen J., Fan Z., Hsu Y. Y., Chen H. M., Zhang H., Liu B., Sci. Adv. 2015, 1, e1500259; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Stamenkovic V., Mun B. S., Mayrhofer K. J., Ross P. N., Markovic N. M., Rossmeisl J., Greeley J., Norskov J. K., Angew. Chem. Int. Ed. 2006, 45, 2897–2901; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2963–2967. [Google Scholar]
- 2.
- 2a. Kibsgaard J., Jaramillo T. F., Besenbacher F., Nat. Chem. 2014, 6, 248–253; [DOI] [PubMed] [Google Scholar]
- 2b. Chen Z., Cummins D., Reinecke B. N., Clark E., Sunkara M. K., Jaramillo T. F., Nano Lett. 2011, 11, 4168–4175; [DOI] [PubMed] [Google Scholar]
- 2c. Li G., Sun Y., Rao J., Wu J., Kumar A., Xu Q. N., Fu C., Liu E., Blake G. R., Werner P., Shao B., Liu K., Parkin S., Liu X., Fahlman M., Liou S.-C., Auffermann G., Zhang J., Felser C., Feng X., Adv. Energy Mater. 2018, 8, 1801258. [Google Scholar]
- 3.
- 3a. Seh Z. W., Kibsgaard J., Dickens C. F., Chorkendorff I., Norskov J. K., Jaramillo T. F., Science 2017, 355, eaad4998; [DOI] [PubMed] [Google Scholar]
- 3b. Pu Z., Amiinu I. S., Kou Z., Li W., Mu S., Angew. Chem. Int. Ed. 2017, 56, 11559–11564; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11717–11722. [Google Scholar]
- 4.
- 4a. Yan B., Stadtmüller B., Haag N., Jakobs S., Seidel J., Jungkenn D., Mathias S., Cinchetti M., Aeschlimann M., Felser C., Nat. Commun. 2015, 6, 10167; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Wiebe J., Meier F., Hashimoto K., Bihlmayer G., Blügel S., Ferriani P., Heinze S., Wiesendanger R., Phys. Rev. B 2005, 72, 193406. [Google Scholar]
- 5. Chen C., He S., Weng H., Zhang W., Zhao L., Liu H., Jia X., Mou D., Liu S., He J., Peng Y., Feng Y., Xie Z., Liu G., Dong X., Zhang J., Wang X., Peng Q., Wang Z., Zhang S., Yang F., Chen C., Xu Z., Dai X., Fang Z., Zhou X. J., Proc. Natl. Acad. Sci. USA 2012, 109, 3694–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Politano A., Chiarello G., Li Z., Fabio V., Wang L., Guo L., Chen X., Boukhvalov D. W., Adv. Funct. Mater. 2018, 28, 1800511. [Google Scholar]
- 7. Rajamathi C. R., Gupta U., Pal K., Kumar N., Yang H., Sun Y., Shekhar C., Yan B., Parkin S., Waghmare U. V., Felser C., Rao C. N. R., ChemPhysChem 2017, 18, 2322–2327. [DOI] [PubMed] [Google Scholar]
- 8. Rajamathi C. R., Gupta U., Kumar N., Yang H., Sun Y., Süß V., Shekhar C., Schmidt M., Yan B., Parkin S., Felser C., Rao C. N. R., Adv. Mater. 2017, 29, 1606202. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Li J., Ma H., Xie Q., Feng S., Ullah S., Li R., Dong J., Li D., Li Y., Chen X.-Q., Sci. China Mater. 2017, 61, 23–29; [Google Scholar]
- 9b. Wang L.-L., Kaminski A., Canfield P. C., Johnson D. D., J. Phys. Chem. C 2018, 122, 705–713; [Google Scholar]
- 9c. Wu Y., Wang L.-L., Mun E., Johnson D. D., Mou D., Huang L., Lee Y., Bud'ko S. L., Canfield P. C., Kaminski A., Nat. Phys. 2016, 12, 667–671. [Google Scholar]
- 10.
- 10a. Wang Y. J., Liang D. D., Ge M., Yang J., Gong J. X., Luo L., Pi L., Zhu W. K., Zhang C. J., Zhang Y. H., J. Phys. Condens. Matter 2018, 30, 155701; [DOI] [PubMed] [Google Scholar]
- 10b. Jo N. H., Wu Y., Wang L.-L., Orth P. P., Downing S. S., Manni S., Mou D., Johnson D. D., Kaminski A., Bud'ko S. L., Canfield P. C., Phys. Rev. B 2017, 96, 165145. [Google Scholar]
- 11.
- 11a. Jiao Y., Zheng Y., Davey K., Qiao S.-Z., Nat. Energy 2016, 1, 16130; [Google Scholar]
- 11b. Greeley J., Jaramillo T. F., Bonde J., Chorkendorff I. B., Norskov J. K., Nat. Mater. 2006, 5, 909–913. [DOI] [PubMed] [Google Scholar]
- 12. Su J., Yang Y., Xia G., Chen J., Jiang P., Chen Q., Nat. Commun. 2017, 8, 14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a.C. Fu, T. Scaffidi, J. Waissman, Y. Sun, R. Saha, S. J. Watzman, A. K. Srivastava, G. Li, W. Schnelle, P. Werner, M. E. Kamminga, S. Sachdev, S. S. P. Parkin, S. A. Hartnoll, C. Felser, J. Gooth, arXiv:1802.09468v1 [cond-mat.mtrl-sci] 2018;
- 13b. Li L., Yu Y., Ye G. J., Ge Q., Ou X., Wu H., Feng D., Chen X. H., Zhang Y., Nat. Nanotechnol. 2014, 9, 372–377; [DOI] [PubMed] [Google Scholar]
- 13c. Zhao Y., Qiao J., Yu Z., Yu P., Xu K., Lau S. P., Zhou W., Liu Z., Wang X., Ji W., Chai Y., Adv. Mater. 2017, 29, 1604230. [DOI] [PubMed] [Google Scholar]
- 14. Wang P., Zhang X., Zhang J., Wan S., Guo S., Lu G., Yao J., Huang X., Nat. Commun. 2017, 8, 14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao Z., Chen Q., Zhang J., Li H., Jiang Y., Shen S., Fu G., Lu B. A., Xie Z., Zheng L., Nat. Commun. 2017, 8, 15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin S., Liu Y., Hu Z., Lu W., Mak C. H., Zeng L., Zhao J., Li Y., Yan F., Tsang Y. H., Zhang X., Lau S. P., Nano Energy 2017, 42, 26–33. [Google Scholar]
- 17. Zheng Y., Jiao Y., Zhu Y., Li L. H., Han Y., Chen Y., Jaroniec M., Qiao S. Z., J. Am. Chem. Soc. 2016, 138, 16174–16181. [DOI] [PubMed] [Google Scholar]
- 18. Lu Q., Wang A. L., Gong Y., Hao W., Cheng H., Chen J., Li B., Yang N., Niu W., Wang J., Yu Y., Zhang X., Chen Y., Fan Z., Wu X. J., Chen J., Luo J., Li S., Gu L., Zhang H., Nat. Chem. 2018, 10, 456. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. McKone J. R., Sadtler B. F., Werlang C. A., Lewis N. S., Gray H. B., ACS Catal. 2013, 3, 166–169; [Google Scholar]
- 19b. Zhang R., Wang X., Yu S., Wen T., Zhu X., Yang F., Sun X., Wang X., Hu W., Adv. Mater. 2017, 29, 1605520; [DOI] [PubMed] [Google Scholar]
- 19c. Ling T., Yan D. Y., Wang H., Jiao Y., Hu Z., Zheng Y., Zheng L., Mao J., Liu H., Du X. W., Jaroniec M., Qiao S. Z., Nat. Commun. 2017, 8, 1509; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Kibsgaard J., Jaramillo T. F., Angew. Chem. Int. Ed. 2014, 53, 14433–14437; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14661–14665. [Google Scholar]
- 20. Lu X., Zhao C., Nat. Commun. 2015, 6, 6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J., Jiao M., Lu L., Barkholtz H. M., Li Y., Wang Y., Jiang L., Wu Z., Liu D. J., Zhuang L., Ma C., Zeng J., Zhang B., Su D., Song P., Xing W., Xu W., Wang Y., Jiang Z., Sun G., Nat. Commun. 2017, 8, 15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okamoto Y., Carter W. J., Hercules D. M., Appl. Spectrosc. 1979, 33, 287–293. [Google Scholar]
- 23.
- 23a. Baetzold R. C., Apai G., Shustorovich E., Jaeger R., Phys. Rev. B 1982, 26, 4022–4027; [Google Scholar]
- 23b. Zhao L., Thomas J. P., Heinig N. F., Abd-Ellah M., Wang X., Leung K. T., J. Mater. Chem. C 2014, 2, 2707. [Google Scholar]
- 24. Mun E., Ko H., Miller G. J., Samolyuk G. D., Bud'ko S. L., Canfield P. C., Phys. Rev. B 2012, 85, 035135. [Google Scholar]
- 25. Shekhar C., Nayak A. K., Sun Y., Schmidt M., Nicklas M., Leermakers I., Zeitler U., Skourski Y., Wosnitza J., Liu Z., Chen Y., Schnelle W., Borrmann H., Grin Y., Felser C., Yan B., Nat. Phys. 2015, 11, 645–649. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary