

Abstract
Objective
This study was undertaken to evaluate glucocorticoid dosages and serologic findings in patients with giant cell arteritis (GCA) flares.
Methods
Patients with GCA were randomly assigned to receive double‐blind dosing with either subcutaneous tocilizumab (TCZ) 162 mg weekly plus 26‐week prednisone taper (TCZ‐QW + Pred‐26), every‐other‐week TCZ plus 26‐week prednisone taper (TCZ‐Q2W + Pred‐26), placebo plus 26‐week prednisone taper (PBO + Pred‐26), or placebo plus 52‐week prednisone taper (PBO + Pred‐52). Outcome measures were prednisone dosage, C‐reactive protein (CRP) level, and erythrocyte sedimentation rate (ESR) at the time of flare.
Results
One hundred patients received TCZ‐QW + Pred‐26, 49 received TCZ‐Q2W + Pred‐26, 50 received PBO + Pred‐26, and 51 received PBO + Pred‐52. Of the 149 TCZ‐treated patients, 36 (24%) experienced flare, 23 (64%) of whom were still receiving prednisone (median dosage 2.0 mg/day). Among 101 PBO + Pred–treated patients, 59 (58%) experienced flare, 45 (76%) of whom were receiving prednisone (median dosage 5.0 mg/day). Many flares occurred while patients were taking >10 mg/day prednisone: 9 (25%) in the TCZ groups and 13 (22%) in the placebo groups. Thirty‐three flares (92%) in TCZ‐treated groups and 20 (34%) in PBO + Pred–treated groups occurred with normal CRP levels. More than half of the PBO + Pred–treated patients had elevated CRP levels without flares. Benefits of the TCZ and prednisone combination over prednisone alone for remission induction were apparent by 8 weeks.
Conclusion
Most GCA flares occurred while patients were still receiving prednisone. Acute‐phase reactant levels were not reliable indicators of flare in patients treated with TCZ plus prednisone or with prednisone alone. The addition of TCZ to prednisone facilitates earlier GCA control.
Introduction
Giant cell arteritis (GCA) is a vasculitis of large‐ and medium‐sized arteries that affects people ≥50 years old 1. Upon being diagnosed as having GCA, patients are treated immediately with high dosages of glucocorticoids to reduce the risk of vision loss and large vessel complications. Long‐term glucocorticoid treatment has traditionally been required to control symptoms and prevent relapse in GCA patients 2, but flares frequently occur 3, 4, 5. Although GCA is the most common primary form of systemic vasculitis in Western countries, there are few data from randomized clinical trials regarding prednisone dosages at disease flares, particularly for patients treated with prednisone for 1 year—a course that approximates the standard of care for many clinicians. Additionally, the usefulness of acute‐phase reactants (APRs) in the clinical assessment of GCA flares has been poorly studied in patients treated with prednisone alone or with tocilizumab (TCZ). Moreover, no randomized clinical trials have been conducted in which clinicians and patients were blinded with regard to prednisone dosages and APR levels. TCZ, a humanized monoclonal antibody against the interleukin‐6 (IL‐6) receptor α, inhibits IL‐6–mediated signaling and inflammatory pathways 6, 7.
In the Giant Cell Arteritis Clinical Research Study (GiACTA), a randomized, double‐blind, placebo (PBO)–controlled phase III study of patients with GCA, TCZ was superior to PBO in the achievement of sustained remission at 1 year 8. TCZ was approved for the treatment of patients with GCA in 2017. Blocking IL‐6 signaling with TCZ reduces levels of APRs such as C‐reactive protein (CRP) and decreases the erythrocyte sedimentation rate (ESR) 7. Consequently, measuring APR levels to quantify systemic inflammation is believed to have limited value in the clinical assessment of disease flares in patients with GCA treated with TCZ 9. GiACTA was the first randomized clinical trial in any disease (to our knowledge) to include a blinded, variable‐dosage prednisone taper. Once patients reduced their daily prednisone dosage, according to protocol, to <20 mg/day, patients and physician‐investigators were blinded with regard to glucocorticoid dosages unless a flare occurred. Disease flares were assessed largely on a clinical basis, irrespective of APR levels, because investigators were blinded with regard to CRP levels, and initially only the laboratory assessor was aware of ESR results.
The design of the GiACTA trial permits a unique opportunity to study prednisone dosages and laboratory features associated with disease flares in GCA patients treated with prednisone alone and those treated with TCZ plus prednisone. These trial data therefore provide guidance for clinical decision‐making within the new and traditional treatment landscapes for GCA.
Patients and methods
Ethics board approval and informed consent
This trial was approved by institutional review boards and/or ethics committees at the appropriate institutions and was conducted in accordance with the Guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent.
Patients and study design
The patient eligibility criteria and study design for the GiACTA trial (ClinicalTrials.gov identifier: NCT01791153) have previously been published 10. Patients were randomly assigned 2:1:1:1 to 4 groups to receive treatment with weekly subcutaneous TCZ 162 mg plus a 26‐week prednisone taper (TCZ‐QW + Pred‐26), every‐other‐week subcutaneous TCZ 162 mg plus a 26‐week prednisone taper (TCZ‐Q2W + Pred‐26), subcutaneous placebo plus a 26‐week prednisone taper (PBO + Pred‐26), or subcutaneous placebo plus a 52‐week prednisone taper (PBO + Pred‐52) 8. Randomization was stratified by baseline prednisone dosage (≤30 mg/day or >30 mg/day). During the study, prednisone was tapered in a double‐blind, protocol‐defined manner 11. Prednisone was initially administered on an open‐label basis at dosages of >20 mg/day and tapered according to weekly, protocol‐defined decrements. Patients and investigators were blinded with regard to prednisone dosages of ≤20 mg/day.
To maintain rigorous blinding of investigators given the anticipated normalization of APR levels with IL‐6 receptor blockade, a separate laboratory assessor was assigned to monitor laboratory parameters (including ESR) independently of the blinded investigator/efficacy assessor, who assessed the patient's GCA and managed the prednisone taper. All investigators were blinded with regard to CRP level. The laboratory assessor was informed of ESR values, but the efficacy assessor was informed only whether the ESR was higher or lower than 30 mm/hour under strict protocol guidelines to ensure patient safety while preserving the blind 8.
Definition of GCA flares
GCA flare was determined by the investigator and defined as the recurrence of signs or symptoms of GCA or an ESR of ≥30 mm/hour attributed by the investigator to GCA even in the absence of any other overt clinical manifestations of active disease. The definition of disease flare included the need for an increase in the prednisone dosage at the time of the clinical event. The symptoms and signs of flare were categorized as follows: GCA signs and symptoms (new‐onset localized headache, scalp tenderness, temporal artery tenderness or decreased pulsation, ischemia‐related vision loss, and otherwise unexplained mouth or jaw pain on mastication) only, polymyalgia rheumatica (PMR) symptoms only, fever (≥38°C) only, visual symptoms (unilateral and bilateral blindness, ischemic optic neuropathy, amaurosis fugax, blurred vision, and diplopia) only, elevated ESR (≥30 mm/hour) attributed to active GCA only, and multiple symptoms (e.g., the occurrence of GCA and PMR symptoms together).
Study analysis
Assessments were performed at each study visit to determine whether a patient's disease was fully controlled and the patient could safely continue the prednisone taper. Remission or flare was determined by the investigator at each study visit. CRP normalization was not included in the definition of remission for this analysis, and, as noted, the investigators were blinded with regard to CRP level. Prednisone dosages and APR values at disease flare were evaluated at the time of first flare. CRP levels and ESR values were examined before flare, at the time of flare, and in the absence of clinical signs or symptoms of flare. If the ESR at the time of flare was unavailable, the last ESR value preceding the flare was used. Remission before baseline was determined by the investigator at each site. Many patients had achieved disease control before the baseline visit because of glucocorticoid use during the 6‐week screening period. The proportions of patients achieving sustained remission, time to first flare, and cumulative prednisone duration and exposure were analyzed according to the patients’ baseline prednisone dosages (≤30 mg/day or >30 mg/day).
Statistical analysis
TCZ and PBO treatment groups were compared using the Cochran‐Mantel‐Haenszel test with adjustment for the stratification factor of starting prednisone dosage (≤30 mg/day or >30 mg/day) for analysis of the proportions of patients in sustained remission and using a Cox proportional hazards model with adjustment for the starting prednisone dosage for time to first flare. The secondary end point of remission rates over time and subgroup analyses by starting prednisone dosage and disease onset were prespecified. All other analyses were performed post hoc and are exploratory, with no adjustment for Type I error control; therefore, we performed a limited number of statistical comparisons for these analyses. Hazard ratios (HRs) and 99% confidence intervals (99% CIs) were calculated.
Results
Baseline characteristics of patients in the GiACTA trial have been reported 8. Briefly, 251 patients were randomly assigned to receive TCZ‐QW + Pred‐26 (n = 100), TCZ‐Q2W + Pred‐26 (n = 50), PBO + Pred‐26 (n = 50), or PBO + Pred‐52 (n = 51). The intent‐to‐treat and safety populations included 250 patients, because 1 patient assigned to the TCZ‐Q2W + Pred‐26 group did not receive the study drug.
Flares
Among the 250 patients included in this analysis, 95 (38%) experienced disease flares following a period of remission during the first 52 weeks of the study. Of these 95 flares, 13 (13.7%) were characterized by symptoms considered typical of GCA only and 13 (13.7%) by PMR symptoms only. No flares following remission were characterized by visual symptoms alone, and none were based on the presence of fever alone. Clinical features at disease flare were not characterized in additional detail. Nine flares (9.5%) were based only on increased ESR attributed to GCA in the absence of an alternative explanation. Only 1 patient in the TCZ‐QW + Pred‐26 group was reported to have a flare after remission without symptoms of flare or an ESR of ≥30 mm/hour (Table 1). All flares responded to increased glucocorticoid dosages.
Table 1.
Prednisone dosages and acute‐phase reactant levels at time of GCA flare*
| Assessment at time of GCA flare | PBO + Pred‐26 (n = 50) | PBO + Pred‐52 (n = 51) | TCZ‐QW + Pred‐26 (n = 100) | TCZ‐Q2W + Pred‐26 (n = 49) |
|---|---|---|---|---|
| Flare experienced after remission | 34 (68.0) | 25 (49.0) | 23 (23.0) | 13 (26.5) |
| Signs and symptoms experienced at time of flare† | ||||
| GCA signs and symptoms only‡ | 2 (5.9) | 1 (4.0) | 5 (21.7) | 5 (38.5) |
| PMR symptoms only | 2 (5.9) | 2 (8.0) | 6 (26.1) | 3 (23.1) |
| Fever (≥38°C) only | 0 | 0 | 0 | 0 |
| Visual symptoms only§ | 0 | 0 | 0 | 0 |
| Elevated ESR (≥30 mm/hour) only | 6 (17.6) | 2 (8.0) | 1 (4.3) | 0 |
| Multiple symptoms | 24 (70.6) | 20 (80.0) | 10 (43.5) | 5 (38.5) |
| No symptoms of flare | 0 | 0 | 1 (4.3) | 0 |
| Receiving steroids at time of first flare†, ¶ | 21 (61.8) | 24 (96) | 17 (73.9) | 6 (46.2) |
| Prednisone dosage at flare, median (range) mg/day | 2.5 (0.0–30.0) | 8.0 (0.0–20.0) | 7.0 (0.0–25.0) | 0.0 (0.0–12.5) |
| First flare experienced while receiving prednisone, by dosage in mg/day† | ||||
| 0 | 13 (38.2) | 1 (4.0) | 6 (26.1) | 7 (53.8) |
| 1–5 | 9 (26.5) | 7 (28.0) | 5 (21.7) | 3 (23.1) |
| >5–10 | 5 (14.7) | 11 (44.0) | 4 (17.4) | 2 (15.4) |
| >10–20 | 6 (17.6) | 6 (24.0) | 6 (26.1) | 1 (7.7) |
| >20–30 | 1 (2.9) | 0 | 2 (8.7) | 0 |
| >30–40 | 0 | 0 | 0 | 0 |
| >50–60 | 0 | 0 | 0 | 0 |
| >60 | 0 | 0 | 0 | 0 |
| CRP level preceding flare, median (range) mg/liter# | 23.1 (1.4–119.0) | 17.3 (0.2–122.0) | 0.4 (0.2–93.2) | 1.0 (0.2–18.1) |
| Presence of elevated CRP at time of flare†, # | 22 (65) | 17 (68) | 1 (4) | 2 (15) |
| Presence of elevated CRP without flare# | 26 (52.0) | 31 (60.8) | 5 (5.0) | 3 (6.1) |
| ESR preceding flare, median (range) mm/hour** | 51.0 (8.0–140.0) | 39.0 (4.0–138.0) | 5.0 (0.0–80.0) | 5.0 (1.0–43.0) |
| Presence of elevated ESR at time of flare†, ** | 27 (79) | 14 (56) | 1 (4) | 3 (23) |
| Presence of elevated ESR without flare | 31 (62.0) | 28 (54.9) | 2 (2.0) | 3 (6.1) |
| Presence of elevated CRP and ESR without flare | 20 (40.0) | 20 (39.2) | 2 (2.0) | 1 (2.0) |
Except where indicated otherwise, values are the number (%) of patients. GCA = giant cell arteritis; PBO + Pred‐26 = placebo plus 26‐week prednisone taper; PBO + Pred‐52 = placebo plus 52‐week prednisone taper; TCZ‐QW + Pred‐26 = tocilizumab once weekly plus 26‐week prednisone taper; TCZ‐Q2W + Pred‐26 = tocilizumab once every 2 weeks plus 26‐week prednisone taper; PMR = polymyalgia rheumatica; ESR = erythrocyte sedimentation rate; CRP = C‐reactive protein.
Values are the number (%) of patients in the corresponding treatment group who experienced flare following remission.
Includes new‐onset localized headache, scalp tenderness, temporal artery tenderness or decreased pulsation, ischemia‐related vision loss, and otherwise unexplained mouth or jaw pain on mastication.
Includes unilateral and bilateral blindness, ischemic optic neuropathy, amaurosis fugax, blurred vision, and diplopia.
Includes prednisone and all other steroids.
Normal ≤10 mg/liter.
Normal <30 mm/hour.
Prednisone dosages at flare
The median prednisone dosage at the time of disease flare was 2.0 mg/day (range 0.0–25.0) for the combined TCZ groups and 5.0 mg/day (0.0–30.0) for the combined PBO groups. Of the 149 patients in the combined TCZ groups, 36 (24%) experienced GCA flares (Table 1); 23 of these 36 flares (63.9%) occurred while patients were still receiving prednisone (8 [22.2%] at a dosage of 1–5 mg/day), and 13 (36.1%) occurred after discontinuation of the prednisone taper. Of the 101 patients in the combined PBO groups, 59 (58%) experienced GCA flares; 45 of these (76.3%) occurred while patients were still receiving prednisone, and 14 (23.7%) occurred after discontinuation of the prednisone taper. Of the 50 patients who received PBO + Pred‐26, 9 of the 34 reported flares (26.4%) occurred at prednisone dosages of 1–5 mg/day. In contrast, two‐thirds of the flares (17 of 25; 68%) in the 51 patients in the PBO + Pred‐52 group occurred at prednisone dosages of >5–20 mg/day. Substantial numbers of disease flares occurred while patients were receiving >10 mg/day prednisone, accounting for 9 (25%) of the flares in the TCZ groups and 13 (22.0%) in the PBO groups. No patients experienced flares while receiving prednisone dosages of >30 mg/day, and only 3 patients experienced flares while receiving dosages of >20–30 mg/day.
Among patients randomly assigned to receive PBO + Pred, 13 (38.2%) of the disease flares in the PBO + Pred‐26 group became manifest only after patients had tapered to a prednisone dosage of 0 mg/day (Table 1). In contrast, only 1 (4.0%) of the disease flares in the PBO + Pred‐52 group was diagnosed in a patient receiving a prednisone dosage of 0 mg/day; the rest of the flares in this group became clinically manifest before the patients tapered to 0 mg/day prednisone.
Relationships between APR level, disease activity, and flare
Flare with normal APR levels
Median CRP and ESR levels preceding GCA flare were lower in the TCZ groups than in the PBO groups (Table 1). This was expected based on the biology of IL‐6 receptor blockade and its downstream effects on APR levels. The overwhelming majority of TCZ‐treated patients had low APR levels during disease flare. In the TCZ groups, 33 of 36 first disease flares (91.7%) occurred with normal CRP levels (≤10 mg/liter) and 32 of 36 flares (88.9%) with normal ESR (<30 mm/hour). In the PBO groups, APR levels remained normal at the time of first disease flares for approximately one‐third of patients; of the 59 first disease flares, 20 (33.9%) were associated with normal CRP levels and 18 (30.5%) with normal ESRs.
Elevated APR levels without flare
More than half of the patients in the PBO groups had CRP elevations without disease flare during 1 year of follow‐up (Table 1). Fifty‐seven of the 101 patients (56.4%) in the combined PBO groups had CRP level elevations (>10 mg/liter) during the 52‐week follow‐up period without disease flare (defined as CRP level or ESR elevated at 2 consecutive visits between weeks 12 and 52 in the absence of clinical manifestations—beyond laboratory markers—of a disease flare). As anticipated, the CRP level was elevated in the absence of flare in only a minority of TCZ‐treated patients: 5 (5.0%) in TCZ‐QW + Pred‐26 and 3 (6.1%) in TCZ‐Q2W + Pred‐26. Few patients in the TCZ groups had elevations of ESR that were not associated with disease flare: 2 patients (2.0%) in the TCZ‐QW + Pred‐26 group and 3 patients (6.1%) in the TCZ‐Q2W + Pred‐26 group. In contrast, more than half the patients in the PBO groups had elevations of ESR that were not reported as disease flares. Elevated CRP level and ESR in the absence of flare was observed in 40 patients (39.6%) in the PBO groups and 3 patients (2.0%) in the TCZ groups (Table 1).
Remission
More than half of all patients (142 of 250; 56.8%) were in remission by the baseline visit because of glucocorticoids received during the 6‐week screening period. Numerically higher proportions of patients in the TCZ groups than in the PBO groups achieved remission between baseline and week 12 (Figure 1A); this was observed for subgroups with newly diagnosed disease (Figure 1B) and relapsing disease (Figure 1C). Proportions of patients achieving remission increased from baseline to week 12 by 28% in the TCZ‐QW + Pred‐26 group and 23% in the TCZ‐Q2W + Pred‐26 group, compared with 2% in the PBO + Pred‐26 group and 14% in the PBO + Pred‐52 group. At week 12, the proportions of patients in remission were 66.0% (n = 33) in the PBO + Pred‐26 group and 64.7% (n = 33) in the PBO + Pred‐52 group, compared with 83.0% (n = 83) in the TCZ‐QW + Pred‐26 group (P = 0.10, versus PBO + Pred‐26; P = 0.03, versus PBO + Pred‐52). The proportion of patients in remission in the TCZ‐Q2W + Pred‐26 group was 81.6% (n = 40), which was not significantly different from the PBO + Pred‐26 group (P = 0.57) or the PBO + Pred‐52 group (P = 0.30).
Figure 1.
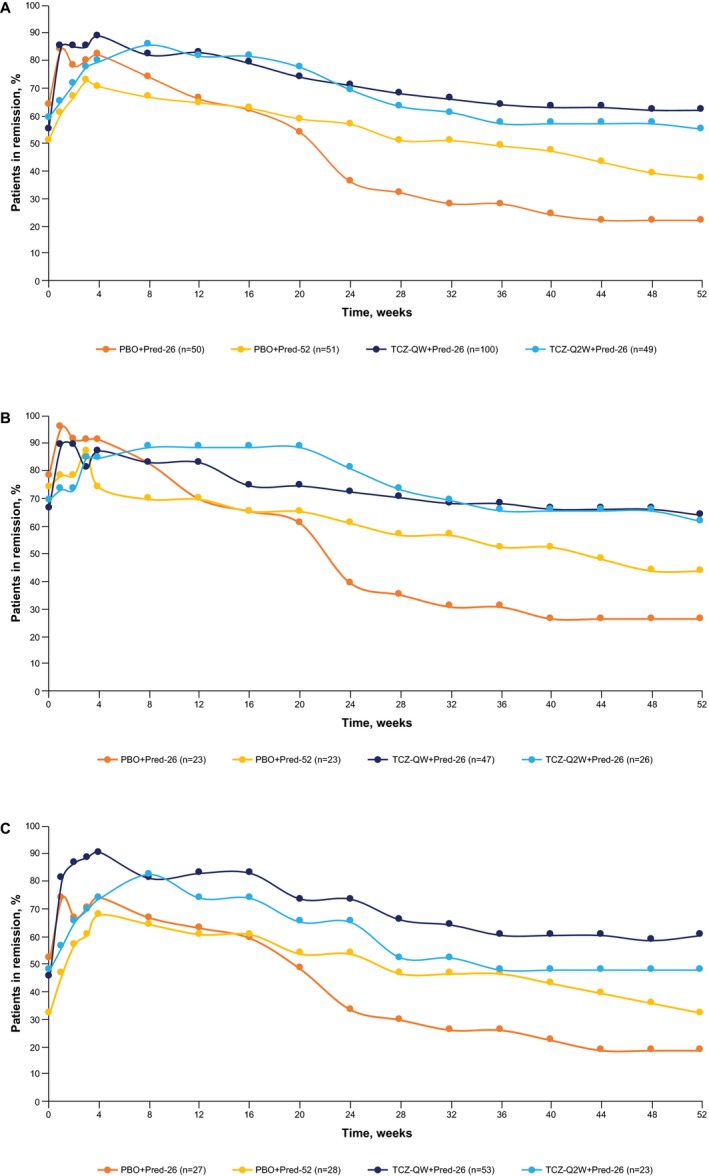
Disease control during the first 52 weeks of treatment in all patients in the intent‐to‐treat population (A), and in patients with newly diagnosed (B) and relapsing (C) giant cell arteritis. Prespecified exploratory analysis of remission rates over time is shown. Remission was defined as absence of flare and did not include C‐reactive protein level in the definition. Patients who withdrew from the study or received escape therapy were excluded from that point. Patients with missing information on remission status were considered not in remission for that time point only. Responders (patients in remission) were analyzed; therefore, values at week 52 are slightly higher than the highest values for sustained remission, which accounts for patients not adhering to the protocol‐defined tapering regimen as nonresponders. PBO + Pred‐26 = placebo plus 26‐week prednisone taper; PBO + Pred‐52 = placebo plus 52‐week prednisone taper; TCZ‐QW + Pred‐26 = tocilizumab once weekly plus 26‐week prednisone taper; TCZ‐Q2W + Pred‐26 = tocilizumab once every 2 weeks plus 26‐week prednisone taper.
Time to first flare according to starting prednisone dosage
Patients in the TCZ groups were more likely than those in the PBO groups to achieve sustained remission at each of the baseline prednisone dosages (Figure 2). Among patients who started at prednisone dosages of >30 mg/day, those in each of the TCZ groups experienced longer times to disease flares than those in either of the PBO + Pred groups (Figure 3A). Among patients who started at prednisone dosages of ≤30 mg/day, a divergence of the PBO + Pred‐26 group from the other 3 groups was evident from week 12, with a shorter time to flare in the PBO + Pred‐26 group than the other groups (Figure 3B). In patients who started at prednisone dosages of ≤30 mg/day, the risk for flare was significantly lower among TCZ‐treated patients than among PBO + Pred‐26–treated patients (HR 0.21 [99% CI 0.08–0.54], P < 0.0001 for TCZ‐QW + Pred‐26 and HR 0.28 [99% CI 0.09–0.86], P = 0.0035 for TCZ‐Q2W + Pred‐26). Among patients who started at prednisone dosages of ≤30 mg/day, the risk for flare did not differ between either of the TCZ groups and the PBO + Pred‐52 group (HR 0.59 [99% CI 0.20–1.73], P = 0.2039 for TCZ‐QW + Pred‐26 and HR 0.76 [99% CI 0.21–2.72], P = 0.5866 for TCZ‐Q2W + Pred‐26).
Figure 2.
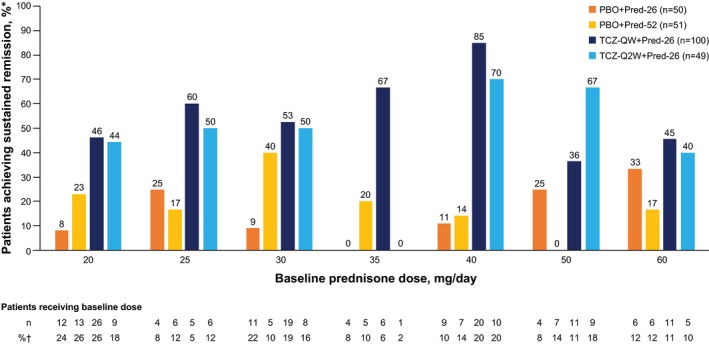
Sustained remission through week 52 according to baseline prednisone dosage. Percentages are based on number of patients receiving baseline dosage (*) and total number of patients in each treatment group at baseline (†). See Figure 1 for definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40876/abstract.
Figure 3.
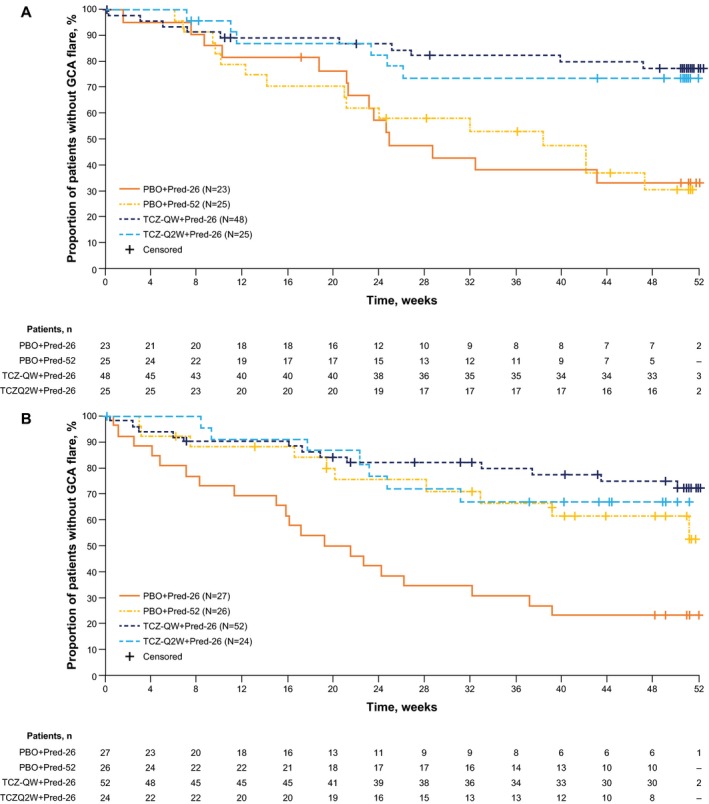
Kaplan‐Meier plot of time to first giant cell arteritis (GCA) flare according to starting prednisone dosage of >30 mg/day (A) and ≤30 mg/day (B) in the intent‐to‐treat population. Patients never in remission were censored at day 1, and patients who withdrew from the study before week 52 were censored from the time of withdrawal. See Figure 1 for definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40876/abstract.
Methotrexate and sustained remission
Only 35 of 250 patients (14%) received concomitant methotrexate (MTX) therapy during the study: 18 in the PBO groups and 17 in the TCZ groups (Table 2). One of the 18 patients (5.6%) receiving MTX in the combined PBO + Pred groups achieved sustained remission, compared with 15 of 83 patients (18.1%) who were not receiving MTX. Seven of 17 patients (41.2%) in the combined TCZ groups achieved sustained remission while receiving MTX, compared with 75 of 132 patients (56.8%) who did not receive MTX.
Table 2.
Methotrexate (MTX) use and sustained remission*
| PBO + Pred‐26 (n = 50) | PBO + Pred‐52 (n = 51) | TCZ‐QW + Pred‐26 (n = 100) | TCZ‐Q2W + Pred‐26 (n = 49) | |
|---|---|---|---|---|
| Received concomitant MTX | 8 (16) | 10 (20) | 11 (11) | 6 (12) |
| Did not receive concomitant MTX | 42 (84) | 41 (80) | 89 (89) | 43 (88) |
| MTX dosage, mean ± SD mg/week | 17.0 ± 6.0 | 15.4 ± 4.2 | 13.7 ± 3.1 | 13.1 ± 5.0 |
| Total MTX dose during 52 weeks, mean ± SD mg | 663.1 ± 434.3 | 635.3 ± 414.7 | 577.1 ± 250.2 | 491.7 ± 376.4 |
| Patients receiving MTX who achieved sustained remission† | 0 (0) | 1 (10) | 4 (36) | 3 (50) |
| Patients not receiving MTX who achieved sustained remission‡ | 7 (17) | 8 (20) | 52 (58) | 23 (54) |
Except where indicated otherwise, values are the number (%) of patients in the corresponding treatment group. See Table 1 for definitions.
Percentage based on number of patients receiving MTX.
Percentage based on number of patients not receiving MTX.
Discussion
The treatment of GCA has long been defined by glucocorticoid treatment. Our analyses of glucocorticoid dosages and APR levels at disease flare provide new information about the natural history of GCA patients treated according to protocol with up to 1 year of prednisone. Given the importance of glucocorticoids in the management of GCA for the past 70 years and the fact that prolonged courses of glucocorticoid treatment have been the only treatment clearly known to be effective, it may seem remarkable that such information was not available earlier. Results from other trials with shorter prednisone tapers 12, 13, 14 and longitudinal studies using data from observational cohorts 15, 16, 17 have implied that the failure rate of glucocorticoid therapy for GCA is high. However, GiACTA is the first randomized clinical trial to use a prednisone‐tapering regimen in GCA for as long as 1 year, and the first trial in any disease to use a variable‐dosage prednisone taper in which patients and investigators were blinded with regard to prednisone dosages. It therefore provides the most rigorous assessment to date of how well—or how poorly—glucocorticoids work in patients with GCA.
Equally important are the insights that these trial data provide into the contemporary management of GCA in the era of TCZ treatment. Three findings of major importance have resulted from this analysis, all of which have the potential to impact current treatment strategies.
First, most disease flares observed in the GiACTA trial occurred while patients were still receiving prednisone and, in many cases, at dosages incompatible with long‐term use. More than one‐fifth of the 95 first disease flares that occurred in the GiACTA trial were observed in patients receiving >10 mg/day prednisone, and 72% occurred while patients were still receiving some dosage of prednisone. This finding is consistent with the fact that GCA patients have endured long courses of prednisone and experienced adverse effects of long‐term glucocorticoid use as a nearly universal feature of their disease management, and with the concept of a lag time between loss of immunosuppressive control of GCA and clinical expression of disease flare. Although it was not formally addressed in this trial, MTX did not show benefits; the benefit of adjunctive MTX in GCA has previously been demonstrated in a meta‐analysis of 3 trials 18, but not with an effect size comparable to that of TCZ.
In the PBO + Pred groups, the slower (52‐week) taper was associated with disease flare at higher dosages of prednisone in patients in the PBO + Pred‐52 group than in patients in the PBO + Pred‐26 group, who were more likely to reach a prednisone dosage of 0 mg/day before experiencing flare. These findings, which may appear counterintuitive, suggest that a lag exists between the reemergence of disease activity and the appearance of clinical manifestations. The data support the concept that subclinical disease activity begins in many patients as they taper to lower daily prednisone dosages, but that they do not experience symptoms until they have discontinued prednisone completely if the prednisone taper is rapid (e.g., 26 weeks versus 52 weeks). Patients undergoing shorter prednisone tapers are therefore more likely to discontinue their prednisone entirely before the disease recurrence manifests as a clinical flare. In contrast, patients undergoing a slower prednisone taper may be more likely to experience clinical disease flare while still receiving prednisone.
Second, APR levels are of little value in monitoring longitudinal disease activity in patients treated with TCZ. CRP levels and ESRs remained low in nearly all TCZ‐treated patients who experienced disease flares. This is not surprising considering the anticipated effects of IL‐6 inhibition on CRP concentrations and ESR. Our data also confirm that APR measurements have shortcomings and the potential to mislead clinicians with regard to patients treated with prednisone alone. Although this point has been made by others 16, 19, it remains underappreciated by many clinicians who manage patients with GCA. Approximately one‐third of all disease flares in the PBO groups occurred while patients had normal CRP levels and ESRs (34% and 31%, respectively). Furthermore, more than half of PBO‐treated patients had elevations of either CRP level or ESR, and more than one‐third had elevations of both APR levels without subsequent clinical disease flares (56.4%, 58.4%, and 39.6%, respectively).
These findings underscore the importance of the clinical assessment—particularly the importance of clinical experience and astute history‐taking—in gauging whether GCA activity might be present. They also highlight the need for advances in the development of clinically useful biomarkers and rigorous correlation between imaging study results and disease activity in longitudinal disease assessments. It is possible that APR level elevations observed in the PBO‐Pred groups that were not followed by disease flares during 52 weeks of follow‐up indicate low‐grade disease activity that remained subclinical (imaging studies were not performed on these patients). A discord between GCA clinical symptoms and vascular changes detected by imaging has previously been described 20, 21. The optimal imaging protocol in large vessel vasculitis in clinical practice is unclear, and the interpretation of large vessel imaging studies in GCA is frequently not straightforward 22, 23, 24.
Third, these analyses demonstrate how swiftly the use of TCZ exerts a beneficial effect in GCA. The primary outcome measure of the trial was maintenance of remission at 52 weeks. However, our findings demonstrate that the effects of TCZ on induction of remission are rapid, which was most obvious in patients who experienced relapse (Figure 1C). In addition, even after 12 weeks, benefits of the combination of TCZ and prednisone over prednisone alone were apparent for the whole patient population (Figure 1A). These findings provide important information about the timing of TCZ use. For clinicians to establish disease control as quickly as possible, avoid acute and chronic complications of poorly controlled disease, and prevent excessive side effects of glucocorticoid use, a treatment strategy that emphasizes initiation of TCZ treatment as early as possible, accompanied by the institution of aggressive prednisone tapers (perhaps even shorter than 6 months) may be appropriate. Longer‐term follow‐up of patients in the GiACTA trial will determine whether such a strategy has a durable effect in a substantial number of patients, even after discontinuation of TCZ at 1 year. The appropriateness of prednisone tapers more aggressive than those used in GiACTA cannot be routinely recommended until carefully conducted longitudinal studies and clinical trials are performed.
The addition of TCZ to glucocorticoid therapy enables faster glucocorticoid tapering than is possible for patients treated with glucocorticoids alone. Nevertheless, treatment with TCZ did not prevent disease flare in all patients. Nearly one‐quarter of patients (24%) randomly assigned to either of the TCZ groups experienced ≥1 disease flare while receiving treatment. One potential contributor to the risk of disease flare in a subset of TCZ‐treated patients is that Th1 cells, which are not directly affected by IL‐6 receptor blockade, may play an important role in some patients with GCA 25, 26. Other explanations for disease recurrence despite ongoing IL‐6 receptor blockade must be examined in other studies. Nevertheless, the fact that GCA flares are possible in patients still taking TCZ underscores the argument that glucocorticoid tapering during GCA remission should continue to be carefully monitored to ensure patient safety and that clinicians must remain alert to the possibility of disease flare. Nevertheless, most patients treated with TCZ were able to discontinue glucocorticoids entirely, which is a highly desirable goal.
The data included in this analysis were from the largest randomized controlled trial of treatment for GCA conducted to date, which highlights the robustness of these findings. However, there are some limitations in these primarily post hoc exploratory analyses, including limited statistical testing. Flares were determined by investigators blinded with regard to CRP and informed only whether the ESR was higher or lower than 30 mm/hour. Although clinical descriptions were consistent with typical symptoms of GCA flare, this approach in the setting of the randomized, double‐blind, PBO‐controlled trial might have led to a lower threshold for the diagnosis of some disease flares, out of concern for patient safety. The definition of remission without CRP that was used for the current analysis was not predefined per protocol except as part of a sensitivity analysis, but it allows for evaluation of remission outside the effect of TCZ on APR levels. Another limitation is the fact that clinical investigators were informed of clinically relevant elevations in ESR in accordance with the dual‐assessor approach previously described 8. However, only 9 patients had flares judged on the basis of ESR alone, and excluding these patients would only reduce the proportion of patients who experienced flare in the PBO + Pred‐26 group from 68% (34 of 50) to 64% (28 of 44). Finally, granular details of the clinical aspects of the disease flares were not collected, and future studies should aim to collect more detailed information.
In conclusion, these data provide important information about the efficacy of glucocorticoid treatment alone and about the occurrence of disease flares in the era of IL‐6 receptor blockade. The addition of TCZ to prednisone in the treatment of GCA allows more patients to achieve remission within the first few months of initiating treatment. This may be important to prevent short‐ and long‐term complications and reduce complications of long‐term glucocorticoid use. Disease flares occur commonly in GCA, even when patients are receiving substantial dosages of prednisone and in patients treated with TCZ. Finally, APR levels have substantial limitations as indicators of disease activity, both in patients treated with TCZ plus glucocorticoids and in those treated with glucocorticoids alone. Astute clinical assessment of patient symptoms and signs remains crucial in the longitudinal management of GCA. Greater emphasis on the identification of useful biomarkers and application of large vessel imaging studies to understand disease activity are important components of the research agenda for GCA.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Stone had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Stone, Tuckwell, Dimonaco, Klearman, Aringer, Blockmans, Brouwer, Cid, Dasgupta, Rech, Salvarani, Schulze‐Koops, Schett, Spiera, Unizony, Collinson.
Acquisition of data
Stone, Tuckwell, Dimonaco, Klearman, Aringer, Blockmans, Brouwer, Cid, Dasgupta, Rech, Salvarani, Schulze‐Koops, Schett, Spiera, Unizony, Collinson.
Analysis and interpretation of data
Stone, Tuckwell, Dimonaco, Klearman, Aringer, Blockmans, Brouwer, Cid, Dasgupta, Rech, Salvarani, Schulze‐Koops, Schett, Spiera, Unizony, Collinson.
Role of the study sponsor
The study was funded by F. Hoffmann‐La Roche Limited, which was involved in the design, conduct, reporting of the study, and interpretation of the results. The first draft of the manuscript was written by Dr. Stone. Third‐party medical writing assistance was provided by Sara Duggan, PhD, and was funded by F. Hoffmann‐La Roche Limited. Publication of this article was contingent upon approval by F. Hoffmann‐La Roche Limited.
Acknowledgments
We thank the teams of trial investigators and subinvestigators, the Roche trial team, and the patients who participated in the trial.
ClinicalTrials.gov identifier: NCT01791153.
Supported by F. Hoffmann‐La Roche Limited.
1John H. Stone, MD, MPH, Sebastian H. Unizony, MD: Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts; 2Katie Tuckwell, PhD, Micki Klearman, MD, PhD: Genentech, San Francisco, California; 3Sophie Dimonaco, MSc, Neil Collinson, PhD: Roche Products Ltd., Welwyn Garden City, UK; 4Martin Aringer, MD: University Medical Center and Technische Universität Dresden, Dresden, Germany; 5Daniel Blockmans, MD: University Hospitals Gasthuisberg, Leuven, Belgium; 6Elisabeth Brouwer, MD, PhD: University of Groningen and University Medical Center Groningen, Groningen, The Netherlands; 7Maria C. Cid, MD: University Hospital Clínic de Barcelona and University of Barcelona, Barcelona, Spain; 8Bhaskar Dasgupta, MBBS, MD, FRCP: Southend University Hospital, NHS Foundation Trust, Southend, UK; 9Juergen Rech, MD, George Schett, MD: Universitätsklinikum Erlangen, Erlangen, Germany; 10Carlo Salvarani, MD: Azienda USL‐IRCCS di Reggio Emilia and Università di Modena and Reggio Emilia, Reggio Emilia, Italy; 11Hendrik Schulze‐Koops, MD, PhD: University of Munich, Munich, Germany; 12Robert Spiera, MD: Hospital for Special Surgery, New York, New York.
Dr. Stone has received consulting fees from Chugai (less than $10,000) and from Roche/Genentech (more than $10,000) and has received research support from Chugai. Dr. Tuckwell owns stock or stock options in Roche/Genentech. Ms Dimonaco owns stock or stock options in Roche. Dr. Klearman has received consulting fees from Roche/Genentech (less than $10,000) and owns stock or stock options in Roche/Genentech. Dr. Aringer has received consulting fees, speaking fees, and/or honoraria from Roche and Chugai (less than $10,000 each). Dr. Blockmans has received consulting fees from Roche (less than $10,000). Dr. Brouwer has received consulting fees, speaking fees, and/or honoraria from Roche (less than $10,000) on behalf of the University Medical Center Groningen, The Netherlands. Dr. Cid has received consulting fees, speaking fees, and/or honoraria from Roche, Novartis, Boehringer‐Ingelheim, GlaxoSmithKline, Vifor, and AbbVie (less than $10,000 each) and research support from Kiniksa. Dr. Dasgupta has received consulting fees from Roche, GlaxoSmithKline, and Sanofi Aventis (less than $10,000 each). Dr. Rech has received consulting fees, speaking fees, and/or honoraria from Bristol‐Myers Squibb, Celgene, Chugai, GlaxoSmithKline, Janssen, Eli Lilly, Novartis, Roche, Sanofi Aventis, and UCB (more than $10,000 each) and research support from Bristol‐Myers Squibb and Celgene. Dr. Salvarani has received consulting fees from Roche (less than $10,000) and research support from Roche. Dr. Schulze‐Koops has received consulting fees, speaking fees, and/or honoraria from Roche and Chugai (more than $10,000 each). Dr. Schett has received consulting fees from AbbVie, Bristol Myers‐Squibb, Celgene, Chugai, GlaxoSmithKline, Eli Lilly, Novartis, Roche, Sanofi Aventis, and UCB (less than $10,000 each). Dr. Spiera has received consulting fees from Roche/Genentech, GlaxoSmithKline, CSL Behring, and Sanofi Aventis (less than $10,000 each) and research support from Roche/Genentech, GlaxoSmithKline, Bristol‐Myers Squibb, Boehringer Ingelheim, Cytori, Chemocentryx, and Corbus. Dr. Unizony has received research support from Roche/Genentech. Dr. Collinson owns stock or stock options in Roche. No other disclosures relevant to this article were reported.
Qualified researchers may request access to data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche's criteria for eligible studies are available (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
References
- 1. Salvarani C, Pipitone N, Versari A, Hunder GG. Clinical features of polymyalgia rheumatica and giant cell arteritis. Nat Rev Rheumatol 2012;8:509–21. [DOI] [PubMed] [Google Scholar]
- 2. Gonzalez‐Gay MA, Martinez‐Dubois C, Agudo M, Pompei O, Blanco R, Llorca J. Giant cell arteritis: epidemiology, diagnosis, and management. Curr Rheumatol Rep 2010;12:436–42. [DOI] [PubMed] [Google Scholar]
- 3. Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double‐blind, placebo‐controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309–18. [DOI] [PubMed] [Google Scholar]
- 4. Hoffman GS, Cid MC, Rendt‐Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid‐induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007;146:621–30. [DOI] [PubMed] [Google Scholar]
- 5. Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puéchal X, et al. Adalimumab for steroid sparing in patients with giant‐cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis 2014;73:2074–81. [DOI] [PubMed] [Google Scholar]
- 6. Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, et al. Tocilizumab inhibits signal transduction mediated by both mIL‐6R and sIL‐6R, but not by the receptors of other members of IL‐6 cytokine family. Int Immunopharmacol 2005;5:1731–40. [DOI] [PubMed] [Google Scholar]
- 7. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin‐6 (IL‐6) and soluble IL‐6 receptor after administration of an anti‐IL‐6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008;112:3959–64. [DOI] [PubMed] [Google Scholar]
- 8. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant‐cell arteritis. N Engl J Med 2017;377:317–28. [DOI] [PubMed] [Google Scholar]
- 9. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 2016;387:1921–7. [DOI] [PubMed] [Google Scholar]
- 10. Unizony SH, Dasgupta B, Fisheleva E, Rowell L, Schett G, Spiera R, et al. Design of the tocilizumab in giant cell arteritis trial. Int J Rheumatol 2013;2013:912562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collinson N, Tuckwell K, Habeck F, Chapman M, Klearman M, Stone JH. Development and implementation of a double‐blind corticosteroid‐tapering regimen for a clinical trial. Int J Rheumatol 2015;2015:589841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spiera RF, Mitnick HJ, Kupersmith M, Richmond M, Spiera H, Peterson MG, et al. A prospective, double‐blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001;19:495–501. [PubMed] [Google Scholar]
- 13. Jover JA, Hernández‐García C, Morado IC, Vargas E, Bañares A, Fernández‐Gutiérrez B. Combined treatment of giant‐cell arteritis with methotrexate and prednisone: a randomized, double‐blind, placebo‐controlled trial. Ann Intern Med 2001;134:106–14. [DOI] [PubMed] [Google Scholar]
- 14. Martínez‐Taboada VM, Rodríguez‐Valverde V, Carreño L, López‐Longo J, Figueroa M, Belzunegui J, et al. A double‐blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis 2008;67:625–30. [DOI] [PubMed] [Google Scholar]
- 15. Alba MA, García‐Martínez A, Prieto‐González S, Tavera‐Bahillo I, Corbera‐Bellalta M, Planas‐Rigol E, et al. Relapses in patients with giant cell arteritis: prevalence, characteristics, and associated clinical findings in a longitudinally followed cohort of 106 patients. Medicine 2014;93:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kermani TA, Warrington KJ, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, et al. Disease relapses among patients with giant cell arteritis: a prospective, longitudinal cohort study. J Rheumatol 2015;42:1213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Restuccia G, Boiardi L, Cavazza A, Catanoso M, Macchioni P, Muratore F, et al. Flares in biopsy‐proven giant cell arteritis in northern Italy: characteristics and predictors in a long‐term follow‐up study. Medicine 2016;95:e3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mahr AD, Jover JA, Spiera RF, Hernández‐García C, Fernández‐Gutiérrez B, LaValley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta‐analysis. Arthritis Rheum 2007;56:2789–97. [DOI] [PubMed] [Google Scholar]
- 19. Salvarani C, Cantini F, Boiardi L, Hunder GG. Laboratory investigations useful in giant cell arteritis and Takayasu's arteritis. Clin Exp Rheumatol 2003;21 Suppl 32:S23–8. [PubMed] [Google Scholar]
- 20. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow‐up temporal artery biopsies in 40 treated patients. Mod Pathol 2017;30:788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Boysson H, Aide N, Liozon E, Lambert M, Parienti JJ, Monteil J, et al. Repetitive 18F‐FDG‐PET/CT in patients with large‐vessel giant‐cell arteritis and controlled disease. Eur J Intern Med 2017;46:66–70. [DOI] [PubMed] [Google Scholar]
- 22. Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F‐fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum 2006;55:131–7. [DOI] [PubMed] [Google Scholar]
- 23. Prieto‐González S, García‐Martínez A, Tavera‐Bahillo I, Hernández‐Rodríguez J, Gutiérrez‐Chacoff J, Alba MA, et al. Effect of glucocorticoid treatment on computed tomography angiography detected large‐vessel inflammation in giant‐cell arteritis: a prospective, longitudinal study. Medicine (Baltimore) 2015;94:e486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grayson PC, Alehashemi S, Bagheri AA, Civelek AC, Cupps TR, Kaplan MJ, et al. F‐fluorodeoxyglucose–positron emission tomography as an imaging biomarker in a prospective, longitudinal cohort of patients with large vessel vasculitis. Arthritis Rheumatol 2018;70:439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watanabe R, Hosgur E, Zhang H, Wen Z, Berry G, Goronzy JJ, et al. Pro‐inflammatory and anti‐inflammatory T cells in giant cell arteritis. Joint Bone Spine 2017;84:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Visvanathan S, Rahman MU, Hoffman GS, Xu S, García‐Martínez A, Segarra M, et al. Tissue and serum markers of inflammation during the follow‐up of patients with giant‐cell arteritis: a prospective longitudinal study. Rheumatology (Oxford) 2011;50:2061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]