

Abstract
A synthetic strategy to incorporate catechol functional groups into benzoxazine thermoset monomers was developed, leading to a family of bioinspired small‐molecule resins and main‐chain polybenzoxazines derived from biologically available phenols. Lap‐shear adhesive testing revealed a polybenzoxazine derivative with greater than 5 times improved shear strength on aluminum substrates compared to a widely studied commercial benzoxazine resin. Derivative synthesis identified the catechol moiety as an important design feature in the adhesive performance and curing behavior of this bioinspired thermoset. Favorable mechanical properties comparable to commercial resin were maintained, and glass transition temperature and char yield under nitrogen were improved. Blending of monomers with bioinspired main‐chain polybenzoxazine derivatives provided formulations with enhanced shear adhesive strengths up to 16 MPa, while alloying with commercial core–shell particle‐toughened epoxy resins led to shear strengths exceeding 20 MPa. These results highlight the utility of bioinspired design and the use of biomolecules in the preparation of high‐performance thermoset resins and adhesives with potential utility in transportation and aerospace industries and applications in advanced composites synthesis.
Keywords: adhesives, bioinspired materials, polybenzoxazines, polymers, surface chemistry
Introduction
Thermoset polymers, cross‐linked networks of repeating organic units, play a vital role in the advanced materials arena due to their high durability and favorable mechanical, chemical, and thermal properties. Among commonly employed classes of these materials are phenolics, epoxies, polyurethanes, polyimides, acrylics, and cyanate esters.1 In recent years, polybenzoxazines have garnered much interest as a promising class of thermoset resins.2 Conventionally prepared from phenol, formaldehyde, and primary amine precursors, 1,3‐benzoxazine monomers are cured by a thermally‐accelerated cationic ring‐opening polymerization without the need for initiators or catalysts (Figure 1 a).3 Thermally cured polybenzoxazines possess a suite of desirable properties, including low volatile formation and near‐zero shrinkage during curing, good thermal stability, and high mechanical strength, making them particularly attractive materials in the transportation and aerospace industries, and for the preparation of high‐performance composites.4
Figure 1.
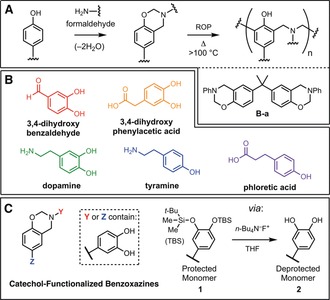
Benzoxazine chemistry and catechol‐derived benzoxazine materials. A) Benzoxazine monomers are readily prepared from phenols, primary amines, and formaldehyde, and undergo thermally‐accelerated ring opening polymerization to yield polybenzoxazines. The commercial benzoxazine resin, B‐a, was used as a benchmark material in this work. B) Biologically occurring phenols were selected as starting materials to introduce novel functionality and properties into designed benzoxazine monomers. C) Bioinspired benzoxazines described in this work contain deprotected catechols prior to curing and are prepared using a silyl ether protecting group strategy.
One of the most attractive features of benzoxazine resins is their rich molecular design flexibility, which enables tailoring of the monomer functionality and the thermoset properties for their end use. Introduction of various functional groups at the amine or phenol fragment in many cases has imbued benzoxazines with interesting and useful properties, including enhanced reactivity, reduced polymerization temperature, additional chemical cross‐linking mechanisms, flame‐retardant characteristics, modified mechanical properties, shape memory and self‐healing behaviors, among others.2a, 5 Many favorable properties of polybenzoxazines have been attributed to complex intermolecular and intramolecular hydrogen bonding within the thermoset network.4, 6 However, a six‐membered intramolecular hydrogen bond between the phenol hydrogen and amine nitrogen in the polybenzoxazine backbone may be of particular relevance to adhesion in that it reduces the material surface energy, weakening interfacial interactions with other materials.7 Few studies have concentrated on enhancing the adhesive performance of polybenzoxazines, and many of these studies emphasize formulation and alloying rather than benzoxazine monomer design to improve adhesion.8
An important recent trend in polymer chemistry research is the preparation of monomers and polymers from bio‐based precursors.9 This interest is driven by both a growing demand for sustainable materials and the rich chemical functionality of biologically‐derived molecules compared to conventional petroleum‐derived building blocks.9a Through advanced microbial engineering, natural and bioengineered metabolic pathways can provide access to an array of monomers and synthetic precursors with diverse structures and properties that are challenging to obtain through synthetic methods alone.10 Furthermore, the development of high throughput genomic engineering tools that integrate automation, machine learning, and molecular biology may enable cost‐effective and industrial‐scale access to the molecular diversity present in living systems.11 This unique pool of potential starting materials includes a range of phenolic molecules that are just beginning to be explored as building blocks for 1,3‐benzoxazine monomers.12
Nature not only provides access to constituents with useful chemical handles for materials synthesis, but also delivers inspiration for synthetic polymers and engineering materials with extraordinary properties and behaviors.13 In our material design, we take inspiration from marine mussels, which have the ability to adhere strongly to a wide range of substrates by secretion of threads terminated by adhesive plaques (the byssus).14 The adhesive proteins found in the plaques are rich in the non‐canonical amino acid 3,4‐dihydroxyphenylalanine (DOPA), which bears a catechol side‐chain.15 The catechol is recognized as a versatile and strong molecular adhesive that can be effective even in wet environments. These mussel adhesive proteins have inspired research on a number of synthetic adhesives for application in dry conditions and in aqueous or biological media, wherein the catechol plays a key role in adhesive and cohesive interactions in the materials.16 Herein we report a family of bioinspired benzoxazine monomers, synthesized in part from phenol and catechol metabolites (Figure 1 b), with enhanced adhesive performance compared to a widely studied and commercially available benzoxazine resin, B‐a (Huntsman Advanced Materials, Figure 1 a).
While mussel‐inspired catechol chemistry has been highlighted in a wide range of synthetic materials, only one recent report describes its application in the context of polybenzoxazines, in which a mono‐benzoxazine resin was prepared from pyrocatechol as the phenol reactant.8f Thermally‐accelerated ring‐opening polymerization unmasked the catechol functionality to provide a thermoset polymer with improved adhesion strength on stainless steel compared to derivatives prepared from the corresponding 1,3‐ or 1,4‐benzenediols. Earlier studies reported using pyrocatechol or the 3‐substituted catechol natural product urushiol to prepare benzoxazine resins which also unveil catechols upon curing, but the adhesive performance of these resins was unexplored.17 In this work, phenol and catechol metabolites were transformed into benzoxazine monomers and polymers with the intention of preparing high‐performance thermoset adhesives. In contrast to previous work, we set out to synthesize benzoxazine monomers with free catechol groups (Figure 1 c), reasoning that in mussel adhesion the catechols in adhesive proteins are unprotected during processing, secretion and solidification of the byssal plaque. Hypothesizing that this would lead to improved adhesive performance, a protecting group strategy was applied to access a family of bioinspired benzoxazines 2 from the corresponding silyl ether protected derivatives 1.
Results and Discussion
Catechol‐Modified Benzoxazine Synthesis
Phenols and catechols both participate in the Mannich reaction commonly employed to prepare 1,3‐benzoxazine monomers, complicating the use of unprotected catechol‐containing precursors in the traditional monomer synthesis. Indeed, catechol derivatives are viable starting materials for the synthesis of monofunctional and bifunctional benzoxazine resins.8f, 17 To access catechol‐modified benzoxazine monomers, a protecting group strategy was developed. Informed by stability studies carried out with a model benzoxazine monomer (C‐m) prepared from para‐cresol and 2‐methoxyethylamine, silyl ethers were identified among common catechol protecting groups, such as cyclic borate, acetals, acetate esters, and benzyl ethers, as promising candidates due to their facile removal under mild conditions by treatment with nucleophilic fluoride reagents (Supporting Information, Figure S7).18 The acid‐sensitive nature of the benzoxazine ring is well documented, precluding our use of acid‐cleavable protecting groups like acetonide and cyclic borate.19 Consistent with previous reports, the benzoxazine ring of C‐m decomposed under catalytic hydrogenation conditions, preventing the use of benzyl ethers.20 While 1,3‐benzoxazines are stable in alkaline aqueous media, the rate of autooxidation of catechols increases under basic conditions if oxygen is not rigorously excluded, which deterred our application of acetate esters.21 Incubation of C‐m with stoichiometric amounts of tetrabutylammonium fluoride (TBAF) or pyrocatechol in tetrahydrofuran resulted in no appreciable degradation, assessed by thin‐layer chromatography (TLC), and intact monomer was recovered after aqueous workup, indicating that both deprotecting agent and unmasked catechol are tolerated under the conditions tested.
Encouraged by this result, tert‐butyldimethylsilyl‐protected (TBS‐protected) monomer 1 a was synthesized incorporating 3,4‐dihydroxyphenylacetic acid and tyramine (Figure 2 a). Treatment of the monomer with a slight excess of TBAF at 4 °C in tetrahydrofuran furnished the catechol‐modified monomer 2 a in high yield after workup. Intact benzoxazine rings and removal of protecting groups were confirmed by 1H‐NMR spectroscopy, in which the characteristic oxazine resonances appear as singlets at 5.33 and 4.57 ppm, and 5.29 and 4.51 ppm for 1 a and 2 a, respectively (red arrows, Figure 2 a). Furthermore, this product immediately produced an intense dark spot on TLC when treated with ferric chloride stain at room temperature, a reagent effective for the detection of 1,2‐dihydroxybenzene moieties.22 This synthetic approach was found to be generalizable for a family of catechol‐modified benzoxazine monomers 2 a–f prepared from biologically occurring phenolic precursors (Figure 2 b, see Supporting Information for experimental details). All products were obtained from the corresponding TBS‐protected derivatives 1 a–f as solids in fair to excellent yields.
Figure 2.
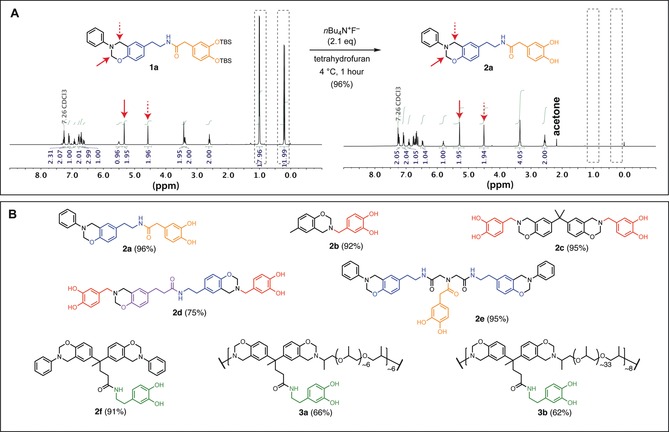
Protecting group strategy used to prepare bioinspired benzoxazine monomers. A) TBS‐protected benzoxazine monomers are readily deprotected with TBAF to provide catechol‐modified benzoxazine monomers, demonstrated in the conversion of 1 a to 2 a. Peaks corresponding to TBS groups disappear after deprotection (grey dashed boxes), and characteristic benzoxazine resonances are present prior to and following deprotection in 1H‐NMR spectra (red arrows). B) This strategy was applied to access a family of bioinspired benzoxazines from biological precursors; yields of the deprotection reactions are shown in parentheses. Colors correspond to the naturally occurring building blocks used to prepare the highlighted fragment, which have the potential to be microbially sourced in the future. (red: 3,4‐dihydroxybenzaldehyde; orange: 3,4‐dihydroxyphenylacetic acid; green: dopamine; blue: tyramine; violet: phloretic acid).
The method also extended to the synthesis of main‐chain polybenzoxazine derivatives 3 a and 3 b, prepared from diphenolic acid, dopamine, and commercial Jeffamines (telechelic amine‐functional polypropylene oxide). Inspired by previous efforts by Sawaryn and colleagues to improve polybenzoxazine mechanical properties by blending with main‐chain variants, these derivatives were prepared as potential performance‐modifying additives for small‐molecule derivatives 2 a–f.23 Notably, 3 b was isolated as a tacky and tough amorphous semi‐solid, exhibiting properties common of pressure sensitive adhesives (see Supporting Information, Figures S8–S10).24
Lap‐shear Adhesion Testing
The adhesive strengths of polybenzoxazines formed by curing of monomers 2 a–f, as well as commercial resin B‐a were surveyed in single‐lap shear on aluminum 6061 (Al 6061) adherends according to ASTM D1002‐10 (Figures 3 a and S11).25 Aluminum was selected due to its widespread use in the transportation and aerospace industries which are promising application areas for benzoxazine resins.2c Monomers were coated onto aluminum struts using a solvent casting method, followed by drying under vacuum. The struts were overlapped in antiparallel fashion, clamped and cured in an air oven to induce ring‐opening polymerization (Figure 3 a, colored boxes). The adhesive strengths were measured by shear testing at room temperature and the failure modes were determined by visual inspection of fractured lap joints (see Supporting Information for details, Figures S12–S14). Commercial B‐a exhibited an adhesive strength of 1.5±0.4 MPa. Addition of 4‐methylcatechol 4 at a 1:1 stoichiometric ratio did not significantly affect the bond strength (1.4±0.4 MPa), indicating that straightforward blending with small‐molecule catechols in similar molar ratios as those in our bioinspired monomers does not enhance B‐a adhesive performance (Figure 3 b). Among the catechol‐modified derivatives 2 a–f, the monovalent derivatives 2 a and 2 b provided the lowest adhesion strengths of 1.5±0.6 and 0.6±0.3 MPa, respectively. We attribute this comparatively poor performance to low crosslinking and consequentially weaker mechanical properties typical of monovalent benzoxazines compared to multivalent derivatives.2a, 26 For bis‐catechol‐bis‐benzoxazine derivatives 2 c and 2 d, a marked increase in adhesive strength compared to B‐a was achieved (4.4±0.1 MPa and 4.6±0.6 MPa, respectively). However, the failed lap joints for samples 2 a–2 d showed evidence of voids with a mixed failure mode, including clear areas of cohesive failure (Figure S12). Mono‐catechol‐bis‐benzoxazine 2 e emerged as a promising high‐strength adhesive candidate, with a lap‐shear strength over five times that of B‐a on degreased Al 6061. While 2 e also exhibited a mixed failure mode, the bond line was more homogeneous than those for 2 a–2 d. Analog 2 f, with equivalent catechol:benzoxazine stoichiometry compared to 2 e, exhibited a significantly lower adhesion strength of 2.8±0.5 MPa, indicating that factors other than benzoxazine‐to‐catechol stoichiometry, such as the monomer scaffold and linker structures, contribute significantly to the thermoset adhesive performance. Blending of B‐a and 2 e in a 1:1 molar ratio provided an adhesion strength that was comparable to those of 2 c and 2 d (5.2±1.1 MPa), demonstrating that adhesion can be tuned by addition of catechol‐modified monomers to resins that lack the catechol functionality.
Figure 3.
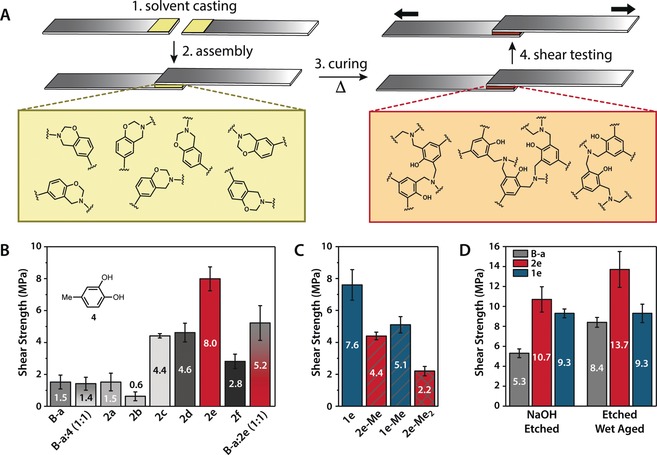
Lap shear adhesion testing of bioinspired benzoxazine thermosets on aluminum 6061. A) Preparation of lap joints by solvent casting monomers onto aluminum adherends and drying (1), lap joint assembly (2), thermal curing in an air oven (3), and shear testing (4). B) Adhesion testing of cured commercial monomer B‐a, catechol modified monomers 2 a–2 f, and 1:1 molar mixtures of B‐a:2 e and B‐a:4‐methylcatechol. Shear testing performed at a rate of 0.5 % strain/minute and at least three lap joints per sample group were tested. Error bars represent standard deviation of adhesive strength. C) Adhesion testing of cured TBS‐protected 1 e and methylated derivatives to probe the importance of the catechol moiety in 2 e for adhesive performance. D) Impact of adherend roughening (etched) and accelerated aging at 63 °C for 14 days on shear adhesive strength.
To investigate the importance of the catechol fragment on the adhesive performance of 2 e, we compared the lap‐shear strength of its TBS‐protected precursor 1 e, measured as 7.6±1.0 MPa (Figure 3 c). The adhesive strength, unexpectedly, was statistically equivalent to that of 2 e (p=0.51, student's t‐test). This result may be explained by a substrate effect leading to deprotection of silyl ethers near the aluminum interface. Preparative alumina‐mediated deprotection of silyl ethers has been previously reported.27 Furthermore, the ability of silyl ethers to undergo migration reactions may unmask phenol and catechol species at the adhesive interface during curing at high temperatures.28 Ring‐opening polymerization of benzoxazines produces backbone phenols that may act as potential acceptors to facilitate silyl ether migration. Partial deprotection of catechols and silyl ether migration in a model precursor was observed after heating on an aluminum surface (see Supporting Information, Figure S25). Additionally, X‐ray photoelectron spectroscopy (XPS) analysis of cured 1 e on the surface of failed lap joints exhibited higher oxygen content and lower carbon content than expected, consistent with deprotection of catechols at the metal‐benzoxazine interface (see Supporting Information, Figure S48). To prevent the possibility of protecting group cleavage or migration during curing, 3‐methoxy (2 e‐Me) and 3,4‐dimethoxy (2 e‐Me2) analogs were synthesized in which one or both of the phenolic hydroxyls were masked as thermally stable methyl ethers, respectively. 2 e‐Me was prepared from the mono‐TBS‐protected 1 e‐Me, which was also applied in lap shear adhesion testing. A progressive decline in adhesive strength was observed for this series of derivatives as catechol hydroxyls were replaced with methoxy groups. The shear strength of 2 e‐Me was 1.8 times lower than 2 e, while the dimethoxy derivative 2 e‐Me2 possessed an adhesive strength only slightly higher than that of B‐a (2.2±0.3 MPa). This result indicates a significant contribution by the catechol moiety to the adhesive performance of 2 e, whether through adhesive interactions with the substrate, or by influencing thermoset crosslinking density and bulk mechanical properties.
The impact of substrate preparation and accelerated aqueous aging on the adhesive performance of 2 e was investigated, benchmarking against B‐a. Protected benzoxazine 1 e was included for comparison, given its similar performance to 2 e on untreated Al 6061. Increasing adherend surface roughness can increase adhesive strength by encouraging mechanical interlocking and increasing substrate surface area available to interact with an applied adhesive.29 The substrate roughness was increased by a previously reported etching protocol involving sonication in aqueous 0.1 n sodium hydroxide.30 We found that alkaline etching enhanced the adhesive strength for all resins tested. Resin 2 e exhibited the highest bond strength of 10.7±1.3 MPa (Figure 3 d), although B‐a exhibited a dramatic 3.5‐fold increase in adhesive strength to 5.3±0.4 MPa. As in untreated substrates, the adhesive strength for 1 e was comparable to 2 e (9.3±0.4 MPa). Accelerated aging of bonded lap joints was carried out in deionized water at 63 °C for 14 days.31 The adhesive strengths of B‐a and 2 e increased by 58 % and 28 % to 8.4±1.8 MPa and 13.7±0.92 MPa, respectively, while the bond strength of 1 e remained unchanged (9.3±1.4 MPa). Although water uptake was not assessed, the increase in bond strength for B‐a and 2 e may be due to plasticization of the cured resins by water, which has been previously reported for benzoxazine composites.2c, 32 The lack of change in adhesion strength for 1 e may represent a decreased water uptake and plasticizing effect compared to that observed for B‐a and 2 e. High shear strength of cured 2 e and 1 e was also observed on sanded 304 stainless steel, indicating that robust adhesion can be achieved on other metal substrates (see Supporting Information, Figure S15).
Flexural Testing and Thermal Stability
The high adhesive strengths of 1 e and 2 e on aluminum prompted our evaluation of their bulk mechanical properties, benchmarking with those of commercially available B‐a. Rectangular samples were prepared for 3‐point flexural testing by compression molding at 205 °C and 550 psi for 2 hours (Figure 4 a, see Supporting Information). The proximity of the melting endotherm (T m=181 °C) and curing exotherm (T max=196 °C), as well as the accelerated curing catalyzed by phenolic hydroxyl groups on the catechol moiety, provided little window for melt processing and degassing of 2 e (Figure 4 b), resulting in specimens riddled with voids (see Supporting Information, Figure S26).33 B‐a and 1 e in comparison possess wide processing windows with well‐separated melting points and curing exotherms. To allow consistent comparisons of polybenzoxazine mechanical properties, however, compression molding was applied for all three resins. Cured B‐a provided red‐orange specimens, while those obtained from 1 e and 2 e were significantly darker brown in color, indicating potential oxidation of catechol functional groups during curing (Figure 4 c).34 The absorbance spectrum of a thin film of cured 2 e exhibited a broad peak from 500–780 nm, consistent with the oxidative polymerization of catechols (Figure S29).35 This peak was absent in the absorbance spectrum of cured B‐a. Fourier transform infrared (FTIR) spectroscopic analysis of cured samples revealed a disappearance of characteristic peaks affiliated with the oxazine ring at 940, 947 and 946 cm−1 for B‐a, 2 e and 1 e, respectively, denoting effective ring‐opening polymerization (Figures S30–S32).36 High‐resolution XPS analysis of C1s and O1s peaks for B‐a and 2 e before and after curing corroborated this result (Figures S39 and S40). Furthermore, the carbonyl content in thermoset 2 e increased in both C1s and O1s high resolution XPS spectra, verifying the partial oxidation of catechol moieties to o‐quinones during thermoset curing.
Figure 4.
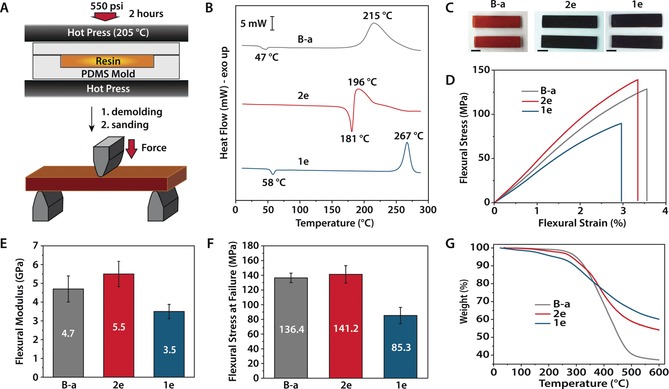
Mechanical and thermal characterization of B‐a, 2 e, and 1 e formulations. A) Preparation of flexural testing samples by compression molding. B) DSC thermograms revealing curing behavior of monomers B‐a, 2 e and 1 e, ΔT=10 °C min−1 in nitrogen. C) Representative appearance of flexural testing samples. Scale bars correspond to 0.5 cm. D) Representative stress‐strain behavior in flexion of cured thermosets. E) Flexural modulus of cured thermosets (N=8, 9 and 6 for B‐a, 2 e and 1 e, respectively). F) Flexural strength of cured thermosets (N=8, 9 and 6 for B‐a, 2 e and 1 e, respectively). G) Thermal gravimetric analysis of cured thermosets prepared by compression molding (N2, heating rate of 10 °C min−1).
Flexural testing by 3‐point bending revealed an elastic response with minimal plastic deformation, terminated by brittle failure (Figure 4 d). The flexural modulus and strength measured for our benchmark material, B‐a, are consistent with values previously reported in literature and provided by the manufacturer at 4.71±0.69 GPa and 136.4±6.5 MPa, respectively (Figures 4 e and f).37 Thermoset 2 e possesses a slightly higher flexural modulus (5.52±0.80 GPa) and similar flexural strength (141.2±11.8 MPa) compared to B‐a, indicating that the strong adhesive properties of 2 e do not come at the cost of favorable mechanical performance. Cured 1 e, in contrast, exhibited substantially lower flexural modulus and strength (3.47±0.38 GPa and 85.3±11.0 MPa, respectively). The bulky silyl ethers in this material likely disrupt hydrogen bonding interactions that contribute to polybenzoxazine mechanical properties, leading to a decline in modulus and strength relative to 2 e and B‐a.6 The elastic moduli measured by 3‐point bending were in close agreement with values determined by nanoindentation measurements (see Supporting Information). Furthermore, the elastic moduli of B‐a and 1 e cured at atmospheric pressure were comparable to those measured for compression molded samples, suggesting that high‐pressure curing does not dramatically influence the mechanical properties for these resins (Figure S38). Differential scanning calorimetry (DSC) of compression molded samples revealed broad glass transitions centered at 147, 183, and 149 °C for B‐a, 2 e, and 1 e, respectively (Figures S33–S35). 2 e cured in a temperature ramp up to 295 °C under nitrogen at 1 atmosphere presented an increased glass transition temperature (T g) of 208 °C in subsequent heating cycles, compared to a T g of 141 °C for B‐a cured under identical conditions (Table S4). This suggests that curing of 2 e at increased temperatures or under inert atmosphere can improve the high temperature performance of this resin.
Another attractive feature of polybenzoxazine materials is their good thermal stability, leading to their development as flame‐resistant materials.2a, 5, 38 The thermal stability of cured 1 e and 2 e were compared to B‐a up to 600 °C in nitrogen by thermal gravimetric analysis of compression‐molded samples (Figure 4 g). Decomposition of polymerized 1 e begins at the lowest temperature in the series, with 10 % weight loss occurring by 295 °C (T 10 %) at a heating rate of 10 °C min−1. The T 10 % values for cured 2 e and B‐a are comparable at 322 and 328 °C, respectively. However, the char yields at 600 °C were higher for both bioinspired derivatives (60 % and 54 % for 1 e and 2 e, respectively) compared to B‐a (37 %), indicating a lower quantity of volatiles formed during decomposition at elevated temperatures. While 1 e produces the greatest char yield in the series, the earlier onset of degradation is potentially prohibitive for applications of this resin at high temperature. The thermal stabilities of cured B‐a and 2 e were also assessed in air up to 600 °C (Figure S52). In this case, 2 e begins to degrade at a lower temperature than B‐a, with T 10 % at 325 °C and 366 °C, respectively. Both resins produce no residue upon heating up to 600 °C, and exhibit maximum rates of weight loss at 540 °C for 2 e and 549 °C for B‐a, interpreted as degradation of char formed by heating the polymers up to this temperature under oxidizing conditions.38a
Enhancing Shear Strength Through Formulation
Lastly, we hypothesized that the promising adhesive performance of 2 e could be further optimized through formulation. In all lap‐shear tests for 2 e we observed a mixed failure mode, indicated by areas of adhesive failure and cohesive failure in the fractured lap joints. We rationalized that blends of 2 e with toughening agents may have improved adhesion strength. Main‐chain polybenzoxazines bearing soft polymer segments have previously been copolymerized with small‐molecule benzoxazine monomers to improve the fracture toughness of polybenzoxazine resins.23 Taking a similar approach, main‐chain derivatives 3 a and 3 b were applied as performance‐modifying additives by blending with 2 e at various weight ratios and measuring adhesive strength on etched aluminum (see Supporting Information, Figures 5 a and b). A 4:3 weight ratio of 2 e:3 a and 2 e:3 b provided the greatest improvement in adhesive strength while maintaining a uniform bond line, with the latter exhibiting an adhesive strength of 16.1±1.6 MPa, a 50 % improvement over 2 e alone. Increasing the loading of 3 a and 3 b beyond this point produced decreased adhesive strengths. This could be caused by dilution of 2 e below a critical threshold as more poly(propylene oxide) (PPO) is added to the formulation in the form of the main‐chain additives. Indeed, pure 3 a and 3 b achieve relatively low adhesive strengths (2.5±0.3 MPa and 0.30±0.04 MPa, respectively). Increased amounts of main‐chain additives would also be expected to influence the mechanical properties of the cured resins as additional soft PPO content is added.23 On the other hand, when 2 e and 3 a were combined in a mass ratio of less than 2:1 the adhesive strengths were unchanged compared to 2 e alone, and the analogous compositions with 3 b produced a sharp decline in adhesive strength (Figure S20). Furthermore, lap joints prepared with 2 e:3 a and 2 e:3 b blends in this regime produced highly heterogeneous bond lines, exhibiting evidence of macroscopic phase separation, and failure modes with areas of adhesive and cohesive failure. This suggests that 3 a and 3 b are not fully compatible with 2 e in these compositions, forming a phase‐separated mixture during curing. Formulations of 4:3 and 1:1 weight ratio, in contrast, provided bond lines with improved macroscopic homogeneity for both main‐chain derivatives, indicating that 3 a and 3 b are compatible with 2 e at higher loadings (see Supporting Information, Figures S21–S22). Also, these formulations exhibited predominantly adhesive failure modes indicating an improvement in the cohesive properties of the cured resin, in some cases at the cost of bond strength.
Figure 5.
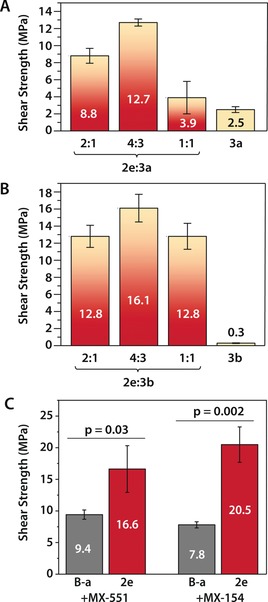
Shear strength of copolymerized blends of 2 e with main‐chain polybenzoxazines and commercial tougheners. Error bars represent standard deviation in adhesive strength for 3 lap joints. A) 2 e copolymerized with 3 a at various weight ratios. B) 2 e copolymerized with 3 b at various weight ratios. C) Lap shear adhesive strength of copolymerized blends of B‐a and 2 e with commercial core–shell rubber‐toughened epoxy additives MX‐551 and MX‐154 (Kaneka, Inc.); the weight ratio of benzoxazine monomer to toughened epoxy additive in each formulation is 20:1.
Commercial additives such as elastomers, thermoplastic polymers, and core–shell rubber microparticles have been previously applied as toughness modifiers for phenol–formaldehyde and polybenzoxazine resins.39 Furthermore, copolymerization of benzoxazines with epoxy thermosets provides alloys with improved mechanical strength compared to polybenzoxazine alone.40 We therefore explored commercially available core–shell rubber toughened liquid epoxy additives Kane Ace MX‐551 and MX‐154 (Kaneka) as performance enhancers for 2 e. MX‐551 and MX‐154 are, respectively cycloaliphatic‐based or bisphenol‐A‐based liquid epoxy resins containing a stable dispersion of poly(styrene‐co‐butadiene) rubber core microparticles with epoxy‐modified shells.41 In the copolymerization of epoxy and conventional benzoxazine monomers, the pendant phenol of ring‐opened benzoxazines acts as a nucleophilic curing agent for epoxy in the system.40 In the case of 2 e, both catechols and ring‐opened benzoxazine phenols can react with epoxy, which we hypothesized would improve the compatibility of 2 e with MX‐551 and MX‐154. Lap‐shear adhesion samples were prepared by mixing 2 e with MX‐551 and MX‐154 at a 20:1 mass ratio (see Supporting Information). In order to determine the importance of the benzoxazine monomer on the adhesive performance, comparative formulations were prepared with B‐a in place of 2 e. Improved adhesive strengths were observed for all compositions compared to either benzoxazine alone, with formulation composed of 2 e and MX‐154 achieving a bond strength of 20.5±2.8 MPa (Figure 5 c). Notably, compositions prepared with monomer 2 e presented a substantial adhesive advantage over those prepared with B‐a, indicating that the adhesive performance is not solely due to the epoxy content of the alloys. This demonstrates that bioinspired 2 e can be formulated with commercial toughening agents with a simple mixing protocol to achieve structural adhesives with excellent bond strength compared to conventional benzoxazine resins. Furthermore, while adhesive failure was observed for alloys of B‐a, a mixed failure mode was observed for blends with 2 e, suggesting that further improvement in bond strength may be achieved by optimization of the adhesive bulk mechanical properties through future formulation efforts (Figure S24).
Conclusion
In summary, we have developed a general approach to prepare a family of novel bioinspired benzoxazine monomers and main‐chain derivatives from biological precursors, enabling the incorporation of deprotected catechols into benzoxazine monomers. These materials undergo thermally accelerated ring‐opening polymerization to provide cured thermosets, and the catechol acts as a curing accelerator. Multivalent catechol‐based bisbenzoxazines exhibited enhanced adhesive strength compared to a widely‐employed commercially available resin B‐a. In particular, bioinspired resin 2 e exhibits superior adhesion up to 5 times greater than B‐a on aluminum while maintaining advantageous bulk mechanical properties, and good thermal stability, highlighting this novel resin as a promising high‐performance thermoset adhesive. The adhesive strength of 2 e can be further improved through adherend surface treatments, formulation with main‐chain benzoxazines 3 a and 3 b, and by blending with commercially available toughened epoxy resins Kane Ace MX‐551 and MX‐154. Lastly, the catechol moiety and benzoxazine architectures were found to be critical to the observed performance of the resins, demonstrating the advantage of biological building block functionality and structure in the design of advanced adhesives. We expect that the versatility of our synthetic strategy will enable future exploration of new benzoxazine monomer designs from a structurally diverse array of phenolic biomolecules with potential utility in a variety of demanding applications in the transportation and aerospace industries.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported with funding from the Defense Advanced Research Projects Agency (DARPA) as part of the Living Foundries program. The views, opinions, and findings presented are those of the authors, and do not represent the official views or policies of the Department of Defense or the U.S. Government. The authors thank Huntsman Advanced Materials for donation of B‐a and Kaneka, Inc. for providing samples of Kane Ace MX‐154 and MX‐551 tougheners for this research. We thank the UC Berkeley College of Chemistry NMR facility, the LBNL Berkeley Catalysis Facility, and QB3 Biomolecular Nanotechnology Center and staff for assistance with analytical and compression molding equipment. The authors wish to acknowledge Dr. C. Michael McGuirk for assistance with thermal gravimetric analysis.
C. J. Higginson, K. G. Malollari, Y. Xu, A. V. Kelleghan, N. G. Ricapito, P. B. Messersmith, Angew. Chem. Int. Ed. 2019, 58, 12271.
References
- 1. Dodiuk H., Goodman S. H., Handbook of Thermoset Plastics, Elsevier Science, Amsterdam, 2013. [Google Scholar]
- 2.
- 2a. Ishida H., Agag T., Handbook of Benzoxazine Resins, Elsevier, Amsterdam, 2011; [Google Scholar]
- 2b. Rimdusit S., Jubsilp C., Tiptipakorn S., Alloys and Composites of Polybenzoxazines: Properties and Applications, Springer, Singapore, 2013; [Google Scholar]
- 2c. Comer A. J., Ray D., Clancy G., Obande W. O., Rosca I., McGrail P. T., Stanley W. F., Compos. Struct. 2019, 213, 261–270. [Google Scholar]
- 3. Han L., Salum M. L., Zhang K., Froimowicz P., Ishida H., J. Polym. Sci. Part A 2017, 55, 3434–3445. [Google Scholar]
- 4. Ishida H., Allen D. J., J. Polym. Sci. Part B 1996, 34, 1019–1030. [Google Scholar]
- 5. Ishida H., Froimowicz P., Advanced and Emerging Polybenzoxazine Science and Technology, Elsevier Science, Amsterdam, 2017. [Google Scholar]
- 6. Kim H.-D., Ishida H., J. Phys. Chem. A 2002, 106, 3271–3280. [Google Scholar]
- 7. Shen D., Liu C., Sebastián R. M., Marquet J., Schönfeld R., J. Appl. Polym. Sci. 2016, 133, 44099. [Google Scholar]
- 8.
- 8a. McGee J. D., Kreiling S., Lehmann S. L., Taden A., U.S. Patent Application No. 12/945,256, 2011;
- 8b. Li H., Gu J., Wang D., Qu C., Zhang Y., J. Adhes. Sci. Technol. 2017, 31, 1796–1806; [Google Scholar]
- 8c. Monisha, Swapnil S., Bimlesh L., Green Mater. 2017, 5, 94–102; [Google Scholar]
- 8d. Liu C., Sun M., Zhang B., Zhang X., Li J., Li Q., J. Appl. Polym. Sci. 2017, 134, 44547; [Google Scholar]
- 8e. Shukla S., Mahata A., Pathak B., Lochab B., RSC Adv. 2015, 5, 78071–78080; [Google Scholar]
- 8f. He Y., Gao S., Lu Z., Polymer 2018, 158, 53–58. [Google Scholar]
- 9.
- 9a. Lligadas G., Tuzun A., Ronda J. C., Galia M., Cadiz V., Polym. Chem. 2014, 5, 6636–6644; [Google Scholar]
- 9b. Rodriguez Arza C., Froimowicz P., Ishida H., RSC Adv. 2015, 5, 97855–97861; [Google Scholar]
- 9c. Puchot L., Verge P., Fouquet T., Vancaeyzeele C., Vidal F., Habibi Y., Green Chem. 2016, 18, 3346–3353; [Google Scholar]
- 9d. Ricapito N. G., Ghobril C., Zhang H., Grinstaff M. W., Putnam D., Chem. Rev. 2016, 116, 2664–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Lee S. Y., Kim H. U., Chae T. U., Cho J. S., Kim J. W., Shin J. H., Kim D. I., Ko Y.-S., Jang W. D., Jang Y.-S., Nat. Catal. 2019, 2, 18–33; [Google Scholar]
- 10b. John G., Nagarajan S., Vemula P. K., Silverman J. R., Pillai C. K. S., Prog. Polym. Sci. 2019, 92, 158–209. [Google Scholar]
- 11. Serber Z., Dean E. J., Manchester S., Gora K., Flashman M., Shellman E., Kimball A., Szyjka S., Frewen B., Treynor T., U.S. Patent No. 9,988,624 B2, 2018.
- 12. Bonnaud L., Chollet B., Dumas L., Peru A. A. M., Flourat A. L., Allais F., Dubois P., Macromol. Chem. Phys. 2019, 220, 1800312. [Google Scholar]
- 13.
- 13a. Wegst U. G. K., Bai H., Saiz E., Tomsia A. P., Ritchie R. O., Nat. Mater. 2015, 14, 23–36; [DOI] [PubMed] [Google Scholar]
- 13b. Munch E., Launey M. E., Alsem D. H., Saiz E., Tomsia A. P., Ritchie R. O., Science 2008, 322, 1516–1520; [DOI] [PubMed] [Google Scholar]
- 13c. Yu Z.-L., Yang N., Zhou L.-C., Ma Z.-Y., Zhu Y.-B., Lu Y.-Y., Qin B., Xing W.-Y., Ma T., Li S.-C., Gao H.-L., Wu H.-A., Yu S.-H., Sci. Adv. 2018, 4, eaat7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Waite J. H., J. Exp. Biol. 2017, 220, 517–530; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Li L., Zeng H., Biotribology, Vol. 5, Wiley, Hoboken, 2016, pp. 44–51. [Google Scholar]
- 15.
- 15a. Papov V. V., Diamond T. V., Biemann K., Waite J. H., J. Biol. Chem. 1995, 270, 20183–20192; [DOI] [PubMed] [Google Scholar]
- 15b. Waite J. H., Qin X., Biochemistry 2001, 40, 2887–2893. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Lee B. P., Messersmith P. B., Israelachvili J. N., Waite J. H., Annu. Rev. Mater. Res. 2011, 41, 99–132; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Brubaker C. E., Messersmith P. B., Langmuir 2012, 28, 2200–2205; [DOI] [PubMed] [Google Scholar]
- 16c. Sparks B. J., Hoff E. F. T., Hayes L. P., Patton D. L., Chem. Mater. 2012, 24, 3633–3642; [Google Scholar]
- 16d. Meredith H. J., Jenkins C. L., Wilker J. J., Adv. Funct. Mater. 2014, 24, 3259–3267; [Google Scholar]
- 16e. Ahn B. K., Das S., Linstadt R., Kaufman Y., Martinez-Rodriguez N. R., Mirshafian R., Kesselman E., Talmon Y., Lipshutz B. H., Israelachvili J. N., Waite J. H., Nat. Commun. 2015, 6, 8663; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16f. Mu Y., Wan X., Macromol. Rapid Commun. 2016, 37, 545–550. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Kotzebue L. R. V., de Oliveira J. R., da Silva J. B., Mazzetto S. E., Ishida H., Lomonaco D., ACS Sustainable Chem. Eng. 2018, 6, 5485–5494; [Google Scholar]
- 17b. Xu H., Lu Z., Zhang G., RSC Adv. 2012, 2, 2768–2772; [Google Scholar]
- 17c. Xu H., Zhang W., Lu Z., Zhang G., RSC Adv. 2013, 3, 3677–3682. [Google Scholar]
- 18.
- 18a. Corey E. J., Venkateswarlu A., J. Am. Chem. Soc. 1972, 94, 6190–6191; [Google Scholar]
- 18b. Lee B. P., Bio-inspired Polymers, The Royal Society of Chemistry, London, 2017, pp. 322–353; [Google Scholar]
- 18c. Wuts P. G. M., Greene T. W., Greene's Protective Groups in Organic Synthesis, Wiley, New York, 2012. [Google Scholar]
- 19. Burke W. J., J. Am. Chem. Soc. 1949, 71, 609–612. [Google Scholar]
- 20. Agag T., Arza C. R., Maurer F. H. J., Ishida H., Macromolecules 2010, 43, 2748–2758. [Google Scholar]
- 21.
- 21a. Heacock R. A., Chem. Rev. 1959, 59, 181–237; [Google Scholar]
- 21b. Maier G. P., Bernt C. M., Butler A., Biomater. Sci. 2018, 6, 332–339; [DOI] [PubMed] [Google Scholar]
- 21c. Yang J., Cohen Stuart M. A., Kamperman M., Chem. Soc. Rev. 2014, 43, 8271–8298. [DOI] [PubMed] [Google Scholar]
- 22. Sharma O. P., Bhat T. K., Singh B., J. Chromatogr. A 1998, 822, 167–171. [Google Scholar]
- 23. Sawaryn C., Landfester K., Taden A., Polymer 2011, 52, 3277–3287. [Google Scholar]
- 24. Benedek I., Feldstein M. M., Fundamentals of Pressure Sensitivity, CRC, Boca Raton, 2008. [Google Scholar]
- 25.ASTM, D1002-10 Standard Test Method for Apparent Shear Strength of Single-Lap-Joint Adhesively Bonded Metal Specimens by Tension Loading (Metal-to-Metal), 2010.
- 26.
- 26a. Riess G., Schwob J. M., Guth G., Roche M., Laude B. in Advances in Polymer Synthesis (Eds.: B. M. Culbertson, J. E. McGrath), Springer US, Boston, MA, 1985, pp. 27–49; [Google Scholar]
- 26b. Ning X., Ishida H., J. Polym. Sci. Part B 1994, 32, 921–927. [Google Scholar]
- 27. Feixas J., Capdevila A., Camps F., Guerrero A., J. Chem. Soc. Chem. Commun. 1992, 1451–1453. [Google Scholar]
- 28. Nishimura Y., Chung J., Muradyan H., Guan Z., J. Am. Chem. Soc. 2017, 139, 14881–14884. [DOI] [PubMed] [Google Scholar]
- 29. Jennings C. W., J. Adhes. 1972, 4, 25–38. [Google Scholar]
- 30. Saleema N., Sarkar D. K., Paynter R. W., Gallant D., Eskandarian M., Appl. Surf. Sci. 2012, 261, 742–748. [Google Scholar]
- 31.R. Jensen, D. DeSchepper, D. Flanagan, G. Chaney, C. Pergantis, U.S. Army Research Laboratory, Adhesives: Test Method, Group Assignment, and Categorization Guide for High-Loading-Rate Applications, ARL-ADHES-QA-001-01, Rev. 2.2, 2016.
- 32.A. J. Comer, D. Ray, G. Clancy, W. Obande, W. Stanley, Proceedings of SAMPE Europe SETEC International Technical Conference and Table Top, Tampere, Finland, 2014.
- 33. Kaya G., Kiskan B., Yagci Y., Macromolecules 2018, 51, 1688–1695. [Google Scholar]
- 34. Martinez M. V., Whitaker J. R., Trends Food Sci. Technol. 1995, 6, 195–200. [Google Scholar]
- 35.
- 35a. Su J., Noro J., Fu J., Wang Q., Silva C., Cavaco-Paulo A., J. Cleaner Prod. 2018, 202, 792–798; [Google Scholar]
- 35b. Pillar E. A., Zhou R., Guzman M. I., J. Phys. Chem. A 2015, 119, 10349–10359. [DOI] [PubMed] [Google Scholar]
- 36. Han L., Iguchi D., Gil P., Heyl T. R., Sedwick V. M., Arza C. R., Ohashi S., Lacks D. J., Ishida H., J. Phys. Chem. A 2017, 121, 6269–6282. [DOI] [PubMed] [Google Scholar]
- 37. Allen D. J., Ishida H., J. Appl. Polym. Sci. 2006, 101, 2798–2809. [Google Scholar]
- 38.
- 38a. Yee Low H., Ishida H., Polymer 1999, 40, 4365–4376; [Google Scholar]
- 38b. Liu J., Safronava N., Lyon R. E., Maia J., Ishida H., Macromolecules 2018, 51, 9982–9991. [Google Scholar]
- 39.
- 39a. Gietl T., Lengsfeld H., Altstädt V., J. Mater. Sci. 2006, 41, 8226–8243; [Google Scholar]
- 39b. Liu W. W., Ma J. J., Zhan M. S., Wang K., J. Appl. Polym. Sci. 2015, 132, 41533. [Google Scholar]
- 40. Ishida H., Allen D. J., Polymer 1996, 37, 4487–4495. [Google Scholar]
- 41. Nakajima M., Zhou H., Chao T.-C., Swarup S., U.S. Patent Application No. 14/887,353, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary