

Abstract
We investigated the role of the intrinsic mevalonate cascade in the neuronal cell death (NCD) induced by the inhibition of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase in rat primary cortical neurons cultured from the brains of 17-d-old fetal SD rats. HMG-CoA reductase inhibitors induced NCD [HMG-CoA reductase inhibitor-induced NCD (H-NCD)] in time- and dose-dependent manners. The apoptotic characteristics were revealed by the formation of the DNA ladder and by the electron microscopical observation. During the progression of H-NCD, p53 was induced followed by the expression of Bax. Although the mevalonate completely inhibited H-NCD, the cholesterol did not. Thus, we examined two major metabolites of mevalonate, geranylgeranyl-pyrophosphate (GGPP) and farnesyl-pyrophosphate (FPP), using a novel liposome system for uptake into the cells. GGPP, not FPP, prohibited H-NCD with inhibition of the induction of p53 and Bax. The inhibition of HMG-CoA reductase decreased the amount of membrane-associated Rho small GTPase families, but not Ras small GTPase, and GGPP restored the blockage by HMG-CoA reductase inhibitor in the translocation or redistribution of Rho small GTPase families to membrane. These data indicated that (1) the inhibition of the intrinsic mevalonate cascade induces the apoptotic NCD with the induction of p53 followed by that of Bax, (2) the inhibition of HMG-CoA reductase concomitantly causes blockage of the translocation or redistribution of Rho small GTPase families, not Ras small GTPase, to membrane, and (3) GGPP, not FPP, is one of the essential metabolites in the mevalonate cascade for protecting neurons from H-NCD.
Keywords: GGPP, mevalonate cascade, HMG-CoA reductase, neuron, cell death, p53, Bax
Mevalonate cascade has a crucial role in the supply of cholesterol and is demonstrated at particularly high concentrations in neuronal tissues (Suzuki, 1972). In the developing brain, increases in cholesterol deposition appear to coincide with the elaboration of axonal and dendritic membranes and with the process of myelination (Kishimoto et al., 1965; Cunzer and Davison, 1968; Bass et al., 1970a,b). The 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which converts HMG-CoA to mevalonate, is a rate-limiting enzyme of the mevalonate cascade (Goldstein and Brown, 1990). It was reported that the activity of HMG-CoA reductase is the highest in the early phase of ontogenetic development of the brain and that the parallel change of its activity with the maturation is observed using neuronal cell cultures (Maltese and Volpe, 1979; Volpe et al., 1985). These data clearly indicate that mevalonate cascade plays critical roles for the differentiation, maturation, and maintenance of neuronal tissues.
Mevalonate cascade has been demonstrated to be involved in many biological phenomenon such as proliferation and apoptosis in addition to differentiation, maturation, and maintenance of cellular functions. Recently, it was reported that HMG-CoA inhibitors induced the apoptosis in not only proliferative cell lines (Reedquist et al., 1995; Miquel et al., 1996; Satoh et al., 1996; Padayatty et al., 1997; Lee et al., 1998), but also neurons (Pavlov et al., 1995).
Mevalonate acts as a precursor of not only cholesterol but also isoprenoids for farnesyl and geranylgeranyl molecules, which have an important signaling function (Casey and Seabra, 1996). It has been demonstrated that the ability of an HMG-CoA reductase inhibitor to interfere with the cell cycle progression and induce the apoptosis could be attributed to its activity in suppressing the isoprenylation of proteins rather than interruption of cholesterol synthesis (Chakrabarti and Engleman, 1991; Ortiz et al., 1995). Geranylgeranyl-pyrophosphate (GGPP) and farnesyl-pyrophosphate (FPP), two components of isoprenoids, were reported to be major products of this pathway. The enzymes that catalyze the covalent attachment to farnesyl and geranylgeranyl moieties to proteins like small GTPases are now well characterized (Casey and Seabra, 1996). It was demonstrated that GGPP is an essential compound through the activation of Rho small GTPase in the proliferation of rat astrocytes (Tanaka et al., 1998), rat thyroid FRTL-5 cells (Hirai et al., 1997; Noguchi et al., 1998; Nakamura et al., 1999), rat smooth muscle cells (Terano et al., 1998), human mesangial cells (Nishimura et al., 1999), and human lymphocytes (Tatsuno et al., 1997). These studies were performed using the proliferative cells to clarify the roles of isoprenoids in the proliferation, and the roles of these isoprenoids in the development and maintenance of nonproliferative cells, especially the neurons, have remained to be elucidated.
In the present study, we directed our focus on the mevalonate cascade, especially the two major isoprenoids of GGPP and FPP, in the cell death of rat cultured cortical neurons. We also tried to clarify the mechanism of neuronal cell death from the view points of cell death-related molecules such as p53 and the Bcl-2 protein family.
MATERIALS AND METHODS
Reagents
GGPP, FPP, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO). Antibodies against Rho A, Rho B, and cdc42 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-rat/mouse Bcl-2 and Bax antibodies were purchased from PharMingen (San Diego, CA). Anti-Ras (NCC-RAS-004) antibody was kindly provided from Dr. S. Hirohashi (National Cancer Center Research Institute, Tokyo, Japan) (Kanai et al., 1987). DMEM nutrient mixture F-12 (DMEM/F-12) was purchased from Life Technologies (Gaithersburg, MD). Horseradish-linked anti-rabbit Ig goat antibody, enhanced chemiluminescence (ECL) detection reagent, and nick DNA labeling kit were purchased from Amersham (Buckinghamshire, UK). [α-32P]dCTP was purchased from NEN Life Science (Boston, MA). Human HMG-CoA reductase cDNA probe, lovastatin, simbastatin, and pravastatin were kindly provided by Dr. S. Kurakata (Sankyo Pharmaceutical Company, Tokyo, Japan).
Preparation of liposomes of isoprenoids
Liposomes containing isoprenoids were prepared as described previously (Hirai et al., 1997; Tatsuno et al., 1997; Tanaka et al., 1998). To make liposomes containing each, an aliquot of a mixture of dipalmitoylphosphatidylcholine (5 μmol) and GGPP or FPP (200 μg) was added to a pear shaped flask, and the solvent was removed by rotary evaporation and a vacuum pump. The dried lipid film was then dispersed in 0.5 ml of PBS. Warming the flask to 50°C facilitates smooth dispersion. The liposomes were sonicated and stored at 4°C.
Cell culture
Rat cortical neurons were cultured as described previously (Tatsuno et al., 1992; Morio et al., 1996) with slight modification. Timed-pregnant SD rats were obtained from Charles River Breeding Labs (Tokyo, Japan), and forebrains were removed from 17-d-old fetal SD rats. Meninges were discarded, and the tissues were dispersed by pipetting. Dissociated cortical cells were seeded onto 24-well culture plates for DNA fragmentation assay, 96-well culture plates for MTT assay, and 100 mm culture dishes (Costar Scientific Corporation, Cambridge, MA) precoated with 10 μg/ml poly-l-lysine at a density of 5 × 105 live cells/cm2 for immunoblotting and Northern blotting. Brain cells were cultured with chemically defined medium (DMEM/F-12 containing 2.5 mml-glutamine, supplemented by 5 μg/ml insulin, 100 μg/ml human transferrin, 3 × 10−4m selenium, 1 × 10−4m putrescine, 2 × 10−8mprogesterone, 1 × 10−12m 17β-estradiol, 37.5 μg/ml bovine serum albumin (fatty acid-free), 1 × 10−8mtriiothyronine, and 1 × 10−7m corticosterone), which contained 1.05 mmCa2+ and 0.7 mmMg2+, in an atmosphere of 95% air and 5% CO2 at 37°C for 10 d before the experiments. The purity of neurons at 14 d after culture was >95%, as determined by immunocytochemistry using the antibody against microtubule-associated protein-2 (MAP-2), a specific marker for neurons, (Boehringer Mannheim GmbH, Germany), and the contamination by astrocytes was <5% by immunocytochemistry with antibody against glial fibrillary acidic protein (GFAP), a specific marker of astrocytes (Boehringer Mannheim).
HMG-CoA reductase inhibitor-induced neuronal cell death
Cortical neurons were incubated with certain concentrations of HMG-CoA reductase inhibitors (lovastatin, simvastatin, and pravastatin) in an atmosphere of 95% air and 5% CO2 at 37°C in chemically defined medium for certain periods. The number of live neurons was determined by cell count using fluorescein diacetate propidium iodide and the MTT assay as described below.
MTT assay. The number of live neurons was determined by the mitochondrial conversion of MTT to formazan as detected by the change of optical density at 570 nm. Neurons were incubated with MTT solution (25 μl of MTT at 2 mg/ml in PBS per well; final concentration, 0.4 mg/ml) during the final 3 hr of the culture. At the end of the culture, the cells were solubilized with 50 μl of 20% SDS in 0.02 N HCl and the color intensity was measured on a microplate reader (TOSOH, Tokyo, Japan) at 570 nm.
Cell-counting assay using fluorescein diacetate propidium iodide stain. Fluorescein diacetate crosses the cell membrane and is hydrolyzed by intracellular esterase to produce a green–yellow fluorescent compound. Neuronal injury curtails fluorescein diacetate staining and facilitates propidium iodide penetration and interaction with DNA to yield a bright red fluorescent complex. The percentage of surviving neurons was calculated by assessing the ratio of fluorescein diacetate/propidium iodide staining in photomicrographs.
DNA fragmentation analysis
The cells were washed with PBS and lysed with lysis buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, and 0.5% Triton X-100) for 20 min at 4°C. The samples were centrifuged at 27,000 × g for 15 min at 4°C. The supernatants were extracted with phenol, phenol/chloroform (vol/vol, 1:1), and chloroform. The DNA was precipitated with 1/10 volume of 3m sodium acetate, pH 5.2, and two volumes of ethanol. The DNA was suspended in TE buffer (10 mm Tris-HCl, pH 8.0, and 1 mm EDTA) and treated with 40 μg/ml RNase A for 1 hr at 37°C. The concentrations of DNA were determined by the absorbance at 260 nm. DNA (20 μg) from the samples was subjected to agarose gel electrophoresis on 2% gel in TAE buffer (40 mm Tris-acetate, pH 8.5, and 2 mm EDTA). Then the gel was stained with 0.5 μg/ml ethidium bromide for 10 min, and DNA was visualized under UV light and photographed.
Morphological analysis by electron microscopy
Neurons were fixed with 3.0% glutaraldehyde in 0.1m phosphate buffer, pH 7.4, at 4°C for 30 min, post-fixed with 1% osmium tetroxide, stained in blocks with uranyl acetate, and embedded in Epon 812. Ultrathin sections were cut on a DuPont (Billerica, MA) MT 6000 ultramicrotome, stained with lead citrate, and examined with an IEN-1200EX electron microscope (JEOL Nihon Denshi, Tokyo, Japan).
Cell fractionation and immunoblotting
For whole-cell lysates, the cells were collected and resuspended in cold lysis buffer (in mm): 50 HEPES, pH 7.0, 2 MgCl2, 250 NaCl, 0.1 EDTA, 0.1 EGTA, 1 DTT, 2 Na2VO4, 10 Na4P2O7, 10 NaF, 0.1% NP-40, 0.5 p-amidinophenyl methanesulfonyl fluoride hydrochloride (p-APMSF), and a protease inhibitor cocktail consisting of 2.5 μg/ml of pepstatin A, 2.5 μg/ml of antipain, 2.5 μg/ml of chymostatin, 0.25 mg/ml of leupeptin, and 0.25 mg/ml of antipain. The cells were allowed to lyse on ice for 60 min. The homogenate was centrifuged for 5 min in a microfuge at 4°C, and the supernatant was frozen in aliquots at −70°C.
For subcellular fractionation, cells were disrupted by sonication in hypotonic buffer (in mm: 5 Tris-HCl, pH 7.0, 5 NaCl, 1 CaCl2, 2 EGTA, 1 MgCl2, and 2 dithiothreitol) containing the protease inhibitor mixture and separated into membrane- and cytosol-containing fractions by centrifugation (100,000 × g, 30 min). Immunoblotting was performed as described previously (Hirai et al., 1997).
Northern blot analysis
The collected neurons were lysed in 25 mm sodium citrate solution, pH 7.0, containing 4 m guanidinium isothiocyanate and 0.1 m 2-mercaptoethanol. This solution was placed on the top of CsCl solution (5.7 mCsCl, pH 7.2, and 0.1 m EDTA) in tubes and centrifuged at 80,000 × g for 36 hr at 16°C. The pellets were dissolved in TE buffer, and RNA was precipitated with 1/10 volume of 3m sodium acetate, pH 5.2, and two volumes of ethanol at 4°C for 20 min. The concentrations of RNA were determined by absorbance at 260 nm. Total RNA samples (20 μg) were electrophoretically separated on 1% formaldehyde agarose gel and transferred onto nitrocellulose membranes for 48 hr. Prehybridization was performed at 42°C for 16 hr in 1% Ficoll, 1% bovine serum albumin, 1% polyvinylpyrolidone, sodium citrate solution (750 mm NaCl and 75 mm sodium citrate, pH 7.0), 50% formamide, 50 mm sodium phosphate, pH 6.5, and 100 μg/ml salmon sperm DNA. Then the membrane was hybridized with 32P-labeled HMG-CoA reductase probe at 42°C for 24 hr and washed by standard protocol.
Statistical analysis
Statistical differences within each experiment were determined by ANOVA, and differences between groups were calculated by the Fisher's exact test. A rejection level of p < 0.05 was considered significant. This analysis was performed on a personal computer using the StatView J4.02 software statistical package (Abacus Concepts, Berkeley, CA).
RESULTS
Development of primary cultured rat cortical neurons
The primary cultured cortical neurons were obtained from the brains of 17-d-old fetal SD rats. The development of these neurons according to morphology, MTT assay, and mRNA expression of HMG-CoA reductase are shown in Figure 1. Although the shape of the neurons immediately after the preparation was round, the neurites were being extended and connected to each other gradually in a time-dependent manner (Fig. 1A). The mitochondrial functions by MTT assay were shown to increase in a time-dependent manner, reaching an equilibrium between days 6 and 10 (Fig. 1B). The mRNA of HMG-CoA reductase was present at day 0, and its level increased to make a plateau at day 10 after the culture (Fig. 1C). In the present study, we used the neurons at day 10, where their development of neurons reached an equilibrium in terms of morphology, MTT assay, and mRNA expression of HMG-CoA reductase.
Fig. 1.

Development of primary cultured rat cortical neurons. A, Morphological observations in neurons immediately (Day 0) and 10 d (Day 10) after beginning the culture. B,Developmental change in mitochondrial function of neurons by MTT assay. Mitochondrial function was assayed by mitochondrial conversion to formazan as detected by the change of optical density (OD) at 570 nm during the final 3 hr of culture, as described in Materials and Methods. Each point represents the mean ± SEM of triplicates.C, Developmental change of mRNA expression of HMG-CoA reductase. In Northern blot, RNA was isolated as indicated in the figure, and the mRNA level for HMG-CoA reductase were analyzed as described in Materials and Methods. The data shown was a representative of three independent experiments.
Neuronal cell death induced by HMG-CoA reductase inhibitors
The time course for neuronal cell death (NCD) induced by 300 μm pravastatin, an inhibitor of HMG-CoA reductase, is shown in Figure 2A. The live cell number was determined by MTT assay. The number of live neurons started to decrease at 24 hr after the addition, was significantly reduced at 36 hr, and progressively continued to decrease during the following 72 hr. Approximately 90% of neurons had died by 96 hr or later (Fig. 2A). In some experiments, we performed the cell counting assay using fluorescein diacetate propidium iodide in parallel with the MTT assay to determine the number of live cells (Fig. 2D). Although small numbers of dead neurons (red-stained round cells) were observed among the live neurons in control (green–yellow cell body) (Fig. 2D), and 300 μm pravastatin induced NCD for almost all neurons (Fig. 2D), there was a linear correlation between the MTT assay and the number of live cells (r2 = 0.987; p< 0.05; data not shown).
Fig. 2.

Neuronal cell death induced by H-NCD.A, Time course of H-NCD. Primary cultured cortical neurons were exposed to 300 μm pravastatin for the indicated periods. B, Dose-dependent effect of neurotoxicity by HMG-CoA inhibitors (pravastatin, simbastatin, and lovastatin). Primary cultured cortical neurons were exposed to the indicated concentrations of pravastatin, simbastatin, and lovastatin for 72 hr. C, Dose-dependent effect of mevalonate to reverse H-NCD. Neurons were incubated with the indicated concentrations of mevalonate in the presence of 300 μm pravastatin. The live cell number was assayed by mitochondrial conversion to formazan as detected by the change of OD at 570 nm during the final 3 hr of culture, as described in Materials and Methods. Each point represents the mean ± SEM of triplicates. D, Morphological observations of H-NCD. Fluorescein diacetate propidium iodide staining 72 hr after addition. Top, Control;Middle, 300 μm pravastatin; andBottom, 0.03 mg/ml of mevalonate in the presence of 300 μm pravastatin. Fluorescein diacetate propidium iodide staining was described in Materials and Methods. The dead neurons were red-stained round cells, and the live neurons had a green–yellow cell body. The data shown was a representative of three independent experiments.
The dose-dependent effect of neurotoxicity by HMG-CoA inhibitors (pravastatin, simbastain, and lovastatin) over a 72 hr period is shown in Figure 2B. Pravastatin progressively caused NCD at concentrations >10 μm in a dose-dependent manner, and >90% of neurons died at 300 μmpravastatin. Although simbastatin and lovastatin, which are less hydrophilic than pravastatin, also induced NCD in a dose-dependent manner, their neurotoxicities were ≥10 times more potent than that of pravastatin (Fig. 2B).
The effect of mevalonate in the presence of pravastatin for 72 hr is shown in Figure 2C. The NCD induced by 300 μm pravastatin was reversed by the addition of mevalonate in a dose-dependent manner and was fully reversed at 0.01 mg/ml in the MTT assay (Fig. 2C). In the cell-counting assay using fluorescein diacetate propidium iodide, it was clearly demonstrated that mevalonate (0.03 mg/ml) completely inhibited NCD induced by 300 μm pravastatin (Fig.2D).
We examined the effect of cholesterol in HMG-CoA reductase inhibitors (H-NCD) and found that neurons were not protected from H-NCD (data not shown). These data clearly demonstrated that the observed H-NCD was attributable to the decrease of mevalonate production occurring as a consequence of HMG-CoA reductase inhibition and that metabolites of mevalonate other than cholesterol might play a critical role in H-NCD.
Roles of the isoprenoids GGPP and FPP in H-NCD
Mevalonate is a donor for the synthesis of several important metabolites involved in the posttranslational modification of proteins. It acts as an isoprenyl precursor for farnesyl and geranylgeranyl molecules, which have an important signaling function. Exogenous FPP and/or GGPP might, therefore, be expected to counteract the effect of pravastatin. Unfortunately, the experimental use of these compounds is limited by their membrane impermeability and sensitivity to thiol reagents present in the culture medium. Therefore, we chose a novel approach to evaluate the role of isoprenoids in H-NCD: the preparation of the liposomes of these isoprenoids.
The NCD induced by pravastatin was mostly reversed by the addition of liposomes containing GGPP in a dose-dependent manner, although neither liposomes containing FPP nor containing vehicle affected the pravastatin-induced NCD (Fig. 3). Such inability of FPP to overcome pravastatin blockage does not reflect impaired entry into the cells, because the incorporation of [3H]FPP in liposomes into neurons was almost equal to that of [3H]GGPP (data not shown). The addition of GGPP to neurons had no effect on their viability in the absence of pravastatin (data not shown). Because GGPP is biosynthetically derived from the single condensation of FPP and isopentenyl-pyrophosphate (IPP), FPP could not be converted to GGPP in pravastatin-treated cells. These results, therefore, indicate that GGPP can protect neurons from H-NCD in the absence of upstream intermediates of cholesterol biosynthesis (Fig. 3).
Fig. 3.

Effect of metabolites of mevalonate in H-NCD. Effect of pravastatin and mevalonate metabolites in H-NCD. Rat cortical neurons were cultured with the indicated concentrations of mevalonate and its metabolites under inhibition by pravastatin (300 μm) for 72 hr. The data were a representative of three independent experiments. Each liposome containing GGPP or FPP was prepared to have the same concentration. GGPP, GGPP in liposomes; FPP, FPP in liposomes;Vehicle, vehicle in liposomes.
Roles of isoprenoids in DNA ladder formation and apoptosis-related molecules during H-NCD
The features of H-NCD were further examined by genomic DNA analysis and by transmission electron microscopy. The genomic DNA extracted from neurons that had undergone H-NCD was analyzed by agarose gel electrophoresis (Fig.4B). Control DNA in neurons without pravastatin already revealed a certain degree of ladder formation in nucleosome-length fragments, a biochemical indicator of apoptosis, and this was consistent with the data from the cell counting assay using fluorescein diacetate propidium iodide, in which small numbers of dead neurons were observed among the live neurons in the control (Fig. 2D). Pravastatin apparently enhanced the ladder formation in nucleosome-length fragments of DNA. In addition, electron microscopy demonstrated that the pravastatin exposure increased the number of dead neurons showing the apoptotic characteristic of compacted nuclei with marginated and condensed chromatin (data not shown). The formation of the DNA ladder and the characteristic features observed by electron microscopy confirmed that H-NCD possessed apoptotic characters. The changes of apoptosis-related molecules such as p53, Bcl-2, and Bax as observed by Western blot analysis are shown in Figure 4C. Both p53 and Bax were dramatically accumulated by pravastatin, whereas the expression of Bcl-2 was detected in the absence of pravastatin, and its level slightly increased in H-NCD.
Fig. 4.
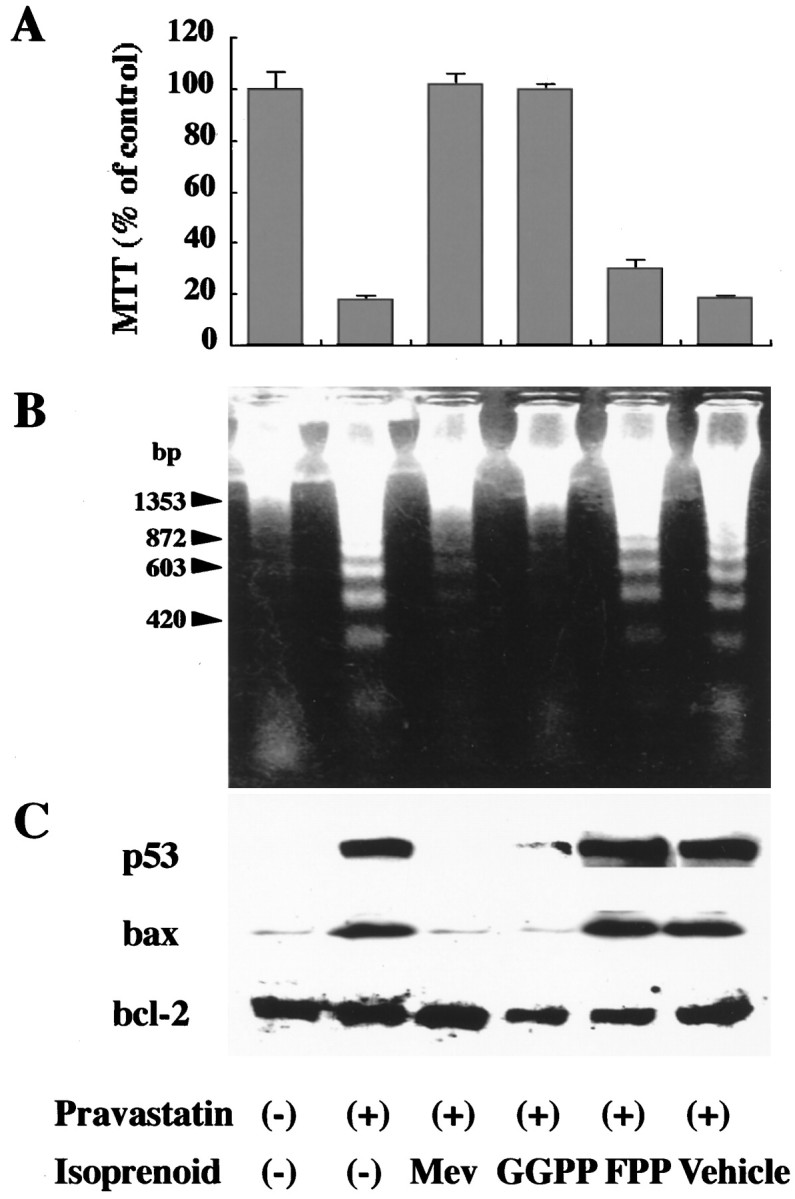
Roles of isoprenoids on DNA ladder formation and apoptosis-related molecules in H-NCD. Rat cortical neurons incubated with mevalonate (0.03 mg/ml), GGPP in liposomes (4 μm), FPP in liposomes (4 μm), or vehicle for 24 hr in the presence of pravastatin (300 μm). A,Viability of neurons determined by MTT assay. The live cell number was assayed by mitochondrial conversion to formazan as detected by the change of OD at 570 nm during the final 3 hr of culture, as described in Materials and Methods. Each column represents the mean ± SEM of triplicates. B, Ladder formation of nucleosome-length fragments of DNA. DNA fragmentation analysis was performed as described in Materials and Methods. The data shown was a representative of three independent experiments. C, Expression of p53, Bcl-2, and Bax. Cell lysates (20 μg) were analyzed by immunoblotting with antibodies against p53, Bcl-2, and Bax, as described in Materials and Methods. The data shown was a representative of three independent experiments.
The DNA ladder formation induced by pravastatin was completely blocked by mevalonate and GGPP, but not FPP, which was consistent with the viability of neurons according to the MTT assay (Fig.4A,B). The expressions of both p53 and Bax induced by pravastatin were also blocked by mevalonate and GGPP (Fig.4C).
Effect of pravastatin and isoprenoids on isoprenylation of small GTPase(s) and their translocation to the membrane during H-NCD
Immunoblot analysis of membrane and cytosolic fractions revealed that pravastatin blocked the translocation or redistribution of Rho A, Rho B, and cdc42 from the cytoplasm to membrane 72 hr after the addition of pravastatin in H-NCD (Fig.5). Moreover, both mevalonate and GGPP reversed the pravastatin-induced blockage of their translocation or redistribution (Fig. 5). Ras small GTPase in membrane, which has been reported to be isoprenylated by FPP, was not affected by treatment with pravastatin. Although the effect was not as striking as for RhoA, RhoB, and cdc42, Ras small GTPase in cytosol was increased by pravastatin (Fig. 5).
Fig. 5.

Roles of mevalonate and isoprenoids in the translocation of Rho GTPase family in H-NCD. Rat cortical neurons incubated with mevalonate (0.03 mg/ml), GGPP in liposomes (4 μm), FPP in liposomes (4 μm), or vehicle for 72 hr in the presence of pravastatin (300 μm). Crude membrane- and cytosol-containing fractions were prepared as described in Materials and Methods. Each lysate (20 μg) was analyzed by immunoblotting with antibodies against Ras, Rho A, Rho B, and cdc42.
DISCUSSION
In the present study, we demonstrated that HMG-CoA reductase mRNA increases during culture, indicating that the mevalonate cascade is activated and maturated during development. Its ontogenetic early development in neuronal tissues has been also demonstrated by others (Maltese and Volpe, 1979; Volpe et al., 1985). These data indicated that the mevalonate cascade may play critical roles during development in the brains.
Statins (HMG CoA reductase inhibitors) induce NCD in a time- and dose-dependent manner, and H-NCD revealed typical features of apoptosis in terms of DNA ladder formation and the morphological changes observed by electron microscopy. This H-NCD was fully restored by the addition of mevalonate, indicating that the observed H-NCD was attributable to the decrease of mevalonate production occurring as a consequence of HMG-CoA reductase inhibition.
We compared the potency of three statins (pravastatin, lovastatin, and simvastatin) in H-NCD. Despite similarities in chemical structure, pravastatin was less potent than lovastatin and simvastatin. Because H-NCD was dependent on the production of mevalonate, their different inhibitory potencies in HMG-CoA reductase of neurons might be responsible for this biological difference in H-NCD. It was reported that sterol synthesis was strongly inhibited by lovastatin and simvastatin but to a much lesser extent by pravastatin in extrahepatic cells (van Vliet et al., 1995). This might also be the case in neurons. Pravastatin is more hydrophilic than simvastatin or lovastatin (Serajuddin et al., 1991). These more hydrophilic drugs probably enter extrahepatic cells.
It was suggested that the metabolite or metabolites of mevalonate other than cholesterol play a critical role in H-NCD. Mevalonate acts as an isoprenyl precursor for farnesyl and geranylgeranyl molecules, and it is well known that GGPP and FPP are two major isoprenoids (Casey and Seabra, 1996). Therefore, we chose a novel approach, that of preparing the liposomes (Hirai et al., 1997; Tatsuno et al., 1997; Noguchi et al., 1998; Terano et al., 1998; Nishimura et al., 1999), and found that GGPP, not FPP, restored H-NCD. GGPP is biosynthetically derived from the single condensation of FPP and IPP. Because IPP could not be synthesized in pravastatin-treated cells, FPP could not be converted to GGPP. This means that GGPP can rescue neurons from H-NCD in the absence of upstream intermediates of cholesterol biosynthesis.
The mechanism of how GGPP protects neurons from H-NCD was unclear in the present study. In the proliferation, it has been demonstrated that the ability of an HMG-CoA reductase inhibitor to interfere with cell cycle progression could be attributed to its activity in suppressing the isoprenylation of proteins such as small GTPases rather than in interrupting cholesterol synthesis (Chakrabarti and Engleman, 1991;Ortiz et al., 1995). In this regard, we recently reported that impairment of geranylgeranylation of cytoskeletal proteins such as Rho proteins is the most likely molecular event in the inhibition of cell cycle progression (Hirai et al., 1997; Tanaka et al., 1998). This may also be the case in neurons. Therefore, we tried to determine the subcellular distribution of Rho and related proteins in H-NCD. We found that (1) pravastatin inhibited the translocation or redistribution of Rho A, Rho B, and cdc42 to membrane, and that (2) mevalonate and GGPP restored the blockage of translocation or redistribution of these Rho proteins. Although Ras small GTPase (one of the farnesylated GTPases) in membrane was not affected, Ras in the cytosolic fraction was slightly accumulated. These data were in good agreement with the protection of mevalonate and GGPP, not FPP, in H-NCD, suggesting that the turnover of Rho small GTPase is more dynamic and that it is continuously translocated or redistributed to membrane, whereas the turnover of Ras is relatively slow. The similar difference in turnover between Rho and Ras was reported in FRTL-5 cells (Noguchi et al., 1998).
The physiological significance of geranylgeranylation for the activation of Rho was well demonstrated in the proliferation using botulinum C3 exoenzyme (Hirai et al., 1997; Tanaka et al., 1998). Botulinum C3 exoenzyme was reported to inactivate Rho small GTPase(s) by ADP ribosylating a specific asparagine residue and thereby interfering with its biological actions, presumably affecting its interaction with putative target protein or proteins (Kikuchi et al., 1988; Narumiya et al., 1988; Nemoto et al., 1991). Therefore, we examined the effect of C3 exoenzyme on the protection of GGPP from H-NCD, and C3 had no effect at concentrations where it was reported to inactivate Rho specifically (data not shown). The inability of C3 exoenzyme to abolish the effect of GGPP does not deny the physiological significance of geranylgeranylation because other geranylgeranylated proteins such as Rac and cdc42 might be critical and/or the combination of inactivation of Rho with those of other geranylgeranylated proteins might be important in H-NCD. In any case, further experiments should be planned to address these questions.
In the present study, we found that HMG-CoA reductase inhibition progressively induces both p53 and Bax, whereas Bcl-2 was present without treatment and then slightly increased with the inhibition. Mevalonate and GGPP protect neurons from H-NCD with the inhibition of the induction of these molecules. The considerable evidence has been accumulating to indicate that the tumor suppressor gene p53 is involved in the regulation of cell death (el-Deiry et al., 1993; Xiong et al., 1993; Symonds et al., 1994). Several studies demonstrated that p53 in neurons is accumulated during neuronal cell death in vitroand in vivo. The neurons derived from the p53 null mice (P53−/−) were reported to be resistant to the excitotoxicity and DNA-damaging agents both in vitro and in vivo(Crumrine et al., 1994; Dessi et al., 1995; Wood and Youle, 1995;Enokido et al., 1996a,b; Morrison et al., 1996; Sakhi et al., 1996;Trimmer et al., 1996; Xiang et al., 1996; Hirata and Cadet, 1997). In addition, overexpression of p53 using adenovirus vector dramatically increased NCD (Xiang et al., 1996). Although these data clearly indicated that p53 plays a critical role in some part of NCD, the downstream of p53 has not been well characterized yet.
bcl-2 is a protooncogene isolated from human B-cell lymphoma (Tsujimoto and Croce, 1986). The overexpression of Bcl-2 in vitro can protect neurons from many kinds of insult (Garcia et al., 1992; Behl et al., 1993; Kane et al., 1993; Zhong et al., 1993a,b; Allsopp et al., 1995; Myers et al., 1995). After the ischemia, the upregulated expression of Bax and the downregulated expression of Bcl-2 were observed in the brain, suggesting that the disproportional expression between Bax and Bcl-2 is critical for NCD because Bax is able to bind to bcl-2 and other related anti-apoptotic family members to inhibit their function (Shimazaki et al., 1994; Chen et al., 1995; Krajewski et al., 1995; Chen et al., 1996). The expression of Bax was reported to be increased by p53 (Selvakumaran et al., 1994; Miyashita and Reed, 1995), and it was clarified that p53 is necessary for Bax-induced NCD (Xiang et al., 1998). It is also possible that p53 activates cell death by directly downregulating Bcl-2 gene expression in neurons (Selvakumaran et al., 1994). Therefore, it is not unreasonable to speculate that the induction of p53 and Bax plays some roles during the process of H-NCD in spite of the fact that direct evidence is still awaited.
Does the disruption of the mevalonate cascade provide one of the common pathways in NCD induced by some pathophysiological conditions? This represents another critical question because H-NCD has typical apoptotic characteristics with the induction of p53 and Bax, which are similar to those of other NCD such as ischemic, glutamate-induced and β-amyloid-induced models. In this regard, we examined the effect of mevalonate and GGPP in glutamate- and β-amyloid-induced neuronal cell death, and neither mevalonate nor GGPP influenced NCD in these models (data not shown), indicating that the disruption of the mevalonate cascade is not involved in these NCD models. However, the possibility that the mevalonate cascade is involved in some types of NCD still remains.
The statins are being widely used clinically for the treatment of hypercholesterolemia. Do statins have some risk of the neurotoxicityin vivo at the clinical doses? In the present study, we demonstrated that pravastatin progressively caused NCD at concentrations of >10 μm, and both simbastatin and lovastatin are 10 times or more potent than that of pravastatin. It was reported that the Cmax of pravastatin in the serum is ∼0.1 μm in the healthy subjects (Pan et al., 1993). In addition, it was demonstrated that pravastatin was not detected in the CSF at the clinical doses (Botti et al., 1991) and that the concentration of lovastatin in CSF comprised ∼20% of that in serum in healthy subjects (Botti et al., 1991). Therefore, there seems to be little possibility that the clinical dose of statins reach its concentration enough to induce the NCD in the brains.
In summary, the NCD induced by the inhibition of HMG-CoA reductase appears to be apoptotic with the induction of p53 and Bax, and GGPP is one of the critical isoprenoids in the mevalonate cascade for protecting neurons from H-NCD probably through the protein geranlygeranylation.
Footnotes
We thank Drs. M. Nishimura and T. Terano for their valuable advice and Y. Tsuchikawa, Y. Okuda, and M. Maemori for their excellent assistance. We also acknowledge Dr. S. Kurakata (Sankyo Pharmaceutical Company, Tokyo, Japan) for the gift of pravastatin and cDNA of HMG-CoA reductase.
Correspondence should be addressed to Dr. Ichiro Tatsuno, Second Department of Internal Medicine, Chiba University School of Medicine, 1-8-1 Inohana, Chuou-ku, Chiba-city Chiba, 260 Japan. E-mail:ichico@intmed02.m.chiba-u.ac.jp.
REFERENCES
- 1.Allsopp TE, Kiselev S, Wyatt S, Davies AM. Role of Bcl-2 in the brain-derived neurotrophic factor survival response. Eur J Neurosci. 1995;7:1266–1272. doi: 10.1111/j.1460-9568.1995.tb01116.x. [DOI] [PubMed] [Google Scholar]
- 2.Bass NH, Netsky MG, Young E. Effect of neonatal maturation on developing cerebrum. I. Microchemical and histological study of cellular differentiation in the rat. Arch Neurol. 1970a;23:289–302. doi: 10.1001/archneur.1970.00480280003001. [DOI] [PubMed] [Google Scholar]
- 3.Bass NH, Netsky MG, Young E. Effect of neonatal malnutrition on developing cerebrum. II. Microchemical and histologic study of myelin formation in the rat. Arch Neurol. 1970b;23:303–313. doi: 10.1001/archneur.1970.00480280017002. [DOI] [PubMed] [Google Scholar]
- 4.Behl C, Hovey LD, Krajewski S, Schubert D, Reed JC. BCL-2 prevents killing of neuronal cells by glutamate but not by amyloid beta protein. Biochem Biophys Res Commun. 1993;197:949–956. doi: 10.1006/bbrc.1993.2571. [DOI] [PubMed] [Google Scholar]
- 5.Botti RE, Triscari J, Pan HY, Zayat J. Concentration of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin Neuropharmacol. 1991;14:256–261. doi: 10.1097/00002826-199106000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 7.Chakrabarti R, Englema EG. Interrelationships between mevalonate metabolism and the mitogenic signaling pathway in T lymphocyte proliferation. J Biol Chem. 1991;266:12216–12222. [PubMed] [Google Scholar]
- 8.Chen J, Graham SH, Chan PH, Lan J, Zhou RL, Simon RP. bcl-2 is expressed in neurons that survive focal ischemia in the rat. NeuroReport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Zhu RL, Nakayama M, Kawaguchi K, Jin K, Stetler RA, Simon RP, Graham SH. Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J Neurochem. 1996;67:64–71. doi: 10.1046/j.1471-4159.1996.67010064.x. [DOI] [PubMed] [Google Scholar]
- 10.Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- 11.Cunzer ML, Davison AN. The lipid composition of rat brain myelin and subcellular fractions during development. Biochem J. 1968;106:1057–1063. doi: 10.1042/bj1060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dessi F, Pollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C. Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem. 1995;64:1980–1987. doi: 10.1046/j.1471-4159.1995.64051980.x. [DOI] [PubMed] [Google Scholar]
- 13.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 14.Enokido Y, Araki T, Aizawa S, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996a;203:1–4. doi: 10.1016/0304-3940(95)12247-8. [DOI] [PubMed] [Google Scholar]
- 15.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996b;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 16.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 18.Hirai A, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Tatsuno I, Oeda T, Tahara K, Terano T, Narumiya S, Kohn LD, Saito Y. Geranylgeranylated rho small GTPase(s) are essential for the degradation of p27Kip1 and facilitate the progression from G1 to S phase in growth-stimulated rat FRTL-5 cells. J Biol Chem. 1997;272:13–16. [PubMed] [Google Scholar]
- 19.Hirata H, Cadet JL. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J Neurochem. 1997;69:780–790. doi: 10.1046/j.1471-4159.1997.69020780.x. [DOI] [PubMed] [Google Scholar]
- 20.Kanai T, Hirohashi S, Noguchi M, Shimoyama Y, Shimosato Y, Noguchi S, Nishimura S, Abe O. Monoclonal antibody highly sensitive for the detection of ras p21 in immunoblotting analysis. Jpn J Cancer Res. 1987;78:1314–1318. [PubMed] [Google Scholar]
- 21.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 22.Kikuchi A, Yamamoto K, Fujita T, Takai Y. ADP-ribosylation of the bovine brain rho protein by botulinum toxin type C1. J Biol Chem. 1988;263:16303–16308. [PubMed] [Google Scholar]
- 23.Kishimoto Y, Davies WE, Radin NS. Developing rat brain: changes in cholesterol, galactolipids and the individual fatty acids of gangliosides and glycerophosphatides. J Lipid Res. 1965;6:532–536. [PubMed] [Google Scholar]
- 24.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, Kang WK, Wang XF, Kim SJ, Trepel JB. Inhibition of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway induces p53-independent transcriptional regulation of p21(WAF1/CIP1) in human prostate carcinoma cells. J Biol Chem. 1998;273:10618–10623. doi: 10.1074/jbc.273.17.10618. [DOI] [PubMed] [Google Scholar]
- 26.Maltese WA, Volpe JJ. Developmental changes in the distribution of 3-hydroxy-3-methylglutaryl coenzyme A reductase among subcellular fractions of rat brain. J Neurochem. 1979;33:107–115. doi: 10.1111/j.1471-4159.1979.tb11712.x. [DOI] [PubMed] [Google Scholar]
- 27.Miquel K, Pradines A, Favre G. Farnesol and geranylgeraniol induce actin cytoskeleton disorganization and apoptosis in A549 lung adenocarcinoma cells. Biochem Biophys Res Commun. 1996;225:869–876. doi: 10.1006/bbrc.1996.1265. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 29.Morio H, Tatsuno I, Hirai A, Tamura Y, Saito Y. Pituitary adenylate cyclase-activating polypeptide protects rat-cultured cortical neurons from glutamate-induced cytotoxicity. Brain Res. 1996;741:82–88. doi: 10.1016/s0006-8993(96)00920-1. [DOI] [PubMed] [Google Scholar]
- 30.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers KM, Fiskum G, Liu Y, Simmens SJ, Bredesen DE, Murphy AN. Bcl-2 protects neural cells from cyanide/aglycemia-induced lipid oxidation, mitochondrial injury, and loss of viability. J Neurochem. 1995;65:2432–2440. doi: 10.1046/j.1471-4159.1995.65062432.x. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura S, Tatsuno I, Noguchi Y, Kitagawa M, Kohn LD, Saito Y, Hirai A. 73-kDa heat shock cognate protein interacts directly with P27Kip1, a cyclin-dependent kinase inhibitor, during G1/S transition. Biochem Biophys Res Commun. 1999;257:340–343. doi: 10.1006/bbrc.1999.0442. [DOI] [PubMed] [Google Scholar]
- 33.Narumiya S, Sekine A, Fujiwara M. Substrate for botulinum ADP-ribosyltransferase, Gb, has an amino acid sequence homologous to a putative rho gene product. J Biol Chem. 1988;263:17255–17257. [PubMed] [Google Scholar]
- 34.Nemoto Y, Namba T, Kozaki S, Narumiya S. Clostridium botulinum C3 ADP-ribosyltransferase gene. Cloning, sequencing, and expression of a functional protein in Escherichia coli. J Biol Chem. 1991;266:19312–19319. [PubMed] [Google Scholar]
- 35.Nishimura M, Tanaka T, Yasuda T, Kurakata S, Kitagawa M, Yamada K, Saito Y, Hirai A. Collagen secretion and growth of mesangial cells require geranylgeranylpyrophosphate. Kidney Int. 1999;55:520–528. doi: 10.1046/j.1523-1755.1999.00292.x. [DOI] [PubMed] [Google Scholar]
- 36.Noguchi Y, Nakamura S, Yasuda T, Kitagawa M, Kohn LD, Saito Y, Hirai A. Newly synthesized Rho A, not Ras, is isoprenylated and translocated to membranes coincident with progression of the G1 to S phase of growth-stimulated rat FRTL-5 cells. J Biol Chem. 1998;273:3649–3653. doi: 10.1074/jbc.273.6.3649. [DOI] [PubMed] [Google Scholar]
- 37.Ortiz MB, Goin M, Gomez dAMB, Hammarstrom S, Jimenez dAL. Mevalonate dependency of the early cell cycle mitogenic response to epidermal growth factor and prostaglandin F2 alpha in Swiss mouse 3T3 cells. J Cell Physiol. 1995;162:139–146. doi: 10.1002/jcp.1041620117. [DOI] [PubMed] [Google Scholar]
- 38.Padayatty SJ, Marcelli M, Shao TC, Cunningham GR. Lovastatin-induced apoptosis in prostate stromal cells. J Clin Endocrinol Metab. 1997;82:1434–1439. doi: 10.1210/jcem.82.5.3960. [DOI] [PubMed] [Google Scholar]
- 39.Pavlov OV, Bobryshev Yu V, Balabanov Yu V, Ashwell K. An in vitro study of the effects of lovastatin on human fetal brain cells. Neurotoxicol Teratol. 1995;17:31–39. doi: 10.1016/0892-0362(95)91641-w. [DOI] [PubMed] [Google Scholar]
- 40.Pan HY, Waclawski AP, Funke PT, Whigan D. Pharmacokinetics of pravastatin in elderly versus young men and women. Ann Pharmacother. 1993;27:1029–1033. doi: 10.1177/106002809302700902. [DOI] [PubMed] [Google Scholar]
- 41.Reedquist KA, Pope TK, Roess DA. Lovastatin inhibits proliferation and differentiation and causes apoptosis in lipopolysaccharide-stimulated murine B cells. Biochem Biophys Res Commun. 1995;211:665–670. doi: 10.1006/bbrc.1995.1863. [DOI] [PubMed] [Google Scholar]
- 42.Sakhi S, Gilmore W, Tran ND, Schreiber SS. p53-deficient mice are protected against adrenalectomy-induced apoptosis. NeuroReport. 1996;8:233–235. doi: 10.1097/00001756-199612200-00047. [DOI] [PubMed] [Google Scholar]
- 43.Satoh T, Isobe H, Ayukawa K, Sakai H, Nawata H. The effects of pravastatin, an HMG-CoA reductase inhibitor, on cell viability and DNA production of rat hepatocytes. Life Sci. 1996;9:1103–1108. doi: 10.1016/0024-3205(96)00427-4. [DOI] [PubMed] [Google Scholar]
- 44.Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, Hoffman B, Liebermann D. Immediate early up-regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9:1791–1798. [PubMed] [Google Scholar]
- 45.Serajuddin AT, Ranadive SA, Mahoney EM. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J Pharmacol Sci. 1991;80:830–834. doi: 10.1002/jps.2600800905. [DOI] [PubMed] [Google Scholar]
- 46.Shimazaki K, Ishida A, Kawai N. Increase in bcl-2 oncoprotein and the tolerance to ischemia-induced neuronal death in the gerbil hippocampus. Neurosci Res. 1994;20:95–99. doi: 10.1016/0168-0102(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki K. Chemistry and metabolism of brain lipids. In: Albers RW, Seigel GJ, Katzman R, Agranoff BW, editors. Basic neurochemistry. Little, Brown; Boston: 1972. pp. 207–227. [Google Scholar]
- 48.Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka T, Tatsuno I, Noguchi Y, Uchida D, Oeda T, Narumiya S, Yasuda T, Higashi H, Kitagawa M, Nakayama K, Saito Y, Hirai A. Activation of cyclin-dependent kinase 2 (Cdk2) in growth-stimulated rat astrocytes. Geranylgeranylated Rho small GTPase(s) are essential for the induction of cyclin E gene expression. J Biol Chem. 1998;273:26772–26778. doi: 10.1074/jbc.273.41.26772. [DOI] [PubMed] [Google Scholar]
- 50.Tatsuno I, Yada T, Vigh S, Hidaka H, Arimura A. Pituitary adenylate cyclase activating polypeptide and vasoactive intestinal peptide increase cytosolic free calcium concentration in cultured rat hippocampal neurons. Endocrinology. 1992;131:73–81. doi: 10.1210/endo.131.1.1319331. [DOI] [PubMed] [Google Scholar]
- 51.Tatsuno I, Tanaka T, Oeda T, Yasuda T, Kitagawa M, Saito Y, Hirai A. Geranylgeranylpyrophosphate, a metabolite of mevalonate, regulates the cell cycle progression and DNA synthesis in human lymphocytes. Biochem Biophys Res Commun. 1997;241:376–382. doi: 10.1006/bbrc.1997.7825. [DOI] [PubMed] [Google Scholar]
- 52.Terano T, Shiina T, Noguchi Y, Tanaka T, Tatsuno I, Saito Y, Yasuda T, Kitagawa M, Hirai A. Geranylgeranylpyrophosphate plays a key role for the G1 to S transition in vascular smooth muscle cells. J Atheroscler Thromb. 1998;5:1–6. doi: 10.5551/jat1994.5.1. [DOI] [PubMed] [Google Scholar]
- 53.Trimmer PA, Smith TS, Jung AB, Bennett JP. Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- 54.Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci USA. 1986;83:5214–5218. doi: 10.1073/pnas.83.14.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Vliet AK, van Thiel GC, Huisman RH, Moshage H, Yap SH, Cohen LH. Different effects of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors on sterol synthesis in various human cell types. Biochim Biophys Acta. 1995;1254:105–111. doi: 10.1016/0005-2760(94)00176-y. [DOI] [PubMed] [Google Scholar]
- 56.Volpe JJ, Goldberg RI, Bhat NR. Cholesterol biosynthesis and its regulation in dissociated cell cultures of fetal rat brain: developmental changes and the role of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Neurochem. 1985;45:536–543. doi: 10.1111/j.1471-4159.1985.tb04021.x. [DOI] [PubMed] [Google Scholar]
- 57.Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 61.Zhong LT, Kane DJ, Bredesen DE. BCL-2 blocks glutamate toxicity in neural cell lines. Brain Res Mol Brain Res. 1993a;19:353–355. doi: 10.1016/0169-328x(93)90139-g. [DOI] [PubMed] [Google Scholar]
- 62.Zhong LT, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993b;90:4533–3537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]