

Abstract
Neurotransmitter receptor function can be influenced by the phosphorylation state of the receptor or of associated proteins. Here we show that kainate receptors expressed in cultured hippocampal neurons can be modulated by Ca2+/calmodulin-dependent phosphatase (calcineurin) and Ca2+/calmodulin-dependent kinase (CaMK). Ca2+ influx through NMDA receptor or voltage-sensitive calcium channels resulted in a transient depression of the kainate receptor current. This calcium-induced depression of the kainate receptor current depended on the activation of the phosphatase calcineurin. The amplitude of the kainate receptor currents returned to the baseline level in ∼9 sec (τ = 3.6 sec), and the recovery of the current amplitude depended on CaMK activity. The effect on kainate receptor currents was dependent on the frequency of NMDA receptor activation. Although low-frequency (0.1 Hz) NMDA application induced depression followed by recovery of the kainate receptor currents, higher frequency (1 Hz) NMDA applications induced a more prolonged depression. Kainate receptors have been shown to modulate synaptic transmission by both presynaptic and postsynaptic mechanisms. Our results suggest that synaptic activity mediated by NMDA receptors, or other routes of Ca2+ influx, may, in turn, modulate the function of kainate receptors.
Keywords: hippocampal neuron, kainate receptor, NMDA receptor, calcineurin, CaMK, receptor modulation, calcium imaging
In the mammalian CNS the neurotransmitter glutamate activates a wide variety of receptors. One class of receptor forms cation-selective ion channels gated by glutamate, the ionotropic glutamate receptor (iGluR). Different subtypes of iGluR have been defined on the basis of the pharmacological properties and the sequence homology between receptor subunits forming the receptor (Hollmann and Heinemann, 1994; Dingledine et al., 1999).
One subclass, kainate receptors, has been shown only recently to participate in synaptic transmission. Postsynaptic activation of kainate receptors has been reported for synapses in the hippocampus (Castillo et al., 1997; Vignes and Collingridge, 1997; Cossart et al., 1998; Frerking et al., 1998), retina (DeVries and Schwartz, 1999), and spinal cord (Li et al., 1999). A presynaptic functional role of kainate receptors has also been reported in the CA1 region of the hippocampus, where activation of these receptors resulted in a decrease of GABA release (Clarke et al., 1997; Rodriguez-Moreno et al., 1997). Kainate receptors have also been implicated in the induction of seizures. In several animal models administration of kainate causes seizures and neurodegeneration (Ben-Ari, 1985), and it has been shown that genetically engineered mice lacking the GluR6 kainate receptor subunit are less susceptible to systemic administration of kainate (Mulle et al., 1998). Thus, by modulating the release of GABA and/or by depolarizing the postsynaptic cells, kainate receptors may have a role in controlling the excitability of neurons under normal as well as pathological conditions (Lerma, 1997).
An important mechanism involved in modulation of synaptic efficacy is the modification of the functional properties of neurotransmitter receptors via phosphorylation (Schulman, 1995). The activity of recombinant kainate receptors expressed in human embryonic kidney (HEK) cells can be modulated by cAMP-dependent protein kinase (PKA) via direct phosphorylation of the receptor (Raymond et al., 1993; Wang et al., 1993; Traynelis and Wahl, 1997).
It has been proposed that some forms of synaptic plasticity may be the result of changes in the phosphorylation state of postsynaptic receptors (Bliss and Collingridge, 1993; Malenka, 1994;Schulman, 1995). This model predicts that particular patterns of synaptic activity induce a rise in the intracellular calcium concentration ([Ca2+]i), which in turn activates kinases and phosphatases capable of modifying glutamate receptor function. Interestingly, although Ca2+ entry is always a requirement, some stimulation patterns appear to induce kinase activity preferentially and lead to synaptic potentiation, whereas others induce mainly phosphatase activity and result in synaptic depression (Schulman, 1995). It has been proposed that the balance between kinase and phosphatase activity depends on the amount of Ca2+ that enters the cells during the different stimulation protocols (Lisman, 1989; Artola and Singer, 1993;Malenka and Nicoll, 1993; but see Neveu and Zucker, 1996).
In the present study we have investigated the modulation of kainate receptors by kinases and phosphatases after Ca2+ entry through NMDA receptors. The goals of this study were to establish whether neuronal kainate receptors can be modulated by phosphorylation and to characterize how the balance between phosphorylation and dephosphorylation of the kainate receptors can be regulated in neuronal cells.
MATERIALS AND METHODS
Neuronal cultures. Newborn [postnatal day 0 (P0)–P1] Sprague Dawley rats were killed, and the hippocampi were dissected in ice-cold Earle's balanced salt solution (EBSS; Life Technologies, Gaithersburg, MD) containing 10 mm HEPES, pH 7.30. The isolated hippocampi were cut into small pieces and enzymatically digested by a 30–60 min incubation at 37°C in a solution containing 500 μm EDTA-NaOH, pH 8.0, 100 μm CaCl2, and 100 μg/ml papain (Worthington, Freehold, NJ) in EBSS. Subsequently the small chunks of partially digested hippocampi were washed three times with 10 FCS medium containing the following: minimum essential medium (MEM; Life Technologies) with 10% fetal calf serum (HyClone, Logan, UT), 20 mm glucose, 0.1% serum extender (Life Technologies), 25 units/ml penicillin, and 25 μg/ml streptomycin. Hippocampi were mechanically dissociated in 10 FCS medium containing 1 mm kynurenate, using a 5 ml disposable pipette. The dissociated tissue was cleared by passing it through a mesh filter (70 μm; Falcon), and neurons were plated at a density of 7–15 × 103 cells/cm2on a monolayer of astrocytes on 12 mm coverslips coated with poly-d-lysine and rat tail collagen. Cultures were maintained in basal medium Eagle (BME; Life Technologies) containing 1% horse serum (HyClone), 20 mm glucose, N2 supplement (Life Technologies), 1 mm sodium pyruvate, 25 units/ml penicillin, and 25 μg/ml streptomycin, at 37°C in the presence of 5% CO2. 5-Fluoro-2′-deoxyuridine, at a final concentration of 33 μm, was added to the cultures 2–4 d after plating. The cultures were fed by replacing one-third of the medium with fresh medium once a week.
Electrophysiology. Electrophysiological experiments were performed 6–14 d after plating. Whole-cell membrane currents were recorded at room temperature using an Axopatch 1-D amplifier. Patch electrodes had an open tip resistance of 3–5 MΩ and were filled with (in mm): 121.5 K gluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES-NaOH, 2 Mg ATP, and 0.5 Li GTP, pH 7.2 (315 mOsm). For several experiments (see Figs. 1-3, 4,7), identical results were obtained with or without 0.5 mmEGTA in the intracellular solution. All the results shown, however, were obtained without EGTA in the intracellular solution to facilitate the comparison with the results of the calcium-imaging experiments, in which EGTA was omitted. In experiments in which the perforated-patch technique was used, the tip of the electrode pipette was filled with normal internal solution, and the pipette was then backfilled with internal solution containing amphotericin B (100 μg/ml; Sigma, St. Louis, MO). In all experiments neurons were continuously superfused with a control solution containing (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 10 glucose, and 10 HEPES-NaOH, pH 7.30.
Fig. 1.
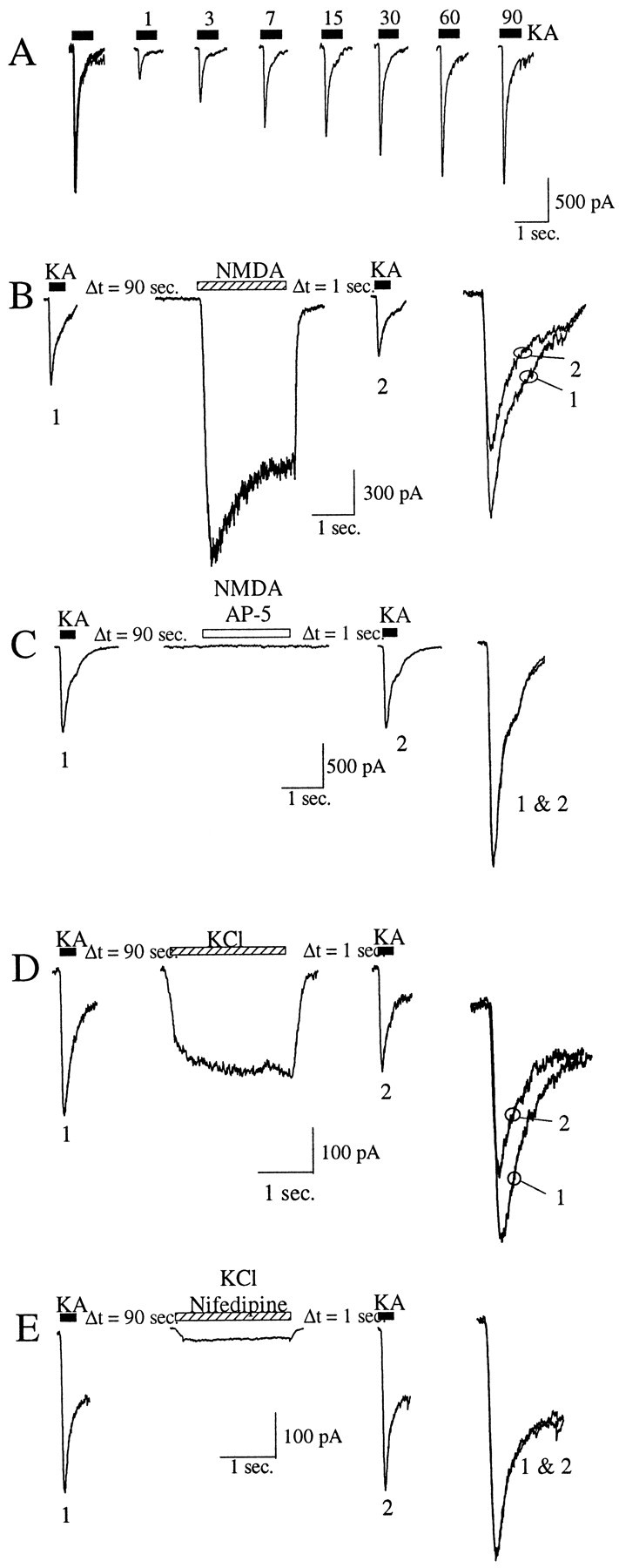
Activation of NMDA receptor or voltage-sensitive calcium channels induces a depression of kainate receptor currents.A, Recovery from desensitization of kainate receptors. Whole-cell currents evoked by 300 μm kainate (blackbars) in control applications (firstseries ofsuperimposedtraces on theleft) and after recovery intervals of 1, 3, 7, 15, 30, 60, or 90 sec (indicated aboveeachtrace) are shown. B,Left, Control (1) and test (2) traces of kainate currents evoked by 300 μm kainate (black bars) before and after activation, respectively, of NMDA receptors by application of 100 μm NMDA and 10 μm glycine (hatched bar). Right, The control and test kainate traces shown superimposed at a larger scale.C, Same experiment as in B but in the presence of 100 μm AP-5 (whitebar). D,Left, Control (1) and test (2) traces of kainate currents evoked by 300 μm kainate (black bars) before and after application, respectively, of 60 mm KCl (hatched bar) in the continuous presence of AP-5 (50 μm). Right, The control and test kainate traces shown superimposed at a larger scale.E, Same experiment as in D but in the presence of 50 μm nifedipine (hatched bar).
Fig. 2.

NMDA receptor activation does not depress the amplitude of AMPA receptor currents. Currents evoked by 1 mm AMPA (black bars) before and after NMDA receptor activation (hatched bar) are shown. In these experiments GYKI 53655 was omitted from the solutions.
Fig. 3.
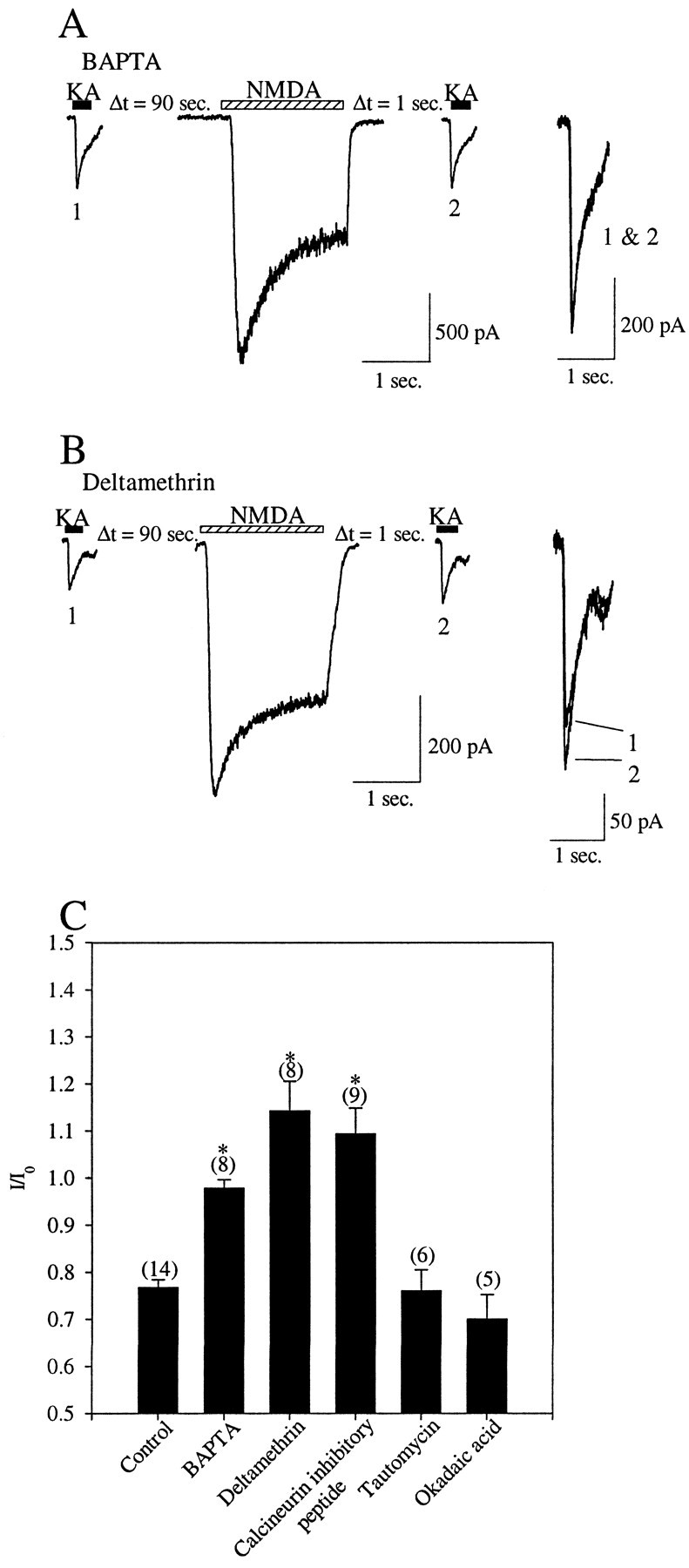
NMDA-dependent depression of kainate receptors requires an increase in [Ca2+]i and activation of calcineurin. A, Left, Currents evoked by 300 μm kainate (black bars) before (1) and after (2) NMDA receptor activation (hatched bar) were recorded with an intracellular electrode containing 10 mm Cs BAPTA.Right, Superimposed kainate currents are shown.B, Left, Currents evoked by 300 μm kainate (black bars) before (1) and after (2) NMDA receptor activation (hatched bar) were recorded with an intracellular electrode containing 500 nm deltamethrin. Right, Superimposed kainate currents are shown. C,Shown are the ratios between the kainate currents recorded after and before NMDA receptor activation (I/I0) and measured when the recording pipette included the following: control recording solution, 10 mm Cs BAPTA, 500 nm deltamethrin, 250 μm calcineurin autoinhibitory peptide, 1 μmtautomycin, and 500 nm okadaic acid. Statistical significance is indicated by * (p < 0.05) and was established by Student's t test. The number of cells examined is indicated inparentheses.
Fig. 4.

Recovery of kainate receptor currents from NMDA-induced depression. A, Right, Kainate currents evoked by 300 μm kainate (white bars) applied 1, 2, 3, 6, 9, or 12 sec after the activation of NMDA receptors (series of superimposedtracesunderblack bar).Left, The superimposed series of control kainate currents. B, The ratio between test and control kainate currents (I/I0) shown as a function of the recovery time after the NMDA receptor stimulation. The data points were fitted to a single exponential. C–F,The ratio between test and control kainate currents (I/I0) shown as a function of the recovery time after the NMDA receptor stimulation in the presence of different kinase inhibitors. In eachpanel, recovery in the absence of inhibitors (same data as in B) is shown for comparison (open circles).C, H-89 (25 μm;filled circles) in the recording electrode (n = 4). D, Chelerythrine (7 μm; filled circles) in the recording electrode (n = 4). E, KN-62 (50 μm; filled circles) in the recording electrode (n = 7). F, Bath application of KN-93 (10 μm; filledcircles; n = 7) or KN-92 (filledtriangles;n = 5).
Fig. 7.
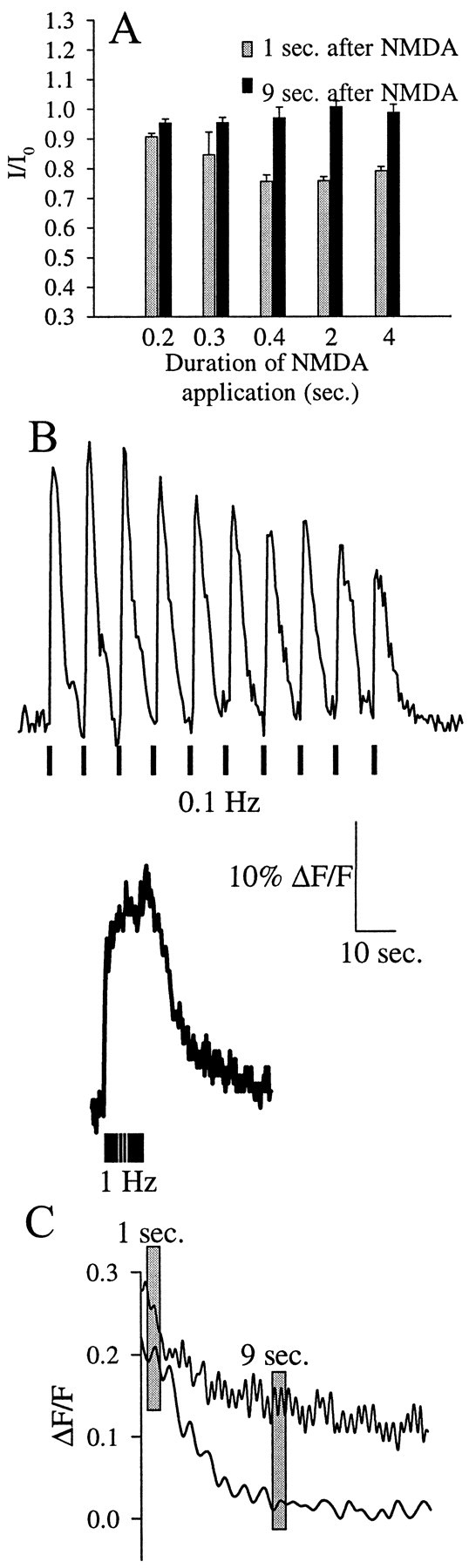
Intracellular calcium dynamics associated with different NMDA application frequencies. A, The ratios between test and control kainate currents (I/I0) measured 1 or 9 sec after different protocols of NMDA application are shown. NMDA applications of different duration were used (0.2, 0.3, 0.4, 2, and 4 sec). The gray bars indicate theI/I0 values measured 1 sec after NMDA application; the black bars are theI/I0 values measured 9 sec after NMDA application.B, Intracellular calcium dynamics was monitored using calcium green-1 as the calcium indicator. NMDA was applied for 400 msec (black bars below traces) at the indicated frequencies [0.1 Hz (top) or 1 Hz (bottom)]. During the 0.1 Hz applications, images were acquired every 500 msec, whereas at the 1 Hz application, frequency images were acquired every 100 msec. C, The calcium decay phases of the two protocols (0.1 and 1 Hz) are shown superimposed. Also shown is the relative timing of the test kainate applications (1 or 9 sec; gray bars).
Rapid agonist application was performed using a three-barreled pipette. The drugs in the reservoirs were gravity driven and controlled by solenoid valves. Solutions were exchanged by lateral displacement of the pipette by means of a piezoelectric bimorph. The central barrel was used to superfuse the cell continuously with the control solution containing 100 μm GYKI 53655 (Eli Lilly), 1 μm TTX, 50 μm picrotoxin, 10 μm glycine, and 0.01% fast green (Sigma). For activation of the kainate receptors one of the lateral barrels was used to apply an external solution containing 100 μm GYKI 53655, 300 μm kainate, 1 μm TTX, and 50 μm picrotoxin. Activation of NMDA receptors was achieved by using the remaining barrel to apply an external solution containing 100 μm NMDA, 10 μm glycine, 1 μm TTX, and 50 μm picrotoxin. The signal was filtered at 2 kHz, digitized at 10 kHz, and stored on a personal computer.
Calcium imaging. Cells were loaded with calcium green-1 AM (3 μm; Molecular Probes, Eugene, OR) in the same extracellular saline used for the electrophysiology experiments, in the presence of 0.02% pluronic F-127. Samples were excited with a xenon lamp, and the excitation wavelength (485 nm) was selected using a polychromatic illumination system (TILL Photonics). Images were acquired using a cooled CCD camera (MicroMax; Princeton Instruments) and Imaging Workbench software (Axon Instruments).
RESULTS
NMDA receptor activation induces a Ca2+-dependent depression of the current carried by kainate receptors
To examine the calcium-dependent modulation of kainate receptors in hippocampal neurons in culture, we measured the whole-cell currents evoked by kainate before and after activation of NMDA receptors. Kainate receptor currents were elicited by fast application of kainate (300 μm) in the continuous presence of the AMPA receptor blocker GYKI 53655 (100 μm) (Paternain et al., 1995). Under these conditions, we observed a kainate-induced desensitizing current that slowly recovered from desensitization (τ1 = 6 sec, 60%; τ2 = 33 sec, 40%) (Fig.1A). A similar time course for kainate receptor recovery from desensitization has been described previously (Wilding and Huettner, 1997; Paternain et al., 1998). After a stable whole-cell recording configuration was obtained, the kainate current was recorded, and 1.5 min later, NMDA (100 μm) was applied for 2 sec followed, 1 sec later, by a test application of kainate. The activation of NMDA receptors caused a depression of 23.3 ± 1.7% (mean reduction ± SEM; n = 14) in the peak amplitude of the kainate receptor test current relative to the control (Fig.1B). The depression of the kainate current was abolished when the experiment was performed in the presence of the NMDA receptor blocker AP-5 [3 ± 5% (mean reduction ± SEM);n = 4] (Fig. 1C).
A similar depression of kainate receptor currents [19.6 ± 2.3% (mean reduction ± SEM); n = 12] was observed when the cells were depolarized by fast application of a solution containing 60 mm KCl, in the presence of the sodium channel blocker TTX (1 μm) and the NMDA receptor blocker AP-5 (50 μm) (Fig. 1D). The inward current associated with the application of the high-KCl solution is primarily caused by the activation of high-voltage-activated calcium channels because it was significantly reduced by 50 μmnifedipine [88.5 ± 15% (mean reduction ± SEM)] (Fig.1E). In the presence of nifedipine, KCl applications did not induce any significant depression of the kainate receptor currents [5 ± 2.5% (mean reduction ± SEM);n = 3] (Fig. 1E).
We asked whether the NMDA-induced depression is specific for kainate receptors. As shown in Figure 2, AMPA receptor currents are identical before and after the activation of NMDA receptors [test current relative to control, 104 ± 3% (± SEM);n = 4], indicating that the NMDA-induced depression specifically targets kainate receptors.
The observation that activation of either NMDA receptors or voltage-sensitive calcium channels can induce a depression of the kainate receptor peak currents suggests that Ca2+ ion influx may be primarily responsible for this phenomenon. As a first step in the characterization of the signaling pathway linking the activation of the NMDA receptors to the depression of the kainate receptor current, we included the calcium chelator BAPTA in the recording electrode buffer. Chelating the intracellular Ca2+ was sufficient to abolish completely the effect of NMDA receptor activation on kainate receptors [2 ± 2% (mean reduction ± SEM);n = 8; p < 0.001, Student'st test in comparison with NMDA application in the absence of BAPTA] (Fig. 3A,C). These results indicate that the NMDA-induced reduction in the amplitude of the kainate current depends on the elevation of [Ca2+]i.
NMDA-induced depression of kainate receptor currents depends on calcineurin activity
The recombinant kainate receptor subunit GluR6 expressed in HEK-293 cells was shown to be phosphorylated by PKA (Raymond et al., 1993; Wang et al., 1993), and phosphorylation increased the amplitude of the kainate currents (Traynelis and Wahl, 1997). Conversely activation of the Ca2+/calmodulin-dependent protein phosphatase calcineurin [also known as phosphoprotein phosphatase 2B (PP2B)] resulted in a depression of the peak current amplitude (Traynelis and Wahl, 1997). We tested whether calcineurin was responsible for the Ca2+-dependent depression of neuronal kainate receptor currents. When the calcineurin inhibitor deltamethrin was included in the recording pipette, the NMDA-induced depression of the kainate current was abolished. Instead, we measured a small increase in kainate current amplitude after NMDA application [114.5 ± 6% (percent of control ± SEM);n = 8; p = 0.02, Student'st test] (Fig. 3B,C). The inclusion of the calcineurin autoinhibitory peptide in the recording electrode (250 μm) (Hashimoto et al., 1990) also completely abolished the NMDA-induced depression of the kainate currents (Fig.3C). In contrast, inclusion of the PP1 and PP2A inhibitors tautomycin (1 μm) or okadaic acid (500 nm) in the recording electrode did not have any effect on the kainate current depression (Fig. 3C). These results suggested that the NMDA-induced depression of the kainate receptor currents is mediated by the phosphatase calcineurin.
Recovery of kainate receptor responses from NMDA-induced depression requires Ca2+/calmodulin-dependent kinase
We then investigated the rate of recovery from the depression by evoking the test kainate current at various time intervals after NMDA receptor activation. As shown in Figure4, A and B, the amplitude of the kainate receptor test current recovered to baseline levels in ∼9 sec after the NMDA receptor activation (τ = 3.6 ± 1.2 sec). Because of our evidence of an involvement of the phosphatase calcineurin in the depression of the kainate receptor current, we hypothesized that recovery from depression might be mediated by a kinase. All known kainate receptor subunits have consensus phosphorylation sites for PKA, PKC, and Ca2+/calmodulin-dependent protein kinase (CaMK) in their putative intracellular regions, and the kainate receptor subunit GluR6 has been shown to be a substrate for PKA when expressed in HEK cells (Raymond et al., 1993; Wang et al., 1993). We tested the effect of the specific PKA inhibitor {N-[2-((p-bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, HCl} (H-89) on the recovery from depression. When included in the recording electrode, the PKA inhibitor did not affect recovery of the kainate receptor current even at a concentration 400-fold higher than the IC50 (Fig.4C). Similarly, the PKC inhibitor chelerythrine did not modify the rate or the extent of the recovery (Fig.4D). However two different inhibitors of the Ca2+/calmodulin-dependent protein kinase, {1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl)-4-phenylpiperazine} (KN-62) and KN-93, blocked the recovery from depression (Fig.4E,F). KN-92, an inactive derivative of KN-93, had no effect on the recovery (Fig. 4F). From these results it can be concluded that the recovery of the kainate receptors to baseline levels after the NMDA-induced depression requires the activity of CaMK.
The experiments we have described so far were performed using the whole-cell configuration. Under these conditions it is possible that we were dialyzing out of the cell the calcium ions, calcium-binding proteins, and calcium-dependent enzymes that are involved in the modulation of kainate receptor function. To study the dynamics of NMDA-induced depression of kainate receptor currents under more physiological conditions, we used perforated patches. As shown in Figure 5, a large NMDA-induced depression was observed when recording in the perforated-patch configuration [46 ± 8.0% (mean reduction ± SEM); n = 5]. Even though the amplitude of the NMDA-induced depression was larger in the perforated patch compared with that measured in the whole-cell configuration, the kinetics of recovery from the depression was faster [τ = 1.4 ± 0.3 (mean ± SEM)]. As a result, 10 sec after the activation of NMDA receptors, the kainate receptor current amplitudes were back at baseline values.
Fig. 5.
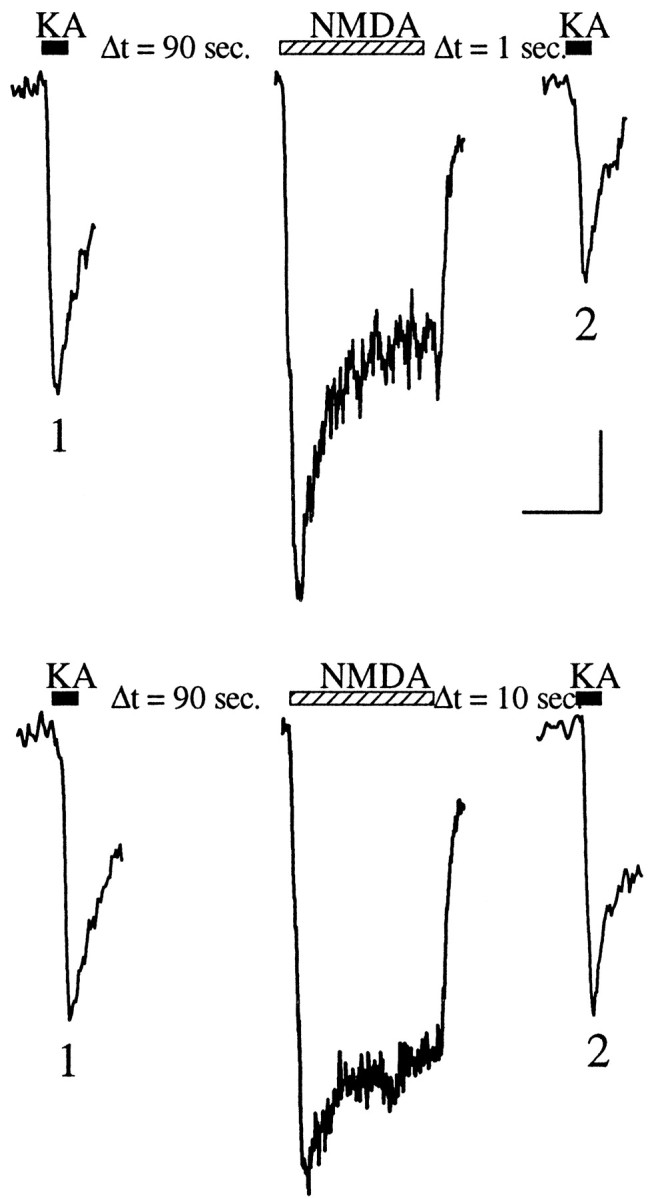
Kainate current depression observed using perforated patches or current-clamp recording. Control (1) and test (2) traces of kainate currents evoked by 300 μm kainate (black bars) before and after activation, respectively, of NMDA receptors by application of 100 μm NMDA in the presence of 10 μm glycine (hatched bars). The different time lags [1 sec (top) or 10 sec (bottom)] between NMDA application and test KA application are indicated. The recordings were performed using the perforated-patch configuration obtained by including amphotericin B in the recording pipette. Calibration: vertical bar, 5 pA for kainate receptor currents, 50 pA for NMDA receptor currents; horizontal bar, 500 msec for kainate receptor currents, 1 sec for NMDA receptor currents.
In vivo, neurons are unlikely to experience the prolonged (2 sec) NMDA receptor activation that we have used to induce the depression of kainate receptor currents. Furthermore, we have activated NMDA receptors under voltage-clamp conditions, which do not correspond to the physiological state of the neurons in vivo, in which receptor activation and ion influx are accompanied by fluctuations of the membrane potential. To study the NMDA-induced depression of kainate receptor currents under more physiological conditions, we have measured the effects of short pulses of NMDA, delivered while in the current-clamp recording mode. As shown in Figure6, five pulses of NMDA, 200 msec each, resulted in a large membrane depolarization while in the current-clamp mode and induced a significant depression of the kainate receptor current [32.5 ± 11.6% (mean reduction ± SEM);n = 4]. After this kind of stimulation, the kainate receptors recover in ∼10 sec (Fig. 6) [τ = 2.2 ± 0.5 (mean ± SEM); n = 4].
Fig. 6.
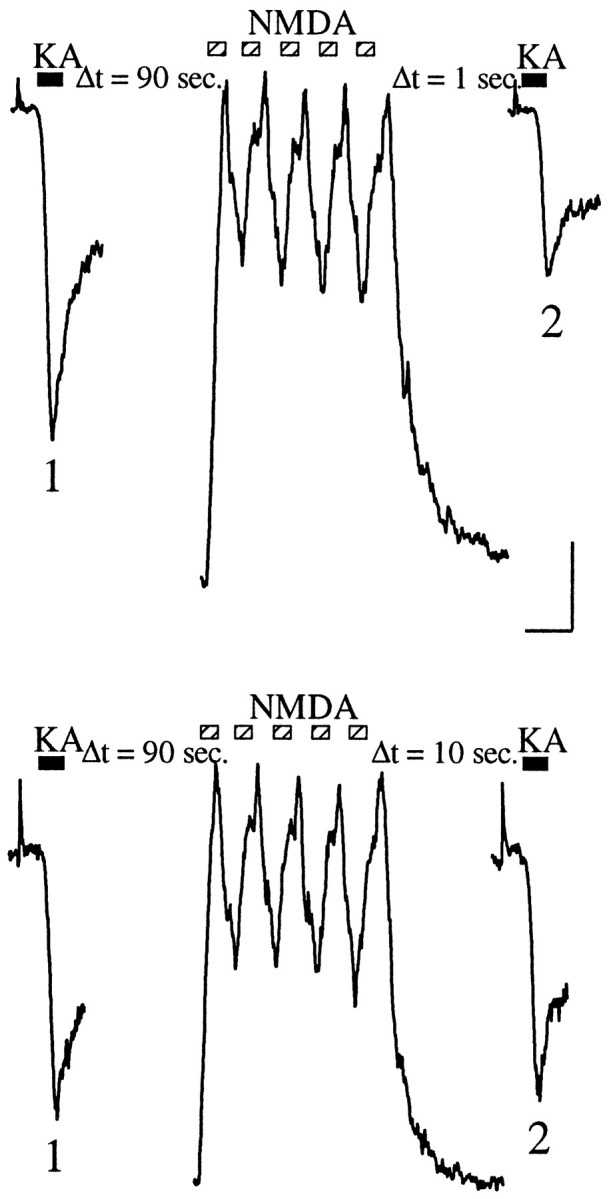
Kainate current depression induced while in current-clamp recording mode. Control (1) and test (2) traces of kainate currents evoked by 300 μm kainate (black bars) before and after activation, respectively, of NMDA receptors by five applications (200 msec each) of 100 μm NMDA in the presence of 10 μm glycine (hatched bars). Kainate currents were recorded while holding the cells at −70 mV in the voltage-clamp mode. NMDA currents were recorded while in current-clamp mode. The different time lags [1 sec (top) or 10 sec (bottom)] between NMDA application and test KA application are indicated. Calibration: vertical bar, 100 pA for kainate receptor currents, 10 mV for NMDA receptor currents; horizontal bar, 250 msec for kainate receptor currents, 500 msec for NMDA receptor currents.
Recovery of kainate receptors from depression can be modulated by the NMDA receptor activation frequency
Both the depression and the recovery from depression of kainate receptors are dependent on the activity of two Ca2+/calmodulin-dependent enzymes, calcineurin and CaMK, respectively. We tested the hypothesis that different patterns of NMDA receptor activation, and therefore Ca2+ influx, may shift the balance between potentiation and depression of kainate receptors.
First, we determined that 400 msec was the shortest NMDA application capable of eliciting depression of the kainate receptor current. As shown previously for longer NMDA application (Figs. 1, 4, 5), the kainate receptor recovers from the 400 msec stimulation in ∼9 sec (Fig. 7A). Increasing the duration of the NMDA pulse up to 4 sec did not substantially alter the extent of kainate receptor depression or recovery (Fig.7A).
We then tested NMDA application protocols capable of eliciting different patterns of intracellular Ca2+rise. As shown in Figure 7B, when NMDA was applied for 400 msec repetitively at two different frequencies (0.1 or 1 Hz), the [Ca2+]i dynamics was different. At the lower NMDA application frequencies, distinct Ca2+ spikes were detectable, whereas when NMDA was applied at 1 Hz, a single plateau of intracellular Ca2+ increase was reached. Also the rate of decay of [Ca2+]i was different at the two application frequencies (at 0.1 Hz, τ = 3.3 ± 0.8 sec; n = 4; at 1 Hz, τ = 14 ± 2 sec; n = 4; p < 0.05). The different NMDA receptor activation frequencies result in similar [Ca2+]i 1 sec after the end of the stimulation but, because of the different decay rates, different [Ca2+]i 9 sec after the end of the stimulation.
We then tested the effects of a train of 10 400 msec NMDA applications. At low frequency (0.1 Hz), this stimulation induced depression of the kainate receptor current (Fig.8A), and the depression was abolished by the calcineurin autoinhibitory peptide (Fig.8E). As with the depression induced by a single 2 sec pulse of NMDA, the depression induced by the low-frequency train fully recovered in 9 sec and was dependent on CaMK (Fig.8B).
Fig. 8.
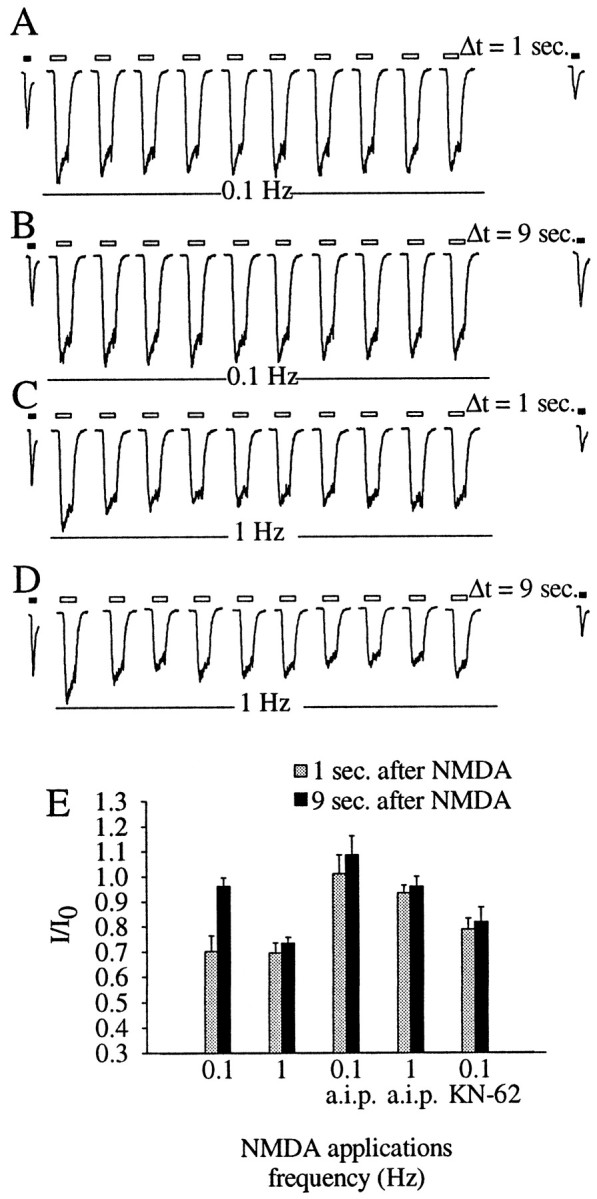
Effect of NMDA application frequency on recovery of the kainate receptors. A–D, Whole-cell currents evoked by 300 μm kainate (black bars) before (first trace on the left) and after (last trace on the right) 10 consecutive applications of 100 μm NMDA for 400 msec (white bars). The different frequencies of NMDA application are indicated. The time interval (Δt) between the last NMDA application and the test kainate application is indicated. E, The ratio between test and control kainate currents (I/I0) measured 1 or 9 sec after NMDA application at 0.1 or 1 Hz. Gray bars indicate theI/I0 values measured 1 sec after NMDA application; the black bars are theI/I0 values measured 9 sec after NMDA application. Where indicated, calcineurin autoinhibitory peptide (a.i.p., 250 μm) or KN-62 (50 μm) was included in the intracellular solution.
A different result was obtained when the 400 msec NMDA pulses were delivered at a higher frequency. First, at 1 Hz we observed that the NMDA receptor currents that follow the first stimulus show a small (22 ± 5%) but significant (p < 0.05) reduction in amplitude, reminiscent of the calcineurin-dependent depression of NMDA receptors described by Jahr and colleagues (Tong et al., 1995). We found that applying NMDA at 1 Hz, the depression was also induced and was mediated by the activation of calcineurin (Fig.8C,E). However, the recovery from the depression at 9 sec was impaired in 10 out of 14 cells tested (Fig. 8D). After the 1 Hz stimulation the kainate receptor current amplitude recovered to control values in 30 sec (data not shown). Thus, different NMDA application protocols, which are associated with different patterns of [Ca2+]i dynamics, can affect the kainate receptor current amplitude to different extents.
DISCUSSION
We have examined the functional interaction between kainate and NMDA receptors. We found that after the activation of NMDA receptors, the Ca2+/CaM-dependent phosphatase calcineurin induces a depression of the kainate receptor current. Previous work on the recombinant kainate receptor GluR6 expressed in HEK cells has shown that this subunit can be phosphorylated and that this results in an increase in peak current amplitude (Raymond et al., 1993; Wang et al., 1993) because of an increased channel open probability (Traynelis and Wahl, 1997). Because of these experiments, our data are best interpreted by assuming that in the resting state the majority of kainate receptors are phosphorylated. After the activation of calcineurin by the NMDA receptor-mediated Ca2+ influx, kainate receptors are dephosphorylated, and this reduces the amplitude of the peak current. This NMDA-induced depression of the kainate receptor current is followed by recovery, which is completed in ∼9 sec. Recovery is blocked by inhibitors of the Ca2+/CaM-dependent protein kinase and is insensitive to blockers of PKC or PKA, indicating that CaMK activity is primarily responsible for the recovery of the kainate receptor currents.
Our results are also consistent with a model in which the modulation of the kainate receptors by NMDA requires phosphorylation and/or dephosphorylation of proteins associated with the receptor and not necessarily of the kainate receptors themselves. The phosphorylation state of the factors associated with the receptors could then influence the channel conductance and/or kinetics. It was shown recently that the kainate receptor subunits GluR6 and KA2 can interact with members of the synapse-associated protein 90/postsynaptic density 95 (SAP90/PSD95) family of proteins and that this interaction controls the desensitization of the kainate receptors (Garcia et al., 1998).
Interestingly, in neuronal cultures a large fraction of kainate receptors is formed by homomeric GluR6 assemblies (Ruano et al., 1995). This observation, together with the sensitivity of recombinant GluR6 to modulation by calcineurin (Traynelis and Wahl, 1997), may suggest that the NMDA-induced depression of kainate receptor currents we have described is caused by the activity of calcineurin on the GluR6-containing kainate receptors. However, we have observed a similar modulation of kainate receptor currents in neurons isolated from genetically engineered mice (Mulle et al., 1998) lacking the GluR6 subunit (A. Ghetti, unpublished observation). This indicates that other kainate receptor subunits could be similarly modulated.
After the activation of NMDA receptors, both the depression and the recovery of the kainate currents are attributable to the activities of two Ca2+/CaM-dependent enzymes, calcineurin and CaMK, respectively. We investigated whether different activation patterns of the NMDA receptor may change the relative effects of these two enzymes on the kainate receptors. By using trains of short NMDA pulses, we have found that the depression of the kainate currents is induced to a similar extent by a single NMDA pulse of at least 400 msec as well as by 10 pulses of 400 msec. This suggests that calcineurin is activated with comparable efficiency after a single as well as multiple short NMDA pulses. The recovery from depression was, however, frequency sensitive in the majority of cells. Stimulation of the NMDA receptors at 1 Hz resulted in a depression of the kainate currents that did not recover after 9 sec but required 30 sec instead. A lower stimulation frequency (0.1 Hz) in the same cells always resulted in complete recovery of the kainate currents after 9 sec. This suggests that in our experimental system the CaMK activity relative to the calcineurin activity is lower after the stimulation at the higher frequency.
The Ca2+-dependent enzymes CaMK and calcineurin may be expected to be activated simultaneously during the NMDA receptor activation and consequent Ca2+ influx. However, the affinity of the two enzymes for Ca2+/CaM differs over fivefold (Hubbard and Klee, 1987; Meyer et al., 1992), and additional factors such as the local enzyme concentration and relative affinity for the substrate are expected to play a role in determining the timing of depression and recovery. In this context, the different intracellular calcium dynamics associated with the low- and high-frequency NMDA application is probably responsible for the observed changes in the relative activity of the two enzymes. Thus the slower decay of intracellular calcium induced by the high-frequency application may result in higher calcineurin activity, and the slower observed recovery may result from the depression.
Postsynaptic kainate receptor currents in the CA3 region of the hippocampus have been observed only after high-frequency stimulation of the mossy fibers (Castillo et al., 1997; Vignes and Collingridge, 1997;Mulle et al., 1998). According to our data, high-frequency stimulations should result in a depression of the kainate receptor currents, a conclusion apparently not in agreement with the results obtained in hippocampal slices. The most likely explanation for this discrepancy is that in those experiments the slices were bathed with NMDA receptor blockers and thus the biochemical pathway responsible for the depression of the kainate receptor currents was not activated. In addition, the amplitude of the NMDA-induced depression is reduced when recording in the whole-cell configuration, which was used in the experiment performed in the slices, compared with the perforated-patch configuration.
These recent studies on kainate receptors in hippocampal slices have highlighted the modulatory function of these receptors in synaptic transmission. Our results indicate that, in turn, synaptic activity, by activating NMDA receptors, may control kainate receptor function. An additional layer of control in the system is provided by the sensitivity of the kainate receptor depression to the activation frequency of NMDA receptors. Postsynaptic kainate receptor responses have been reported in the hippocampus, the retina, and the spinal cord, and in at least one case they have been shown to be sensitive to the stimulation frequency (Castillo et al., 1997; Vignes and Collingridge, 1997). Additional work will be required to establish the role of this form of glutamate receptor modulation in synaptic computation and plasticity.
Footnotes
This work was supported by National Institutes of Health Grant NS28709 to S.F.H.; A.G. was sponsored by a fellowship from Fondazione Telethon. We thank Mary Ann Pilla for technical assistance and Anis Contractor, Tim Green, Robert Petroski, Juan Pina-Crespo, and Geoff Swanson for discussions and critical reading of this manuscript.
Correspondence should be addressed to Dr. Andrea Ghetti, Molecular Neurobiology Laboratory, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, CA 92037. E-mail:ghetti@axp1.salk.edu.
REFERENCES
- 1.Artola A, Singer W. Long term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Collingridge GL. A synaptic model of memory: long term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 4.Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- 5.Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboji RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- 6.Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben-Ari E. GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci. 1998;1:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- 7.DeVries SH, Schwartz EA. Kainate receptors mediate synaptic transmission between cones and Off bipolar cells in a mammalian retina. Nature. 1999;397:157–160. doi: 10.1038/16462. [DOI] [PubMed] [Google Scholar]
- 8.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 9.Frerking M, Malenka RC, Nicoll RA. Synaptic activation of kainate receptors on hippocampal interneurons. Nat Neurosci. 1998;1:479–486. doi: 10.1038/2194. [DOI] [PubMed] [Google Scholar]
- 10.Garcia EP, Metha S, Blair LA, Wells DG, Shang J, Fukushima T, Fallon JR, Garner CC, Marshall J. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron. 1998;21:727–739. doi: 10.1016/s0896-6273(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 11.Hashimoto Y, Perrino BA, Soderling TR. Identification of an autoinhibitory domain in calcineurin. J Biol Chem. 1990;265:1924–1927. [PubMed] [Google Scholar]
- 12.Hollmann M, Heinemann SF. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 13.Hubbard MJ, Klee CB. Calmodulin binding by calcineurin. J Biol Chem. 1987;262:15062–15070. [PubMed] [Google Scholar]
- 14.Lerma J. Kainate reveals its targets. Neuron. 1997;19:1155–1158. doi: 10.1016/s0896-6273(00)80407-9. [DOI] [PubMed] [Google Scholar]
- 15.Li P, Wilding TJ, Calejesan AA, Huettner JE, Zhuo M. Kainate-receptor-mediated sensory transmission in mammalian spinal cord. Nature. 1999;397:161–164. doi: 10.1038/16469. [DOI] [PubMed] [Google Scholar]
- 16.Lisman J. A mechanism for the Hebb and anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 18.Malenka RC, Nicoll RA. NMDA receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 19.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 20.Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 21.Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTP and LTD. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- 22.Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- 23.Paternain AV, Rodriguez-Moreno A, Villarroel A, Lerma J. Activation and desensitization properties of native and recombinant kainate receptors. Neuropharmacology. 1998;37:1249–1259. doi: 10.1016/s0028-3908(98)00098-7. [DOI] [PubMed] [Google Scholar]
- 24.Raymond LA, Blackstone CD, Huganir RL. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature. 1993;361:637–641. doi: 10.1038/361637a0. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- 26.Ruano D, Lambolez B, Rossier J, Paternain AV, Lerma J. Kainate receptor subunits expressed in single cultured hippocampal neurons: molecular and functional variants by RNA editing. Neuron. 1995;14:1009–1017. doi: 10.1016/0896-6273(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 27.Schulman H. Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol. 1995;5:375–381. doi: 10.1016/0959-4388(95)80051-4. [DOI] [PubMed] [Google Scholar]
- 28.Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 29.Traynelis SF, Wahl P. Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J Physiol (Lond) 1997;503:513–531. doi: 10.1111/j.1469-7793.1997.513bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- 31.Wang L-Y, Taverna FA, Huang X-P, MacDonald JF, Hampson DR. Phosphorylation and modulation of a kainate receptor (GluR6) by cAMP-dependent protein kinase. Science. 1993;259:1173–1175. doi: 10.1126/science.8382377. [DOI] [PubMed] [Google Scholar]
- 32.Wang L-Y, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- 33.Wilding TJ, Huettner JE. Activation and desensitization of hippocampal kainate receptors. J Neurosci. 1997;17:2713–2721. doi: 10.1523/JNEUROSCI.17-08-02713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]