

Abstract
Release of mitochondrial cytochrome c into the cytosol is a critical step in apoptosis. We have reported that early release of cytochrome c in vivo occurs after permanent focal cerebral ischemia (FCI) and is mediated by the mitochondrial antioxidant manganese superoxide dismutase (SOD). However, the role of reactive oxygen species produced after ischemia–reperfusion in the mitochondrial apoptosis process is still unknown, although overexpression of copper/zinc-SOD (SOD1), a cytosolic isoenzyme, protects against ischemia–reperfusion. We now hypothesize that the overexpression of SOD1 also prevents apoptosis after FCI. To address this issue, we examined the subcellular distribution of the cytochrome c protein in both wild-type mice and in SOD1 transgenic (Tg) mice after transient FCI. Cytosolic cytochrome c was detected as early as 2 hr after reperfusion, and correspondingly, mitochondrial cytochrome c was significantly reduced after FCI. Cytosolic cytochrome c was significantly lower in the SOD1 Tg mice compared with wild types 2 (p < 0.0001) and 4 (p < 0.05) hr after FCI. Apaf-1, which interacts with cytochrome c and activates caspases, was constitutively expressed in both groups of animals, with no alteration after FCI. Double staining with cytochrome c immunohistochemistry and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling showed a spatial relationship between cytosolic cytochrome c expression and DNA fragmentation. A significant amount of DNA laddering was detected 24 hr after ischemia and was reduced in SOD1 Tg mice. These data suggest that SOD1 blocks cytosolic release of cytochrome c and could thereby reduce apoptosis after transient FCI.
Keywords: cerebral ischemia, cytochrome c, copper zinc-superoxide dismutase, apoptosis, mitochondrial injury, reactive oxygen species, caspase
Cytochrome c, a water-soluble peripheral membrane protein of the mitochondria, is an essential component of the mitochondrial respiratory chain (Boyer et al., 1977). Its function is to transport electrons from the coenzyme QH2-cytochrome c reductase complex to the cytochrome c oxidase complex in the electron transport chain. Growing evidence suggests the critical role of cytochrome c in apoptosis (Liu et al., 1996; Kluck et al., 1997; Yang et al., 1997). Mitochondria are involved in apoptosis by releasing cytochrome c to the cytoplasm in which it activates caspases by interacting with some cytosolic factors, including Apaf-1, a protein homologous to Caenorhabditis elegans CED-4 (Zou et al., 1997; Yoshida et al., 1998). Released cytochrome c interacts with Apaf-1 and caspase-9, both of which play an essential role in the cytochrome c-dependent mitochondrial pathway of apoptosis (Zou et al., 1997, 1999; Hakem et al., 1998; Kuida et al., 1998) by activating caspases such as caspase-2, -3, -6, -7, -8, and -10 (Slee et al., 1999), which results in apoptosis. Bcl-2, a mitochondrial outer membrane protein, inhibits cytochrome c translocation, thereby blocking caspase activation and apoptosis (Kluck et al., 1997; Yang et al., 1997). Although the mechanism by which Bcl-2 prevents cytochrome c release has not been established, a recent in vitro study showed that overexpression of Bcl-2 prevents superoxide production and then blocks cytochrome c release and apoptosis (Cai and Jones, 1998), suggesting that the antioxidant function of Bcl-2 contributes to the inhibition of cytochrome c release and subsequent apoptosis. In fact, we have shown that deficiency in the mitochondrial antioxidant manganese superoxide dismutase (SOD) results in a marked increase in the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after focal cerebral ischemia (FCI) (Fujimura et al., 1999a), again suggesting the regulatory role of mitochondrial antioxidants in the cytochrome c-dependent apoptotic pathway. Besides the effect of the mitochondrial proteins, recent evidence shows that early release of mitochondrial cytochrome c is mediated by a variety of cytosolic factors, such as Bax, Bid, and caspase-8 (Jürgensmeier et al., 1998; Li et al., 1998; Narita et al., 1998). However, the role of the cytosolic antioxidant system in the early release of cytochrome c is unknown.
The antioxidant enzyme is one of the major mechanisms by which cells counteract the deleterious effects of reactive oxygen species (ROS) after cerebral ischemia and reperfusion. We have shown that copper/zinc-SOD (SOD1), a cytosolic antioxidant isoenzyme, is highly protective against ischemia–reperfusion injury after focal (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997) and global cerebral ischemia (Murakami et al., 1997; Chan et al., 1998). Using SOD1 knock-out mice, we have suggested the protective role of SOD1 against DNA-damaged neuronal cell death after ischemia–reperfusion (Kondo et al., 1997). However, it has not yet been determined whether SOD1, an endogenous cytosolic antioxidant, could affect mitochondrial cytochrome c release to the cytosol, thereby preventing apoptosis after transient FCI. Using both wild-type mice and transgenic (Tg) mice that overexpress SOD1 (Epstein et al., 1987), the present study was designed to clarify this critical issue by examining the early release of mitochondrial cytochrome c to the cytosol and DNA fragmentation after transient FCI in which apoptosis, as well as necrosis, are assumed to participate (Li et al., 1995; Du et al., 1996; Hara et al., 1997;Endres et al., 1998; Fujimura et al., 1998, 1999b; Namura et al., 1998).
MATERIALS AND METHODS
SOD1 Tg mice. Heterozygous SOD1 Tg mice of the SOD1 TGHS/SF-218–3 strain with a CD-1 background, carrying human SOD1 genes with a threefold increase in copper/zinc-SOD, were derived from the founder stock described previously (Epstein et al., 1987). They were further bred with CD-1 wild-type mice to generate heterozygous mice. The SOD1 Tg mice were identified by quantitative demonstration of SOD1 using nondenaturing gel electrophoresis, followed by nitroblue tetrazolium staining (Epstein et al., 1987). There were no differences in the phenotypes, including the anatomy of the circle of Willis between the SOD1 Tg mice and their wild-type littermates (Yang et al., 1994). There was no difference in the regional cerebral blood flow before and after FCI between the SOD1 Tg and wild-type mice (Chan et al., 1993). A part of the ischemic middle cerebral artery (MCA) territory cortex, including the cortical penumbra, was reported to have been rescued in SOD1 Tg mice after transient FCI (Yang et al., 1994).
Focal cerebral ischemia. Adult male SOD1 Tg mice and non-Tg littermates (35–40 gm) were subjected to transient FCI by intraluminal MCA blockade with a nylon suture as described previously (Yang et al., 1994). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases, samples for analysis being taken immediately after cannulation, 10 min after occlusion, and 10 min after reperfusion. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics Ltd., Essex, UK). After a midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. A 11.0 mm 5–0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 min of MCA occlusion, blood flow was restored by the withdrawal of the nylon suture.
Histological assessment. The experimental animals were killed 0, 1, 2, 4, 8, and 24 hr after 60 min of MCA occlusion. The brains were removed, rapidly frozen in −20°C 2-methylbutane, and stored at −80°C. They were sectioned with a cryostat into a thickness of 20 μm from the anterior side to the posterior side and stained with cresyl violet.
Western blot analysis. Protein extraction of both the mitochondrial and cytosolic fractions was performed as described previously (Fujimura et al., 1998). Approximately 50 mg of fresh brain tissue (nonfrozen) from the MCA territory of the ischemic side and from the nonischemic area of the corresponding contralateral brain were cut into pieces after 0, 1, 2, 4, 8, and 24 hr of reperfusion and gently homogenized by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ) in 7 vol of cold suspension buffer (20 mm HEPES-KOH, pH 7.5, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 0.1 mm PMSF, 2 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml pepstatin, and 12.5 μg/ml N-acetyl-leu-leu-norleucinal). The homogenates were centrifuged at 750 × g at 4°C and then at 8000 × g for 20 min at 4°C. The 8000 × gpellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000 × g for 60 min at 4°C. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA), and 3.6 μg of protein from the cytosolic fraction and 2.2 μg from the mitochondrial fraction were loaded per lane. To analyze the direct relationship between SOD1 overexpression and cytosolic cytochrome c release, we used the same series of samples for SOD1 and cytochrome c detection. The primary antibodies were either a 1:1000 dilution of rabbit anti-mouse SOD1 polyclonal (StressGen, Victoria, Canada), 1:1000 dilution of anti-human SOD1 monoclonal (PharMingen, San Diego, CA), 1:1000 dilution of rabbit cytochrome c polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Apaf-1 polyclonal (Chemicon, Temecula, CA), and 1 μg/ml 20E8C12 cytochrome oxidase (COX) subunit IV monoclonal (Molecular Probes, Eugene, OR). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG using enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech, Buckinghamshire, UK). A densitometric analysis of the cytochrome c expression was made of the results of the mitochondrial fraction and of the cytosolic fraction of the ischemic brain from both wild-type and SOD1 Tg mice. The film was scanned with a GS-700 imaging densitometer (Bio-Rad), and the results were quantified using Multi-Analyst software (Bio-Rad). Western blot analysis of β-actin was performed with horseradish peroxidase-conjugated anti-mouse IgG reagents (Amersham Pharmacia Biotech). To confirm the total amount of cytochrome c in the nonischemic mitochondria from both wild-type and SOD1 Tg mice, Western blot analyses of mitochondrial cytochrome c, as well as COX, were performed.
Immunohistochemistry. Anesthetized animals were perfused with 10 U/ml heparin and subsequently with 4% formaldehyde in 0.1m PBS, pH 7.4, 2 hr after transient FCI as described previously (Fujimura et al., 1999a). The brains were removed, post-fixed for 12 hr, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. The sections were incubated with a blocking solution and reacted with anti-cytochrome c polyclonal antibody (Santa Cruz Biotechnology) at a dilution of 1:100. Immunohistochemistry was performed using the avidin-biotin technique, and then the nuclei were counterstained with methyl green solution for 10 min. As a negative control, sections were incubated without primary antibodies. For histological assessment, alternate slices from each brain section were stained with cresyl violet.
Double-labeling with cytochrome c immunohistochemistry and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling. To clarify the spatial relationship between cytosolic cytochrome c expression and DNA damage, we performed double staining of the cytochrome c antibody and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) as described previously (Kawase et al., 1999). Animals were perfused with 10 U/ml heparin and subsequently with 4% formaldehyde in 0.1m PBS, pH 7.4, 24 hr after transient FCI. The fixed sections were immunostained with the cytochrome c antibody as described above, and the sections were mounted on glass slides (Superfrost; Fisher Scientific, Pittsburgh, PA), passed through ethanol (70, 95 and 100%), and then immersed in chloroform for 5 min. The sections were rehydrated by passage through a decreasing ethanol series, rinsed with water, and processed with TUNEL. The slides were placed in 1× terminal deoxynucleotidyl transferase (TdT) buffer (Life Technologies, Gaithersburg, MD) for 15 min, followed by reaction with TdT enzyme (Life Technologies) and biotinylated 16-dUTP (Boehringer Mannheim, Indianapolis, IN) at 37°C for 60 min. The slides were then washed two times in SSC (150 mm sodium chloride and 15 mm sodium citrate, pH 7.4) for 15 min, followed by a washing in PBS two times for 15 min. Avidin-biotin horseradish peroxidase solution (ABC kit; Vector Laboratories, Burlingame, CA) was applied to the sections for 30 min. Staining was visualized using 0.025% diaminobenzidine and 0.075% H2O2 with nickel sulfate. The slides were rinsed with water, stained with methyl green for 10 min, dehydrated, and mounted.
Gel electrophoresis. Genomic DNA gel electrophoresis was performed as described previously (Fujimura et al., 1999b). Animals were killed 24 hr after 60 min of MCA occlusion. Thirty to 50 mg wet weight of brain tissue from the ischemic MCA territory were taken from the third 2 mm section along with homologous tissue from the contralateral side after the brain was cut coronally. Samples were incubated overnight in 0.6 ml of lysis buffer (0.5% SDS, 10 mm Tris-HCl, and 0.1 mEDTA) with 0.6 mg of proteinase K (Boehringer Mannheim) at 55°C. The DNA was extracted with equal volumes of phenol and phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated overnight in 0.2 m sodium chloride in 100% ethanol at −80°C. The DNA was washed twice with 75% ethanol, air dried, and resuspended in DNase-free water (Sigma, St. Louis, MO). The DNA concentration was measured using To-Pro-1 dye (Molecular Probes). Gel electrophoresis for detecting DNA laddering was performed according to the manufacturer's instructions (Trevigen, Gaithersburg, MD), as described previously (Fujimura et al., 1999b). Before electrophoresis, 1 μg of DNA was incubated with 50 μg/ml DNase-free RNase (Boehringer Mannheim) for 30 min at 37°C. Then the samples were reacted with Klenow enzyme (Trevigen) and dNTP (Trevigen) in 1× Klenow buffer (Trevigen) for 10 min at room temperature. Samples were mixed with loading buffer and subjected to electrophoresis on a 1.5% agarose gel. Then the gel was washed with 0.25 m HCl, 0.4m NaOH or 0.8 m NaCl, and 0.5 m Tris buffer, pH 7.5. DNA was transferred to a nylon membrane overnight in 10× SSC. The membrane was first blocked by 5% powdered milk (Bio-Rad) in PBS for 30 min and incubated with Strept-horseradish peroxidase conjugate (Trevigen) for 30 min. Finally, the bands were visualized by the chemiluminescence method using PeroxyGlow (Trevigen), and the films were exposed to x-ray film.
RESULTS
SOD1 expression is not modified after transient FCI in either wild-type or Tg mice
As shown in Figure 1, endogenous mouse SOD1 was constitutively expressed in both wild-type and Tg mice and was not modified until 24 hr after transient FCI (Fig.1A, top lane). Because anti-mouse SOD1 antibody used in the present study had a cross-reactivity against human SOD1, characteristic bands of human SOD1, which are products of the transgene, were also seen in the SOD1 Tg mice (Fig.1A, top lane) but not in the wild-type mice. The use of anti-human SOD1 antibody resulted in the detection of characteristic single bands of human SOD1 only in the Tg mice that were not modified after 1–24 hr of transient FCI (Fig.1B, top lane). A consistent amount of β-actin is seen in the bottom panels (Fig.1A,B).
Fig. 1.

Western blot analysis of mouse (A) and human (B) SOD1 expression in wild-type (Wt) and SOD1 Tg mice before and after transient FCI. Protein (6.5 μg) from the cytosolic fraction was loaded per lane. Constitutive expression of endogenous mouse SOD1 was seen in both the wild-type and Tg mice and was not modified after FCI (A, top panel). Human SOD1 was also detected by anti-mouse antibody in SOD1 Tg mice (A, top panel), as shown by thetop bands, but not in the wild-type mice. Human SOD1 was not detected in the wild-type mice, but it was detected in the SOD1 Tg mice before and after FCI. There was no change in the human SOD1 level before and after FCI in the SOD1 Tg mice (B, top lane). A consistent amount of β-actin expression is shown in the bottom lanes. C, Control.
Western blot analysis demonstrating the early release of mitochondrial cytochrome c to the cytosol after transient FCI
As shown in Figure2A, cytochrome c immunoreactivity was evident as a single band of molecular mass 15 kDa (Fujimura et al., 1999a) in the cytosolic fraction in the ischemic brain in the wild-type mice as early as 2 hr after reperfusion, whereas it was barely detected in the normal control brain. The characteristic single band in the ischemic sample was sustained until 24 hr after reperfusion (Fig. 2A). These data not only confirm the specificity of the polyclonal antibody for cytochrome c used in this study but also show that cytosolic localization of cytochrome c was significantly increased after transient FCI. The mitochondrial fraction of cytochrome c was also examined in the wild-type animals 4 hr after transient FCI. As shown in Figure 2B, a significant amount of mitochondrial cytochrome c was detected in the nonischemic brain and decreased after FCI (Fig. 2B,top panel). Correspondingly, the cytosolic fraction from the same sample showed a marked increase of cytochrome c in the ischemic brain (Fig. 2B, top panel). COX was strongly expressed in the mitochondrial fraction, but not in the cytosolic fraction, in both the ischemic and nonischemic brains (Fig. 2B, middle panel).
Fig. 2.

Western blot analysis of cytosolic (A) and mitochondrial (B) cytochrome c from wild-type mice. A, Cytochrome c from the cytosolic fraction in the nonischemic control brain (lane C) and in the ischemic brains (lanes 1–24). Protein (4.1 μg) was loaded per lane. Cytochrome c immunoreactivity was evident as a single band of molecular mass 15 kDa in the cytosolic fraction in the ischemic brain as early as 2 hr after transient FCI (lane 2), whereas it was barely detected in the normal control brain. Cytosolic cytochrome c was sustained until 24 hr after transient FCI. The results of the β-actin analysis are shown in the bottom panel as an internal control. The results shown are representative of three independent studies. B, Western blot analysis of the mitochondrial and cytosolic fractions 4 hr after reperfusion. A significant amount of mitochondrial cytochrome c was detected in the control samples (top lane C under Mitochondria) and was decreased after transient FCI (top lane I underMitochondria). Correspondingly, cytosolic cytochrome c from the same animal showed an increase after transient FCI. Contrarily, COX showed no alteration after FCI (middle panel). Approximately 1.8 μg of protein from the mitochondrial fraction and 6.0 μg of protein from the cytosolic fraction were loaded per lane. C, Nonischemic control;I, ischemic brain 4 hr after reperfusion;CL, control sample of 0.8 μg of mitochondrial protein. A consistent amount of β-actin expression is shown in thebottom lane. The results shown are representative of three independent studies
Early release of mitochondrial cytochrome c to the cytosol is significantly reduced in SOD1 Tg mice compared with wild-type mice after transient FCI
The amount of cytosolic cytochrome c was compared between SOD1 Tg mice and wild-type mice 2 (Fig.3A) and 4 (Fig. 3B) hr after transient FCI. The results shown are derived from one of the nonischemic control samples and three of the ischemic samples from different animals in each group. There was no difference in the β-actin level between the wild-type and SOD1 Tg mice (data not shown). The mean optical density of the characteristic bands from the SOD1 Tg mice was significantly lower than that from the wild-type mice both 2 (wild type, 2.3 ± 1.1; Tg, 0.24 ± 0.016) and 4 (wild type, 2.3 ± 1.1; Tg, 0.55 + 0.38) hr after transient FCI (p < 0.0001 at 2 hr; p < 0.05 at 4 hr) (Fig. 4), indicating that cytosolic localization of cytochrome c was significantly decreased in the Tg mice compared with the wild-type mice after transient FCI. To confirm the total amount of nonischemic mitochondrial cytochrome c in wild-type and SOD1 Tg mice, we performed Western blot analysis using nonischemic mitochondrial fractions from both wild-type and SOD1 Tg mice. A similar amount of cytochrome c (Fig.5, top panel) as well as COX (bottom panel) were seen in the nonischemic mitochondrial fraction from both wild-type (lanes 1–4) and SOD1 Tg (lanes 5–8) mice. These results indicate that the total amount of mitochondrial cytochrome c was similar between wild-type and SOD1 Tg mice.
Fig. 3.

Western blot analysis of cytosolic cytochrome c in wild-type mice (Wt) and SOD1 Tg mice 2 (A) and 4 (B) hr after transient FCI. The data shown are from three different animals of each group. Protein (6.8 μg) was loaded per lane. C, Nonischemic control brain; I, ischemic brain. The mean optical density of the characteristic bands from the SOD1 Tg mice was significantly lower both 2 (A) and 4 (B) hr after transient FCI compared with that of the wild-type mice, indicating that cytosolic redistribution of cytochrome c was significantly decreased in SOD1 Tg mice.
Fig. 4.

The results of the densitometric analysis of cytosolic cytochrome c in wild-type (Wt) and SOD1 Tg mice 2 and 4 hr after reperfusion. The mean optical density of the characteristic bands from the SOD1 Tg mice was significantly lower than that from the wild-type mice, indicating that cytosolic redistribution of cytochrome c was significantly decreased in SOD1 Tg mice. *p < 0.05; **p < 0.0001 compared with wild-type mice at the same time point; †p < 0.01 compared with normal control; ‡p < 0.001 compared with contralateral side using Student's t test for statistical analysis.
Fig. 5.

Western blot analysis of mitochondrial cytochrome c from nonischemic brains in wild-type and SOD1 Tg mice. Approximately 1.8 μg of protein from the mitochondrial fraction was loaded per lane. A similar amount of cytochrome c (top panel), as well as COX (bottom panel), was seen in the nonischemic mitochondrial fraction from both wild-type (lanes 1–4) and SOD1 Tg (lanes 5–8) mice, suggesting that the total amount of constitutive cytochrome c expression is not modified by the overexpression of SOD1 in Tg mice.
Apaf-1 was constitutively expressed in the cytosol and was not modified after transient FCI in both wild-type and SOD1 Tg mice
As shown in Figure 6, a characteristic band of Apaf-1 is evident in the cytosolic fraction in the nonischemic control brains (top panel). There was no significant modification of Apaf-1 expression after 1–24 hr of transient FCI, and there was no difference in Apaf-1 expression between the wild-type and SOD1 Tg mice during these time points. A consistent amount of β-actin is shown in the bottom panel (Fig.6).
Fig. 6.

Western blot analysis of Apaf-1 in the cytosolic fraction in wild-type (Wt) and SOD1 Tg mice before and after transient FCI (top panel). Protein (6.5 μg) from the cytosolic fraction was loaded per lane. A similar amount of Apaf-1 was seen in the cytosolic fraction from both wild-type and SOD1 Tg mice before FCI and was not modified after 1–24 hr of transient FCI. There was no difference in the postischemic expression of Apaf-1 between the wild-type and Tg mice. A consistent amount of β-actin is shown in the bottom panel.C, Control.
Cytosolic expression of cytochrome c was detected by immunohistochemistry in the ischemic brain after transient FCI and was decreased in SOD1 Tg mice
Cytochrome c protein expression before and after transient FCI was also analyzed by immunohistochemistry in both the wild-type (Fig.7A,C,E,G) and SOD1 Tg (Fig.7B,D,F,H) mice. Homogeneous cytoplasmic immunoreactivity of cytochrome c was visible 2 hr after transient FCI in the wild-type mice in the entire MCA territory, including the ischemic cortex (Fig. 7C,arrows), lateral caudate putamen (G,arrows), and cortical penumbra (E,arrows). Both the number of cytochrome c-positive cells and cytosolic immunoreactivity were profoundly reduced in the SOD1 Tg mice 2 hr after FCI in entire regions, such as the ischemic cortex (Fig.7D, arrow), lateral caudate putamen (H, arrow), and cortical penumbra (F,arrow) compared with the wild-type mice. There was no immunoreactivity in the contralateral hemisphere in either the wild-type (Fig. 7A) or the Tg (Fig. 7B) mice. The absence of immunoreactivity in the nonischemic brain (Fig.7A,B), which is consistent with our previous studies (Fujimura et al., 1998, 1999a), is considered to be because of the thorough fixation of the brain with formaldehyde, which prevented the antibody from reaching the mitochondrial intermembrane space, but not the cytosol. In fact, immunohistochemistry with frozen sections resulted in dotted cytoplasmic immunoreactivity of cytochrome c in control brains (data not shown). To analyze the relationship between cytosolic accumulation of cytochrome c and DNA fragmentation, we performed double staining of cytochrome c and TUNEL 2 and 24 hr after transient FCI in the wild-type mice. Two hours after transient FCI, cytosolic cytochrome c expression was seen as a browncolor, whereas there were no TUNEL-positive cells in the ischemic brain (Fig. 8B). Twenty-four hours after transient FCI, a significant number of the cells that sustained cytosolic cytochrome c immunoreactivity were seen in the ischemic brain, most of which showed densely labeled nuclei, chromatin condensation, and apoptotic bodies by TUNEL (Fig. 8C,arrows). The number of TUNEL-positive cells was consistent with our previous report (Kondo et al., 1997). Because the typical apoptotic cells showed shrunken morphology, cytosolic expression of cytochrome c was seen as a thin brown staining in some cells (Fig. 8C, arrows). There were no TUNEL-positive cells or cytosolic cytochrome c immunoreactivity in the control specimens (Fig. 8A).
Fig. 7.
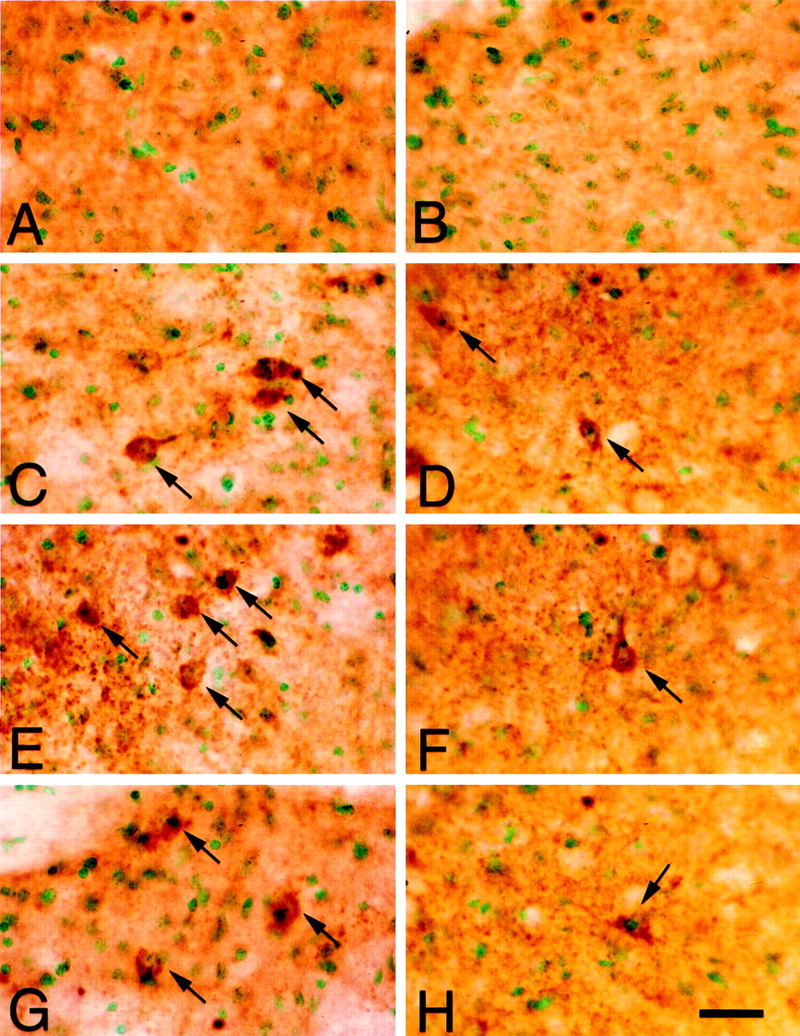
Cytochrome c immunostaining with methyl green counterstaining in coronal brain sections from wild-type (A, C, E,G) and SOD1 Tg (B, D,F, H) mice 2 hr after transient FCI. Cytochrome c immunoreactivity was barely seen in the nonischemic specimens from both the wild-type (A) and SOD1 Tg (B) mice. Two hours after reperfusion, homogeneous cytoplasmic immunoreactivity of cytochrome c was visible in the entire MCA territory, including the ischemic cortex (C, D, arrows), cortical penumbra (E, F, arrows), and lateral caudate putamen (G, H,arrows). Both the number of cytochrome c-positive cells and the cytosolic immunoreactivity were profoundly reduced in the SOD1 Tg mice (D, F, H) compared with wild-type mice (C, E,G). Scale bar, 0.02 mm.
Fig. 8.

Double staining with cytochrome c immunohistochemistry and TUNEL in wild-type mice 2 and 24 hr after transient FCI. Two hours after transient FCI, cytosolic cytochrome c expression was seen as brown in color, whereas there were no TUNEL-positive cells in the ischemic brain (B). A significant amount of TUNEL-positive cells was seen in the entire ischemic MCA territory 24 hr after FCI (C, black areas). At this time point, most of the TUNEL-positive cells showed cytosolic immunoreactivity of cytochrome c (C, brown areas;arrows). There were no TUNEL-positive cells or cytosolic cytochrome c immunoreactivity in the control specimens (A). Scale bar, 0.02 mm.
DNA laddering was detected by genomic DNA gel electrophoresis
To detect the occurrence of intranucleosomal DNA fragmentation, we analyzed DNA from both the ischemic brain and the homologous sample on the contralateral side. DNA laddering was absent in the control tissue from both the wild-type and SOD1 Tg mice. A significant amount of DNA laddering was detected 24 hr after ischemia and was decreased in the SOD1 Tg mice. This result is in complete accordance with the data published previously (Fujimura et al., 1999c).
Physiological data and cerebral infarction
Physiological parameters showed no significant differences in mean arterial blood pressure and arterial blood gas analysis between each group. The preischemic physiological values were as follows (wild-type and SOD1 Tg, respectively): mean arterial blood pressure, 71 ± 3.4 and 72 + 6.6 mmHg; PaO2, 160 ± 20 and 170 + 7.1 mmHg; PaCO2, 33 ± 4.6 and 31 ± 3.5 mmHg; pH, 7.3 ± 0.06 and 7.4 ± 0.02 (values are mean ± SD; n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hr after reperfusion, and extended to the entire MCA territory at 4 hr by cresyl violet staining (data not shown). The time-dependent increase of infarction in mouse brain using the intraluminal suture blockade is consistent with previous reports that used the same focal stroke model in mice (Yang et al., 1994; Kondo et al., 1997).
DISCUSSION
The current study provides the first evidence that overexpression of SOD1, a cytosolic antioxidant isoenzyme, prevents the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after focal ischemia and reperfusion. Our findings are as follows. First, mitochondrial cytochrome c was reduced in the ischemic brain 2 hr after transient FCI in wild-type mice, and the cytosolic fraction showed a corresponding increase of cytochrome c in the ischemic brain during reperfusion. Second, a postischemic increase of cytosolic cytochrome c was significantly reduced 2 and 4 hr after transient FCI in the Tg mice that overexpressed SOD1 compared with the wild-type mice. Third, a significant amount of intranucleosomal DNA fragmentation was detected by genomic DNA gel electrophoresis 24 hr after FCI in the wild-type mice and was reduced in the SOD1 Tg mice. Finally, the spatial relationship of the cytosolic accumulation of cytochrome c and the occurrence of DNA fragmentation was indicated by double staining with cytochrome c immunohistochemistry and TUNEL 24 hr after transient FCI. Taken together, these results indicate that the early release of mitochondrial cytochrome c may contribute to the occurrence of DNA fragmentation after transient FCI and that the cytosolic antioxidant SOD1 has a potential role in preventing the early release of cytochrome c from the mitochondria, thereby reducing subsequent DNA fragmentation. However, we do not rule out the possibility that some other pathway also contributes to the protective effect of SOD1 against DNA fragmentation and infarction after transient FCI. Our recent study showed that overexpression of SOD1 prevents the early decrease of apurinic–apyrimidinic endonuclease, a DNA base excision repair protein, and subsequent DNA fragmentation after transient FCI in mice (Fujimura et al., 1999c).
We reported previously that there was no difference in the anatomy of the blood vessels (Yang et al., 1994) and of regional cerebral blood flow (Chan et al., 1993) between wild-type mice and SOD1 Tg mice. Physiological parameters, such as mean arterial pressure and arterial blood gas, showed no difference between the two groups in the present study. Therefore, we believe that the MCA territory area on the lesion side was subjected to a similar level of the ischemic condition in both groups. Based on this concept, we made it a rule to cut the anatomical MCA territory for Western blot analysis and DNA gel electrophoresis to compare cytochrome c expression and the amount of DNA fragmentation between wild-type and SOD1 Tg mice. As a result, the SOD1 Tg mice showed much less accumulation of cytosolic cytochrome c than did the wild-type mice 2 hr after transient FCI (Figs. 3, 4). Using the same samples, we confirmed the significant overexpression of human SOD1, which is a product of the transgene, in the Tg mice before and after transient FCI. These results indicate that the overexpression of SOD1 directly corresponds to the lower cytosolic cytochrome c expression in the SOD1 Tg mice after transient FCI.
Because ischemia–reperfusion are known to modify a variety of protein expressions that play a role in the pathophysiology of cerebral infarction, we examined the protein levels in SOD1 and Apaf-1, a protein homologous to Caenorhabditis elegans CED-4, which interact with cytochrome c and play critical roles in downstream caspase activation in vitro (Li et al., 1997; Zou et al., 1997; Yoshida et al., 1998). A similar level of endogenous mouse SOD1 was detected in both the wild-type and Tg mice and was not altered during reperfusion. A significant amount of human SOD1, which is a product of the transgene in the TGHS/SF-218–3 strain used in this study, was detected only in the SOD1 Tg mice and was not modified after 1–24 hr of transient FCI. These results indicate that the Tg mice maintained the overexpression of the SOD1 protein during the entire time course in the present study and that the differences in cytochrome c release and the amount of DNA fragmentation reflect the accumulative effect of SOD1 overexpression. We also showed for the first time that Apaf-1 was constitutively expressed in adult mouse brain and did not change after 1–24 hr of transient FCI (Fig. 6). Furthermore, there was no difference in Apaf-1 expression before and after FCI between the wild-type and SOD1 Tg mice, suggesting that the difference shown in DNA fragmentation (Fujimura et al., 1999c) was not caused by the modification of the Apaf-1 level but was caused by, at least in part, the release of less mitochondrial cytochrome c after transient FCI.
The exact mechanism by which SOD1 blocks the release of mitochondrial cytochrome c to the cytosol is unknown. There is increasing evidence that the release of mitochondrial cytochrome c is mediated by several cytosolic factors as follows. First, mitochondrial translocation of cytosolic Bax, which has been reported to induce cytochrome c from isolated mitochondria and to exacerbate apoptosis (Jürgensmeier et al., 1998; Narita et al., 1998), was implicated in cytochrome c release after hypoxia-reoxygenation in vitro (Saikumar et al., 1998). Second, cytosolic caspase-8, which plays a significant role in the CD95–Fas pathway of apoptosis, was shown to cleave the cytosolic factor Bid, which then targets mitochondria, thereby inducing cytochrome c release, triggering a subsequent mitochondrial cascade (Li et al., 1998). Thus, overexpression of the cytosolic antioxidant SOD1 might prevent cytochrome c release after FCI by altering the activation of these cytosolic factors, including caspase-8, Bid, and Bax. Alternatively, overexpression of SOD1 in the cytosol may compensate for mitochondrial oxidative stress, thereby preventing the early release of cytochrome c after FCI. In fact, the mitochondria are sites at which ROS are produced after cerebral ischemia (Piantadosi and Zhang, 1996), and overproduction of mitochondrial ROS by manganese SOD deficiency results in a marked increase of cytochrome c release and subsequent DNA fragmentation after FCI in vivo (Fujimura et al., 1999a). Finally, cytochrome c release after nerve growth factor deprivation in sympathetic neurons was shown to be a reversible event (Martinou et al., 1999). Whether overexpression of SOD1 may have a role in synthesizing new cytochrome c during reperfusion to replace that which was released is not clear at present. Further evaluations of cytosolic factors as described above and of mitochondrial ROS production in wild-type and SOD1 Tg mice after transient FCI would address these important issues. At the same time, we do not rule out the possibility that the lower level of nonspecific mitochondrial membrane damage in SOD1 Tg mice could contribute in part to the reduction of cytochrome c release in the present study. A future study using electron microscopy would address this important issue.
Increasing evidence suggests that an active process similar to apoptosis contributes to the death of neurons (Li et al., 1995; Du et al., 1996; Hara et al., 1997; Endres et al., 1998; Fujimura et al., 1998, 1999b; Namura et al., 1998) and to the expansion of the lesion after focal ischemia and reperfusion (Du et al., 1996). Intranucleosomal DNA fragmentation was detected by genomic DNA gel electrophoresis (Du et al., 1996; Endres et al., 1998; Fujimura et al., 1999b), and TUNEL showed a significant number of neurons exhibiting chromatin condensation and apoptotic bodies (Li et al., 1995; Fujimura et al., 1998). However, a recent study demonstrated that most of the DNA fragmentation produced after either focal or global ischemia in adult rats has staggered ends with a 3′ recess of 8–10 nucleotides, which is distinct from the DNA fragmentation seen during neonatal apoptosis (MacManus et al., 1999). The electron microscopic study byColbourne et al. (1999) also showed evidence against apoptosis, such as organelle swelling and the lack of apoptotic bodies, after global cerebral ischemia in gerbils. We do not completely rule out the possibility that DNA-damaged cell death seen in the present study might be distinct from that seen in typical apoptosis in embryonic neurons. Nevertheless, the protective effect of caspase inhibitors on DNA damage and neuronal cell death after transient FCI in mice (Hara et al., 1997;Endres et al., 1998) and transient global ischemia in rats (Chen et al., 1998) strongly suggests that the biochemical cascade, including caspase activation and subsequent DNA fragmentation, plays a major role in neuronal cell death after ischemia–reperfusion. In fact, it was reported that release of mitochondrial cytochrome c and caspase-9 occurred after ischemia–reperfusion (Fujimura et al., 1998; Krajewski et al., 1999). Our results, which show that overexpression of SOD1 in mice blocked the early release of mitochondrial cytochrome c and consistently prevented subsequent DNA fragmentation, may also suggest that the mitochondrial pathway, including cytochrome c release, caspase activation, and DNA fragmentation, plays a role in reperfusion injury after FCI in mice. Because caspase-3 has been shown to directly activate DNase, thereby leading to DNA fragmentation (Enari et al., 1998), we believe that the mitochondrial pathway plays a major role in DNA-damaged cell death after transient FCI in mice, even when its characteristics are distinct from typical apoptosis seen during the developmental stage. Future morphological studies with an electron microscope and/or DNA analysis using ligation-mediated PCR methods (MacManus et al., 1999) in mice after transient FCI would address these critical issues.
Footnotes
This study was supported by National Institutes of Health Grants NS14543, NS25372, NS36147, NS37530, and NS38653 and contract NO1 NS82386. P.H.C. is a recipient of the Jacob Javits Neuroscience Investigator Award. We are grateful to Dr. Charles J. Epstein (Department of Pediatrics, University of California, San Francisco, School of Medicine) for continuous collaboration by providing breeding pairs of SOD1 transgenic mice. We thank Jane O. Kim for her assistance with the Western blot analysis, Cheryl Christensen for her editorial assistance, and Liza Reola and Bernard Calagui for their technical assistance.
Drs. Fujimura and Morita-Fujimura contributed equally to this study.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University, 701B Welch Road, #148, Palo Alto, CA 94304. E-mail: phchan@leland.stanford.edu.
REFERENCES
- 1.Boyer PD, Chance B, Ernster L, Mitchell P, Racker E, Slater EC. Oxidative phosphorylation and photophosphorylation. Annu Rev Biochem. 1977;46:955–1026. doi: 10.1146/annurev.bi.46.070177.004515. [DOI] [PubMed] [Google Scholar]
- 2.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 3.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 4.Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 5.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 9. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391 1998. 43 50[Erratum (1998) 393:396]. [DOI] [PubMed] [Google Scholar]
- 10.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Groner Y. Transgenic mice with increased CuZn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA. 1987;84:8044–8048. doi: 10.1073/pnas.84.22.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999a;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Early decrease of apurinic/apyrimidinic endonuclease expression after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999b;19:495–501. doi: 10.1097/00004647-199905000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Fujimura M, Morita-Fujimura Y, Narasimhan P, Copin J-C, Kawase M, Chan PH. Copper, zinc-superoxide dismutase prevents the early decrease of apurinic/apyrimidinic endonuclease and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 1999c;30:2408–2415. doi: 10.1161/01.str.30.11.2408. [DOI] [PubMed] [Google Scholar]
- 16.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 17.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawase M, Fujimura M, Morita-Fujimura Y, Chan PH. Reduction of apurinic/apyrimidinic endonuclease expression after transient global cerebral ischemia in rats. Implication of the failure of DNA repair in neuronal apoptosis. Stroke. 1999;30:441–449. doi: 10.1161/01.str.30.2.441. [DOI] [PubMed] [Google Scholar]
- 20.Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis [see comments]. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 22.Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesan GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 26.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 29.MacManus JP, Fliss H, Preston E, Rasquinha I, Tuor U. Cerebral ischemia produces laddered DNA fragments distinct from cardiac ischemia and archetypal apoptosis. J Cereb Blood Flow Metab. 1999;19:502–510. doi: 10.1097/00004647-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami K, Kondo T, Epstein CJ, Chan PH. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke. 1997;28:1797–1804. doi: 10.1161/01.str.28.9.1797. [DOI] [PubMed] [Google Scholar]
- 32.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. [DOI] [PubMed] [Google Scholar]
- 35.Saikumar P, Dong Z, Patel Y, Hall K, Hopfer U, Weinberg JM, Venka-tachalam MA. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 1998;17:3401–3415. doi: 10.1038/sj.onc.1202590. [DOI] [PubMed] [Google Scholar]
- 36.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 40.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4: participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 41.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]