

Abstract
There are several lines of evidence suggesting that, in addition to neurotrophins, member(s) of glial cell line-derived neurotrophic factor (GDNF) family play important roles in the development of sympathetic neurons. However, the mechanism regulating the responsiveness of the neurons to GDNF family members is not known. Previously, we reported on the cooperative roles of bone morphogenetic protein-2 (BMP2) and retinoic acid (RA) in the enhancement of neurotrophin-3 (NT3) responsiveness in cultured sympathetic neurons dissociated from perinatal rat superior cervical ganglia (SCG). In the present study, we further examined the effects of BMP2 and RA on the regulation of the responsiveness of SCG neurons to GDNF family members. Consequently, we found that RA alone induced the responsiveness of SCG neurons specifically to GDNF by upregulating the ligand-specifying receptor for GDNF (GFRα-1) at both the mRNA and protein levels. The expression levels of mRNAs for other ligand-specifying receptors for GDNF family (GFRα-2 and GFRα-3) were unaffected by RA. Although the upregulation of signal-transducing receptor Ret by the RA treatment was rather small, this treatment significantly increased the efficacy of tyrosine phosphorylation of Ret by GDNF. Experiments using synthetic retinoids suggested that RA acts through α-type of nuclear retinoic acid receptor to exert the induction of GDNF responsiveness. On the other hand, BMP2, which had no significant effect by itself on the GDNF responsiveness, promoted the action of RA to upregulate GFRα-1 and enhance the GDNF responsiveness. These results indicate that RA and BMP2 play important roles in the induction of GDNF responsiveness, as well as NT3 responsiveness, of developing SCG neurons.
Keywords: neurotrophic factor, GDNF, GFRα-1, BMP2, retinoic acid, RARα, SCG neurons
Responsiveness to particular neurotrophic factors determines the survival and the functional fate of developing neuronal cells. Therefore, acquisition of neurotrophic factor responsiveness marks an important step during differentiation of neuronal cells. Neurotrophin (Bothwell, 1995) and glial cell line-derived neurotrophic factor (GDNF) (Olson, 1997) families are two major gene families of neurotrophic factors. Although the mechanisms for the acquisition of responsiveness to members of neurotrophin family have been well investigated (Wyatt and Davies, 1993; Robinson et al., 1996; Holst et al., 1997; Kobayashi et al., 1998; Zhang et al., 1998;Wyatt et al., 1999), the mechanism for that of the GDNF family is not well known.
Mature sympathetic neurons of rat superior cervical ganglia (SCG) depend for their survival on the target-derived nerve growth factor (NGF) that interacts with TrkA receptor. However, preceding the onset of TrkA expression, developing SCG neurons express Ret and TrkC that transduce signals from GDNF and neurotrophin-3 (NT3), respectively. Gene disruption studies elsewhere also suggest the roles of both GDNF and NT3 in the development of SCG neurons. For example, disruption of Ret leads to severe degeneration of sympathetic ganglia, as well as loss of the enteric nervous system (Schuchardt et al., 1994; Durbec et al., 1996). GDNF knock-out mice also show shrinkage of SCG by one-third (Moore et al., 1996), a similar extent to that caused by the gene disruption of NT3 (Ernfors et al., 1994). On the other hand, studies using in vitro culture system indicate that GDNF and NT3, when added separately, support survival of the SCG neurons from perinatal rats to a lower extent: 5–20% of the plated neurons. This is much less than that supported by NGF (>80% of the plated neurons) (Trupp et al., 1995; Kobayashi et al., 1998). These studies suggest that environmental factor(s) are necessary for SCG neurons to maintain responsiveness to GDNF and NT3. They suggest also that these environmental factors are missing from or are already deceased in thein vitro culture system of SCG neurons from perinatal animals.
In the search for such environmental factors, we previously found cooperative roles of bone morphogenetic protein-2 (BMP2) and retinoic acid (RA) in the enhancement of NT3 responsiveness in SCG neurons (Kobayashi et al., 1998). Alteration of the Trk receptor expression from TrkA to TrkC by BMP2–RA indicates that cultured SCG neurons from perinatal rats keep the plasticity of responsiveness to neurotrophic factors and thus serve as a suitable model to study the mechanism of the developmental acquisition of the neurotrophic factor responsiveness. In the present study, we further examined the action of BMP2–RA on the regulation of responsiveness to GDNF family members in the cultured SCG neurons. The results indicated that RA is the primary factor acting through the α-type of nuclear retinoic acid receptor (RARα) to induce the GDNF responsiveness by upregulating GFRα-1, the ligand-specifying receptor for GDNF, whereas BMP2, which had almost no effect by itself, promoted the action of RA.
MATERIALS AND METHODS
Materials. Mouse 2.5S NGF, all-trans RA, GDNF, and neurturin were purchased from Upstate Biotechnology (Lake Placid, NY), Sigma (St. Louis, MO), Alomone (Tel Aviv, Israel), and Pepro Tech (London, UK), respectively. Human recombinant BMP2 was kindly provided by Yamanouchi Pharmaceuticals Co. Ltd. (Tokyo, Japan). Synthetic retinoids (Ro 41–6055, RARα agonist; Ro 41–5253, RARα antagonist; Ro 19–0645, RARβ agonist) (for review, see Apfel et al., 1992) were generously provided by Dr. M. Klaus of Hoffmann-La Roche (Basel, Switzerland).
Cell culture. Sympathetic neurons were cultured according to the method described previously (Kobayashi et al., 1998). SCG were dissected from Wistar rats at embryonic day 17 (E17) and sequentially digested with collagenase (3 mg/ml; Sigma) for 30 min and trypsin (2 mg/ml; Difco, Detroit, MI) for 30 min at 37°C. After centrifugal washing, cells were dissociated by trituration. Single-cell suspension was plated onto 35 mm culture dishes or 12-well plates precoated with poly-d-lysine (0.1 mg/ml; Sigma) and laminin (1 μg/cm2; Collaborative Research, Bedford, MA). Within a single experiment, the density of plated cells in all dishes or wells was kept constant (approximately equal to one ganglion per square centimeter). On the next day of seeding, the culture medium was replenished, and cytosine-β-d-arabinofuranoside (10 μm; Sigma) was added for 3 d to prevent proliferation of non-neuronal cells. SCG neurons were cultured in the basal culture medium supplemented with NGF (40 ng/ml), GDNF (40 ng/ml), or neurturin (40 ng/ml). The basal culture medium (serum-free) consisted of Ham's F-12 medium containing BSA (0.1 mg/ml), streptomycin (0.1 mg/ml), penicillin (50 U/ml), and N2 supplement. Schwann cells were prepared from newborn rat sciatic nerves as described previously (Matsuoka et al., 1991).
RNA preparation and reverse transcription-PCR. To assess the effects of RA and BMP2 on the gene expression in cultured SCG neurons, we performed a series of reverse transcription (RT)-PCR experiments (Kobayashi et al., 1998). Total RNA was prepared from cultured SCG neurons according to the method of Chomczynski and Sacchi (1987) with slight modifications as described previously (Kobayashi et al., 1998). Aliquots (0.1 μg) of the total RNA samples were treated with DNaseI (Takara, Tokyo, Japan) at 37°C for 30 min. Single-strand cDNA was synthesized with M-MLV reverse transcriptase (Life Technologies, Rockville, MD), 0.5 mmdNTPs, and 0.5 μm random hexamer at 37°C for 1 hr. PCR was performed in a total volume of 50 μl containing cDNA, 50 mm KCl, 10 mm Tris-HCl, pH 8.3, 1.5 mmMgCl2, 0.2 mm dNTPs, 0.5 μm each 5′ and 3′ primers, 0.1 μCi/μl [α-32P]dCTP, and 0.02 U/μl AmpliTaq Gold DNA polymerase (Perkin-Elmer, Branchburg, NJ). Samples were subjected to 16–39 cycles of PCR according to the following scheme: 94°C for 1 min, 57°C (for β-actin and GFRα-3) or 64°C (for GFRα-1, GFRα-2, and Ret) for 1 min, and 72°C for 1 min. The PCR products were electrophoresed on polyacrylamide gel, and radioactivity incorporated into cDNA fragments was quantified with Bio-Imaging Analyzer (Fuji Photo Film, Tokyo, Japan) (Kobayashi et al., 1998). β-Actin mRNA in the same sample was amplified by RT-PCR as an internal standard. To ensure that the amounts of PCR products were proportional to the amounts of corresponding mRNAs in the cultured neurons, various numbers of amplification cycles were tried, and only the data from cycles within the logarithmic amplification period was processed further. The nucleotide positions of amplified cDNA fragments were as follows: GFRα-1, 697–1178 (GenBank accession number U59486); GFRα-2, 430–832 (GenBank accession number AF005226); RARα, 510–999 (GenBank accession number U15211); RARβ, 455–943 (GenBank accession number AJ002942); GDNF, 71–363 (GenBank accession numberL15305); neurturin, 790–890 (GenBank accession number U78109); and β-actin, 222–440 (GenBank accession number J00691). Numbers denote nucleotide positions in the corresponding cDNAs registered in GenBank, the European Molecular Biology Laboratory, and the DNA Data Bank of Japan databases. As for Ret mRNA, it is reported that at least three different Ret isoforms are expressed in kidneybecause of the alternative splicing at its 5′ regions (Lorenzo et al., 1995; Ivanchuk et al., 1997). However, our RT-PCR experiments using primers designed at exon 2 and exon 6 of Ret gene indicated that only the full-length form of Ret mRNA containing all exons between exon 2 and exon 6 (Ivanchuk et al., 1997) is expressed in E17 rat SCG neurons (data not shown). Therefore, the following primers were designed for quantification of Ret mRNA: upstream primer, 5′-GATGCCCCTGGA-GAAGTGCCC (exon 2); and downstream primer, 5′-GGCCAATG-ACACTCTCCCTCTCTC (exon 3). Nucleotide sequences of primer pairs used for amplification of other cDNAs are as follows: GFRα-3 upstream primer, 5′-CAACTCAG-GAACAGCTCTCT; and downstream primer, 5′-TCIGAGTCTGGT-TTGAGCAT (degenerate primers designed from sequences of human and mouse GFRα-3 cDNAs; GenBank accession numbers AF051767 and AF020305, respectively). Authenticity of the amplified products (GFRα-1, GFRα-2, GFRα-3, RARα, RARβ, and Ret) was confirmed by DNA sequencing after cloning into plasmid vectors (PCR II from Invitrogen Inc./Funakoshi Co. Ltd., Tokyo, Japan; and T-eazy, from Promega, Tokyo, Japan).
Bioassay of neuron survival. SCG neurons from E17 rats were plated onto 12-well plates (Corning-Coaster Japan, Tokyo) and cultured as described above. The neurons were treated with 10−7m RA and/or 10 ng/ml BMP2 in the presence or absence of 40 ng/ml GDNF for 4 d. At appropriate stages of culture, the number of viable neurons (cells with round and phase-bright soma and neurites) within 2 × 3 mm rectangles in each well was counted (Kobayashi et al., 1998). The number of surviving neurons was expressed as percentage of the number of neurons surviving in the presence of 40 ng/ml NGF at 24 hr after plating (≥85% of the plated neurons survived in the presence of NGF at 24 hr).
Immunoprecipitation and Western blotting. SCG neurons were pretreated without or with 10−7m RA in the presence of NGF for 4 d. After 2 hr deprivation of NGF with NGF-free medium, cells were stimulated with 20 ng/ml GDNF for 10 min at room temperature and rinsed with ice-cold TBS (150 mm NaCl and 20 mm Tris-HCl, pH 8.0) containing 0.1 mmNa3VO4. Treated cells were lysed in lysis buffer (1% Triton X-100, 150 mmNaCl, 10 mm NaF, 5 mm EDTA, 2 mmNa3VO4, 1 mm PMSF, 0.2 mmNa2MoO4, 20 nm okadaic acid, 10 mmβ-glycerophosphate, and 20 mm Tris-HCl, pH 7.5) and immunoprecipitated with anti-Ret antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and protein-G Sepharose. Immunoprecipitates were washed four times with lysis buffer and then solubilized by boiling for 5 min in SDS sample buffer. Samples were electrophoresed on 7% SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore, Tokyo, Japan). Tyrosine phosphorylation of Ret was detected by probing with anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology). The amount of Ret protein in each lane was determined by reprobing the same membrane with anti-Ret antibody. For Western blotting of GFRα-1, we used mouse monoclonal anti-GFRα-1 (Transduction Laboratories, Lexington, KY) at 1:5000 dilution. Detection was accomplished by using horseradish peroxidase-conjugated secondary antibody in conjunction with chemiluminescence reagents (SuperSignal West Dura; Pierce, Rockford, IL). If necessary, the levels of chemiluminescence were quantified with Molecular Imager (Bio-Rad, Tokyo, Japan).
RESULTS
RA and BMP2 synergistically induced GDNF responsiveness in developing sympathetic neurons
Neurotrophic factors of the GDNF family include GDNF, neurturin, persephin, and artemin and are known to promote the survival of a variety of neurons, including midbrain dopaminergic, spinal motor, sensory, and sympathetic neurons with different spectra (Lin et al., 1993; Henderson et al., 1994; Arenas et al., 1995; Beck et al., 1995; Trupp et al., 1995; Kotzbauer et al., 1996; Baloh et al., 1998; Milbrandt et al., 1998). To examine the ability of RA and BMP2 to regulate GDNF responsiveness, cells dissociated from SCG of E17 rats were treated with RA and/or BMP2 in the presence or absence of GDNF for 4 d. The number of surviving neurons were then counted and expressed as a percentage of the number of neurons surviving in the presence of 40 ng/ml NGF at 24 hr after plating. As shown in Figure1, A and B, only 10% of the neurons survived in the presence of GDNF alone at 4 d in culture. However, 40% of the SCG neurons survived on GDNF when they were treated with RA for 4 d. As shown in Figure2, the RA treatment did not change the half-maximal effective concentration of GDNF (EC50, ≃15 ng/ml) to support survival of SCG neurons but rather increased the maximum response of these neurons to GDNF (maximum percent survival, 40%). On the other hand, in the absence of RA, neurturin supported the survival of SCG neurons to the same extent as GDNF (10%). Treatment of SCG neurons with RA did not affect either the half-maximal effective concentration of neurturin (EC50, ≃20 ng/ml) or the maximum survival response to neurturin. These results indicate that RA enhanced specifically the GDNF responsiveness of the E17 SCG neurons in culture.
Fig. 1.
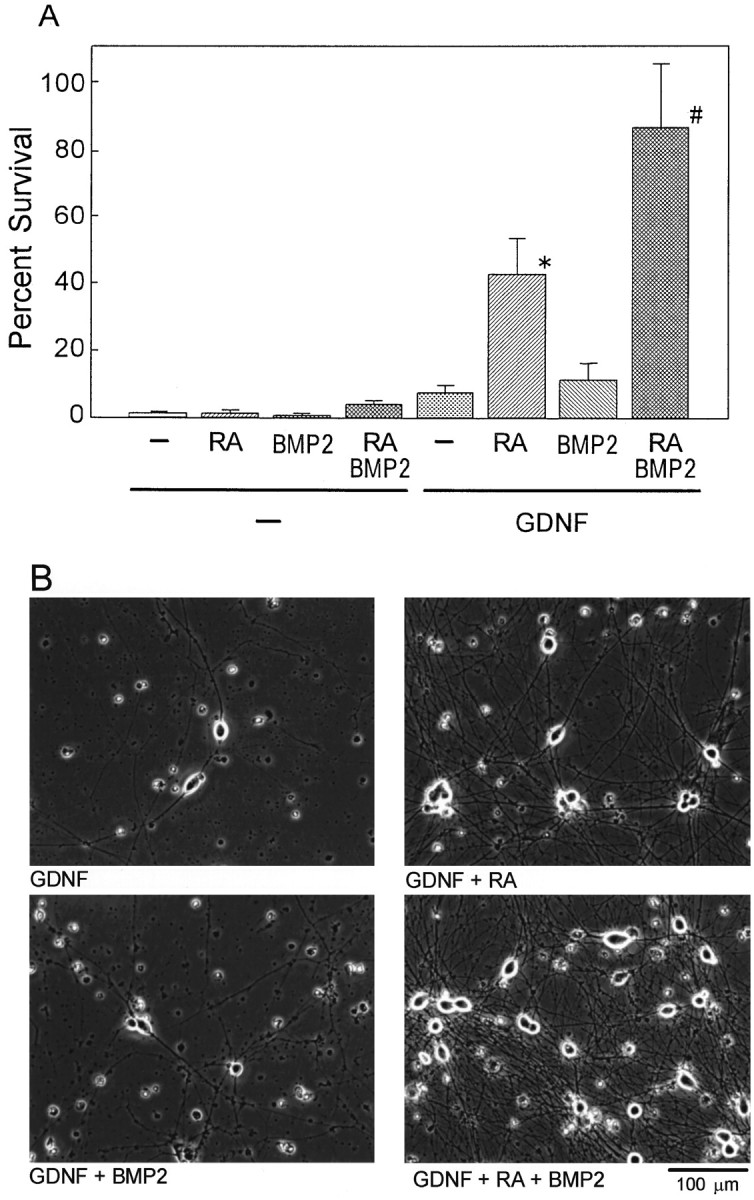
Synergistic action of RA and BMP2 on the induction of GDNF responsiveness of SCG neurons for their survival. Neurons dissociated from E17 rat SCG were treated with 10−7m RA and/or 10 ng/ml BMP2 in the absence or presence of 40 ng/ml GDNF for 4 d before numbers of surviving neurons were counted (A) and photomicrographs were taken (B). The number of viable neurons (cells with round and phase-bright soma and neurites) within 2 × 3 mm rectangles in each well was counted and expressed as percentage of the number of neurons surviving in the presence of 40 ng/ml NGF at 24 hr after plating. Each column represents the mean and SD (n = 4–6). *p < 0.0001 compared between GDNF plus RA and GDNF alone; #p < 0.0001 compared between GDNF plus RA plus BMP2 and GDNF plus RA (ANOVA, with Boneferroni correction for multiple comparison). Note that BMP2, which had no effect by itself, promoted the action of RA to enhance the GDNF-dependent survival. Scale bar (in B), 100 μm.
Fig. 2.

Effects of RA on the induction of responsiveness to GDNF and neurturin (NTN). SCG neurons were cultured in the presence of the indicated concentrations of GDNF or neurturin without or with 10−7m RA for 4 d before neuronal survival was examined. The number of viable neurons was counted and expressed as in Figure1B. Each value represents the mean and SD (n = 3). *p < 0.05, **p < 0.01 compared between RA-treated and untreated neurons (Student's t test).
Although the treatment of the E17 SCG neurons with BMP2 alone did not affect their responsiveness to GDNF, BMP2 further increased the effect of RA (Fig. 1A). Thus, >80% of the E17 SCG neurons survived on GDNF after the cotreatment with RA and BMP2 for 4 d. These results indicate that RA and BMP2 synergistically enhanced the GDNF responsiveness of the SCG neurons. Additionally, SCG neurons cotreated with RA plus BMP2 and surviving on GDNF formed an extensive network of neurites (Fig. 1B), showing little difference in morphological structure compared with those surviving on NGF.
We then examined the effects of RA and BMP2 on the GDNF responsiveness of SCG neurons prepared from postnatal day 2 (P2) animals. Survival response of the P2 SCG neurons in the presence of GDNF alone at 4 d in culture (≤10%) was similar to or less than that of the E17 neurons, whereas only 20–25% or <40% of the P2 SCG neurons survived on GDNF when they were treated for 4 d with RA (10−7m) alone or RA and BMP2 (10 ng/ml), respectively. These results indicate that, although most of the SCG neurons respond to RA and BMP2 by the increase of GDNF-dependent survival at E17, subpopulation of the RA–BMP2 unresponsive neurons exists, and the size of this subpopulation increases with development of the SCG neurons. These results are consistent with the developmental loss of the BMP2–RA sensitivity of SCG neurons for the induction of NT3 responsiveness (Kobayashi et al., 1998). The higher BMP2–RA sensitivity of the SCG neurons at the earlier stage of the neuron development highlights the important roles of the RA and BMPs in the acquisition of the initial neurotrophic factor responsiveness of the SCG neurons.
RA and BMP2 upregulate GFRα-1 mRNA expression
Neurotrophic factors of GDNF family signal through multicomponent receptors that consist of Ret receptor tyrosine kinase and one of the glycosyl-phosphatidylinositol-linked ligand-binding subunits, GFRαs; GFRα-1 is the preferred ligand-binding subunit for GDNF as GFRα-2 is for neurturin (Jing et al., 1996; Trupp et al., 1996; Baloh et al., 1997; Jing et al., 1997; Klein et al., 1997). To assess the potential effect of RA on the expression of GDNF receptors, a series of RT-PCR experiments were performed with [α-32P]dCTP and total RNA prepared from the cultured SCG neurons prepared from E17 rats. PCR primers were designed for receptors of GDNF family, GFRα-1, GFRα-2, GFRα-3, and Ret. PCR primers were designed also for β-actin as an internal standard. PCR products were separated by PAGE and quantitated with an image analyzer. As shown in Figure3A, the treatment with RA alone exerted a remarkable increase in the level of GFRα-1 mRNA in a concentration-dependent manner with a maximal induction at 10−7m. The RA induction of GFRα-1 mRNA levels was promoted further by BMP2, although BMP2 alone had almost no effect on GFRα-1 mRNA levels (Fig.3B). BMP2 at 10 ng/ml was sufficient to promote the effect of RA on the induction of the GFRα-1 mRNA. On the other hand, RA and BMP2 had little effect on the expression of GFRα-2 and GFRα-3 mRNA in E17 SCG neurons (Fig. 3C,D). As shown in Figure 4, RA up to 10−7m increased the level of Ret mRNA slightly up to twofold (Fig.4A). However, BMP2 alone or BMP2 added with RA had no significant effects on the level of Ret mRNA. In addition, the synergistic action of RA and BMP2 on the induction of GFRα-1 was less potent in SCG neurons prepared from P2 animals compared with those prepared from E17 animals (data not shown). These results were consistent with the above results that BMP2 promoted the effects of RA on the induction GDNF responsiveness. Therefore, it is strongly suggested that RA and BMP2 enhanced the GDNF responsiveness of the E17 SCG neurons through upregulation of GFRα-1, a ligand-specifying component, whereas Ret, the signal-transducing component of the GDNF receptor, seems to be expressed at a level sufficient to maintain survival of SCG neurons.
Fig. 3.
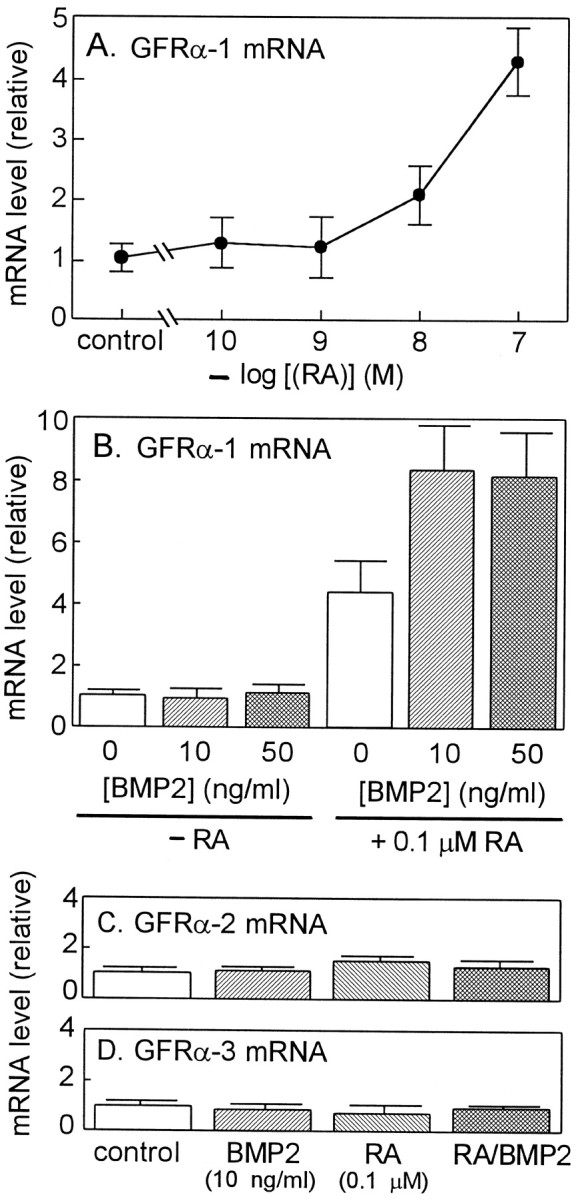
Effects of RA and BMP2 on the levels of GFR mRNAs in cultured SCG neurons. A, B, SCG neurons were treated with various concentrations of RA in the presence of 40 ng/ml NGF for 4 d before GFRα-1 mRNA levels were measured by RT-PCR. B, SCG neurons were treated with the indicated concentrations of BMP2 and 10−7m RA in the presence of 40 ng/ml NGF for 4 d before GFRα-1 mRNA levels were measured. C, D, SCG neurons were treated without or with 10−7m RA and 10 ng/ml BMP2 in the presence of 40 ng/ml NGF for 4 d before GFRα-2 and GFRα-3 mRNA levels were measured. Levels of mRNAs for GFRα-1, GFRα-2, and GFRα-3 were measured by quantitative RT-PCR as described in Materials and Methods. Eachcolumn represents the mean and range of two independent cultures. Note that among three GFRs, RA and RA–BMP2 treatments specifically and remarkably induced the level of GFRα-1 mRNA. Each value represents the mean and range of two independent RNA samples from the same series of culture. Each panel is representative of two independent experiments (independent series of culture) that gave similar results.
Fig. 4.

Effects of RA and BMP2 on the levels of Ret mRNA in cultured SCG neurons. A, SCG neurons were treated with various concentrations of RA in the presence of 40 ng/ml NGF for 4 d before Ret mRNA levels were measured by RT-PCR.B, SCG neurons were treated without or with 10−7m RA and 10 ng/ml BMP2 in the presence of 40 ng/ml NGF for 4 d before Ret mRNA levels were measured. Each value represents the mean and range of two independent RNA samples. Each panel is representative of two independent experiments that gave similar results.
Previously, it was reported that RA induces the expression of TrkB receptors, which mediates biological action of BDNF (Kaplan et al., 1993; Kobayashi et al., 1994) and suppress the expression of TrkA, which mediates biological action of NGF (Kobayashi et al., 1994; Wyatt et al., 1999). BMP2, however, abolished the effects of RA on the induction of TrkB (M. Kobayashi, unpublished observation) and promoted the effects of RA on the suppression of TrkA (Kobayashi et al., 1998) in cultured SCG neurons, suggesting that the combination of RA and BMP2 specifies the responsiveness of SCG neurons to neurotrophic factors (NT3 and GDNF) that play important roles in the development of these neurons at the embryonic period.
RA increases Ret tyrosine-phosphorylation through upregulation of GFRα-1 protein
To examine the consequence of the RA induction of GFRα-1 mRNA on protein level, we performed Western blotting experiments by using anti-GFRα-1 antibody. As shown in Figure5A, GFRα-1 protein was induced remarkably within 1 d of the treatment with RA. The induced level of the GFRα-1 protein was almost the same between 1 and 4 d of the treatment. Therefore, the survival of the SCG neurons in the presence of RA but in the absence of NGF from the beginning of culture seems to correspond to the quick induction of the GFRα-1 protein by RA.
Fig. 5.
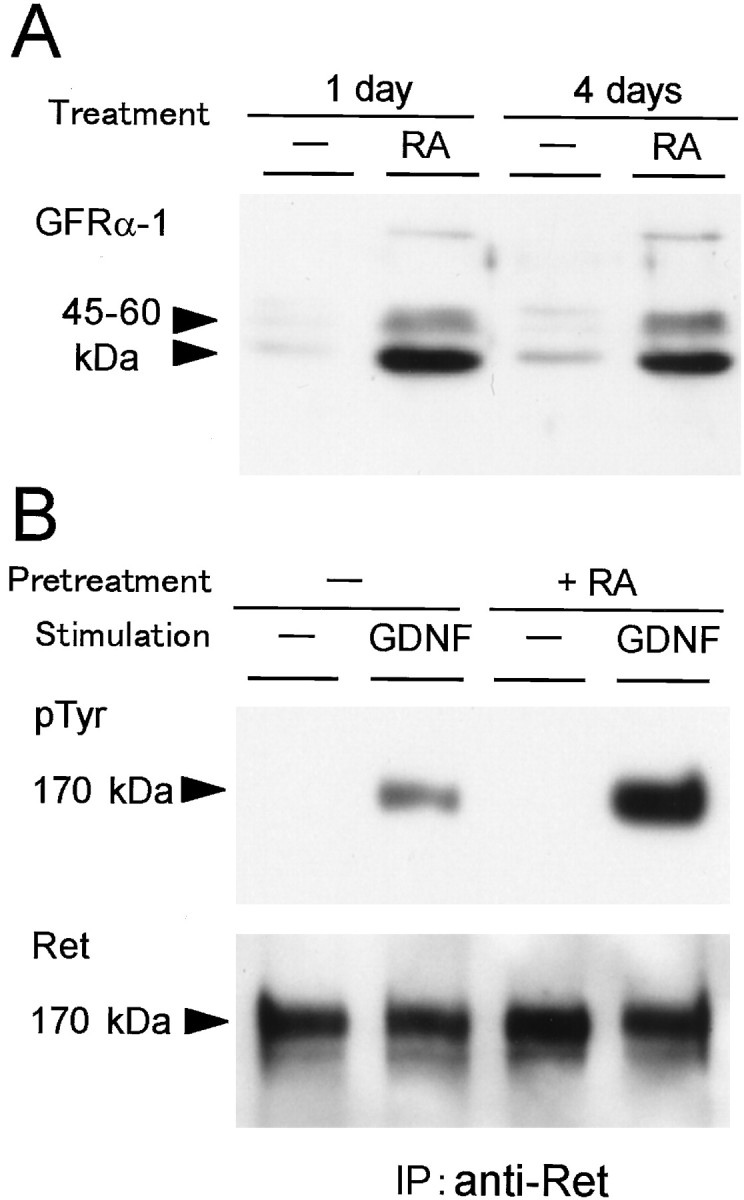
Effects of the RA treatment on the levels of GFRα-1protein and GDNF-induced Ret tyrosine phosphorylation in cultured SCG neurons. A, SCG neurons were treated without or with 10−7m RA in the presence of 40 ng/ml NGF for 1 or 4 d before cell lysate was prepared. The cell lysate (7 μg of protein) was subjected to Western blotting with anti-GFRα-1 antibody. B, SCG neurons were pretreated without or with 10−7mRA in the presence of 40 ng/ml NGF for 4 d. Then, SCG neurons were stimulated with 50 ng/ml GDNF for 5 min before cell lysate was prepared. The cell lysate was immunoprecipitated with anti-Ret antibody and subjected to Western blotting with anti-phosphotyrosine antibody (top panel). Blotted membrane was reprobed with anti-Ret antibody to evaluate the amounts of recovered Ret protein (bottom panel).
It is expected that the induced level of GFRα-1 expression would facilitate the GDNF signaling through Ret. Therefore, we measured the level of the GDNF-triggered Ret tyrosine phosphorylation in the RA-treated SCG neurons. SCG neurons were pretreated with 10−7m RA for 4 d before stimulation with GDNF. Then, cell lysate was prepared and immunoprecipitated with anti-Ret antibody for Western blotting with anti-phosphotyrosine antibody. As shown in Figure 5B, the GDNF-triggered Ret tyrosine phosphorylation was increased 3.0-fold by pretreatment with RA (Fig. 5B, top panel). In this experiment, the amount of recovered Ret protein was increased slightly (1.3-fold) (Fig. 5B, bottom panel) in accordance with the slight increase of Ret mRNA level by the RA pretreatment (Fig. 4). Consequently, the extent of tyrosine phosphorylation per Ret molecule was increased by 2.4-fold. These results clearly indicate that the efficacy of Ret tyrosine phosphorylation was increased by the RA treatment. Therefore, it is strongly suggested that the increased levels of GFRα-1 in the RA-treated SCG neurons contribute to the facilitation of GDNF signaling through Ret in addition to the slightly increased expression of Ret itself.
RA induced the GFRα-1 mRNA levels through RARα
The action of RA on regulation of various genes is thought to be mediated by nuclear retinoid receptors that act as transcription factors for the target genes (Mangelsdorf et al., 1995; Chambon, 1996). Two families of nuclear retinoid receptors have been identified so far: RARs and the retinoid X receptors (RXRs). All-trans RA (RA) activates mainly RARs, whereas RXRs preferentially bind 9-cis RA and serve as a common coreceptor of the nuclear receptor dimer. The RAR family consists of three subtypes of receptors: α, β, and γ. To identify the subtype of retinoid receptor involved in the RA-induced GDNF responsiveness and upregulation of GFRα-1 mRNA, we examined the effects of several synthetic retinoids on cultured SCG neurons.
It has already been shown that Ro 40–6055 preferentially binds to and activates RARα and thus behaves as a RARα agonist, whereas Ro 41–5253 specifically binds to RARα and prevent its activation, behaving as an RARα antagonist (Apfel et al., 1992). In contrast, Ro 19–0645 preferentially binds to and activates RARβ and so behaves as a RARβ agonist (Apfel et al., 1992).
E17 SCG neurons were treated with 3 × 10−8m RA, 3 × 10−8m Ro 40–6055, 3 × 10−8m Ro 19–0645, or 3 × 10−7m Ro 41–5253 in the absence or presence of 40 ng/ml GDNF for 4 d before the number of surviving neurons was counted. As shown in Figure6, either Ro 40–6055 (RARα agonist) or RA alone showed little neurotrophic effect on the survival of SCG neurons. However, Ro 40–6055 enhanced the GDNF responsiveness of SCG neurons to a similar extent as RA. The enhancement of GDNF responsiveness by either RA or Ro 40–6055 was counteracted by the 10-fold excess concentration of Ro 41–5253 (RARα antagonist). In contrast, Ro 19–0645 (RARβ agonist) did not alter the GDNF responsiveness of SCG neurons. These results suggest that RA enhanced the GDNF responsiveness of SCG neurons through activation of RARα.
Fig. 6.

Effect of synthetic retinoids on the induction of GDNF responsiveness of the SCG neurons. SCG neurons were treated with 3 × 10−8m RA, Ro 40–6055 (RARα agonist), Ro 19–0645 (RARβ agonist), or 3 × 10−7m Ro 41–5253 (α antagonist) in the absence or presence of 40 ng/ml GDNF for 4 d before surviving neurons were counted. Each value represents the mean and SD (n = 4–8). *p < 0.0001 compared with GDNF alone (ANOVA, with Boneferroni correction for multiple comparison).
Next, we examined the effects of the synthetic retinoids on GFRα-1 mRNA levels. E17 SCG neurons were treated with 10−7m RA, 10−7m Ro 40–6055, or 10−7m Ro 19–0645 in the presence of 40 ng/ml NGF before total RNA was extracted. As expected, Ro 40–6055 and RA increased the GFRα-1 mRNA levels by threefold to fourfold, but Ro 19–0645 did not affect the GFRα-1 mRNA levels (Fig.7). These results further strengthen our speculation that RARα mediates the action of RA on the induction of GDNF responsiveness in developing SCG neurons.
Fig. 7.

Effect of synthetic retinoids on the level of GFRα-1 mRNA in the cultured SCG neurons. SCG neurons were treated with 10−7m RA, Ro 40–6055 (RARα agonist), or Ro 19–0645 (RARβ agonist) in the presence of 40 ng/ml NGF for 4 d. Level of GFRα-1 mRNA was measured by RT-PCR. Eachcolumn represents the mean and range of two independent RNA samples. Note that RA and RARα agonist but not RARβ agonist increased the GFRα-1 mRNA level. This is representative of two independent experiments that gave similar results.
Regulation of RAR expressions by RA and BMP2
It is shown in Figure 1 that BMP2 promoted the effect of RA on the enhancement of the GDNF responsiveness; therefore, it is now reasonable for us to speculate that BMP2 strengthens the effect of RA by upregulating the expression of RARs. To identify the molecular species of RARs expressed in the cultured E17 SCG neurons, we designed primer pairs specific to RARα and RARβ and performed a series of RT-PCR experiments. Subsequent sequence analysis revealed that both RARα and RARβ are expressed in the E17 SCG neurons. Then, we treated the E17 SCG neurons with BMP2 and/or RA. As shown in Figure8, BMP2 increased the RARα mRNA levels by 2.7-fold but reduced the RARβ mRNA level significantly. This result is comparable with the effects of BMP2 on the promotion of the RA action in the E17 SCG neurons. Moreover, RA also increased the RARα and RARβ mRNA levels by fourfold and twofold, respectively, in accordance with the concept that the RARs belong to a superfamily of ligand-inducible transcriptional regulators (Martin et al., 1990; Wu et al., 1992). More interestingly, the combination of BMP2 and RA synergistically increased the RARα mRNA levels by almost eightfold, whereas BMP2 and RA counteracted on the RARβ mRNA level. These results suggest that the molecular mechanism of the promoting effect of BMP2 on the action of RA on SCG neurons includes BMP2/RA-induced upregulation of RARα.
Fig. 8.
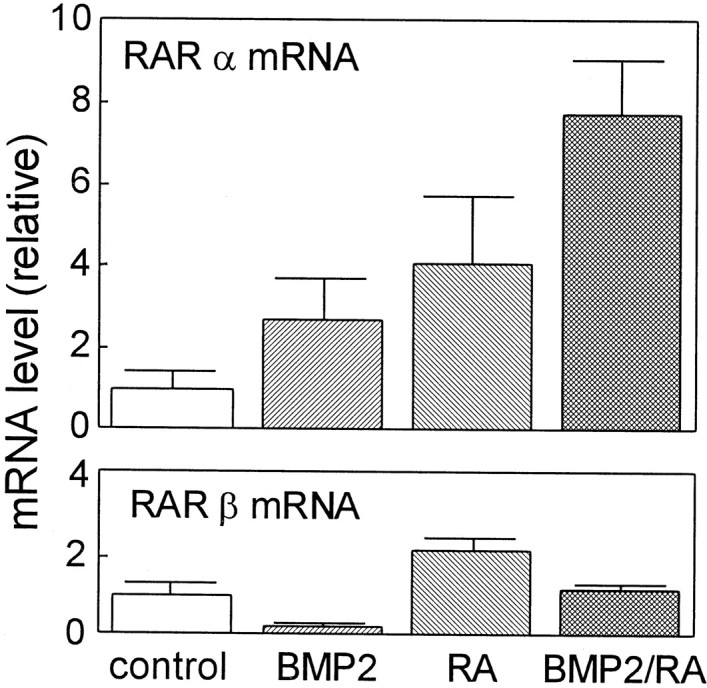
Effects of RA and BMP2 on the levels of RARα and RARβ mRNAs in cultured SCG neurons. SCG neurons were treated with 10−7m RA and/or 10 ng/ml BMP2 in the presence of 40 ng/ml NGF for 4 d. Levels of RARα and RARβ mRNAs were measured by RT-PCR. Each column represents the mean and range of two independent RNA samples. Eachpanel is a representative of two independent experiments that gave similar results.
Regulation of GDNF expression in Schwann cells by RA and BMP2
In PNS, GDNF is expressed mainly by non-neuronal cells, such as Schwann cells (Henderson et al., 1994; Trupp et al., 1995). Therefore, we examined the regulation of the GDNF mRNA expression in Schwann cells as a possible source of GDNF for sympathetic neurons. Schwann cells prepared from newborn rat sciatic nerves were cultured as described byMatsuoka et al. (1991), and levels of GDNF and neurturin mRNA were determined by RT-PCR (Fig. 9). Treatment of Schwann cells with RA (10−7m) for 4 d increased the GDNF mRNA level by 3.5-fold. Interestingly, the combined treatment with RA and BMP2 (10 ng/ml) increased the GDNF mRNA levels by 5.5-fold, whereas treatment with BMP2 alone had almost no effect. BMP2 and/or RA did not affect levels of neurturin mRNA in Schwann cells. These results indicate that RA and BMP2 possess the ability to induce GDNF expression in Schwann cells.
Fig. 9.

Effects of RA and BMP2 on the levels of GDNF and neurturin mRNA levels in cultured Schwann cells. Schwann cells were treated with 10−7m RA and/or 10 ng/ml BMP2 for 4 d. Levels of GDNF and neurturin mRNAs were measured by RT-PCR. Each column represents the mean and range of two independent RNA samples. Each panel is representative of two independent experiments that gave similar results.
DISCUSSION
The acquisition of neurotrophic factor responsiveness of neuronal cells is thought to be controlled by environmental cues. We have shown previously that BMP2 and RA have synergistic effects on the induction of NT3 responsiveness in the developing SCG neurons (Kobayashi et al., 1998). In this study, we showed that RA and BMP2 also have synergistic effects on the induction of GDNF responsiveness in these neurons. Our studies describe a mechanism by which responsiveness of developing neurons to neurotrophic factors is induced by a combination of RA and BMP2 stimulating the expression of neurotrophic factor receptors. We speculate that the loss of the GDNF and NT3 responsiveness of SCG neurons isolated from E17 rats is a reflection of the removal of the environmental signals that maintain the receptors for GDNF and NT3 attributable to the transfer to in vitro culture system. Our results clearly indicate that RA and BMP2 can substitute for such environmental signals, although these signals in vivo may decline toward adulthood.
RA and BMPs are known to possess profound effects in a variety of biological processes, including differentiation of neural crest-derived cells (Kaplan et al., 1993; Henion and Weston, 1994; Kobayashi et al., 1994, 1998; Holst et al., 1997; Mehler et al., 1997; Zhang et al., 1998; Wyatt et al., 1999). For example, RA supports survival and proliferation of neural crest cells (Henion and Weston, 1994). Together with previous studies (Kobayashi et al., 1994, 1998; Holst et al., 1997; Zhang et al., 1998; Wyatt et al., 1999), the present results indicate that RA acts not only during an early stage of neural crest development but also at a later stage of neuronal differentiation by regulating the expression of receptors for neurotrophic factors and hence responsiveness to neurotrophic factors (NT3 and GDNF) that play important roles in the development of these neurons at the embryonic period. For the induction of GDNF responsiveness in cultured SCG neurons from E17 rats, RA markedly induced the expression of ligand-specifying receptor component GFRα-1 and increased the efficacy of GDNF-triggered Ret tyrosine phosphorylation. Effect of RA on the upregulation of Ret expression in cultured SCG neurons was rather small, although the significant ability of RA to upregulate the expression of Ret in tumorigenic neuroblastoma cells has been reported previously (Tahira et al., 1991; Bunone et al., 1995; Patrone et al., 1997). The expression of Ret starts from the early stage of neural crest development (Lo and Anderson, 1995; Tsuzuki et al., 1995; Durbec et al., 1996) and continues even after transfer to in vitroculture system, as shown in this study. Therefore, the regulatory mechanism of Ret expression in nontumorigenic neural crest cells and SCG neurons should be carefully examined further. Our results rather imply that GFRαs, as well as ligand-specifying neurotrophin receptors, Trks, serve as the primary targets of environmental cues that are regulating specific neurotrophic factor responsiveness of the developing SCG neurons.
Our results on the actions of synthetic retinoids suggest that RARα mediates the action of RA on the induction of GDNF responsiveness. It is also plausible that RARα mediates other actions of RA on the rat SCG neurons, such as the suppression of NGF responsiveness and the promotion of the BMP2-induced NT3 responsiveness (Kobayashi et al., 1998). This notion is consistent with recent observations that the differential regulation of Trk mRNAs in developing mouse and chick sympathetic neurons is also achieved by activation of RARα (Holst et al., 1997; Wyatt et al., 1999). In vivo roles for RA in the development of SCG neurons may be further verified by specific tissue expression of retinalaldehyde dehydorogenase-2, which mediates endogenous RA synthesis (Niederreither et al., 1997; Corcoran and Maden, 1999) and RARα during development.
Recently, it was revealed that BMPs play important roles in the neuronal differentiation of rodent and chick neural crest-derived cells. In rodent neural crest cell culture, BMP4 and BMP7 increase the number of Ret- and Mash1-positive neurons (Shah et al., 1996), whereas in chick neural crest cell culture, BMPs induce adrenergic properties, such as tyrosine hydroxylase expression (Reissmann et al., 1996; Varley and Maxwell, 1996). Recently, it was shown that BMP2 possesses the ability to induce the expression of TrkC in cultured SCG neurons (Kobayashi et al., 1998; Zhang et al., 1998). It is thought that BMPs are supplied from dorsal aorta besides which migrating neural crest cells starts to form sympathetic ganglion (Reissmann et al., 1996; Shah et al., 1996), although embryonic SCG also express BMPs (Kobayashi et al., 1998). Recent studies indicate that BMPs are involved in the development of neural cell types also in the CNS (Gross et al., 1996;Li et al., 1998; Mabie et al., 1999). It is intriguing to analyze the effects of BMP and RA on the neurotrophic factor responsiveness of the CNS neurons.
There are two possible mechanisms for the synergistic action of RA and BMP2 on the induction of GDNF responsiveness of SCG neurons. One possibility is that the expression of receptors for RA and BMP2 are upregulated by BMP2 and RA, respectively. It is already known that RA stimulates the expression of retinoid receptors, including RARα. In the present study, we found that BMP2 and RA cooperatively increased the expression of RARα, strengthening the first possibility. In addition, we have suggested previously the induction of type II receptors for BMP2 by RA (Kobayashi et al., 1998). As a second possibility, the synergistic action of RA and BMP2 may include the convergence of intracellular signaling pathways. Recently, it was reported that Smad3, a downstream protein in the TGFβ signaling pathway, acts as a specific coactivator for the ligand-induced transactivation of nuclear vitamin D receptor, (Yanagisawa et al., 1999). In the crosstalk of distinct signaling pathways, which includes nuclear receptors, roles of transcription coactivator proteins such as SRC-1 and p300/CBP are reported (Kamei et al., 1996; Yanagisawa et al., 1999). Our preliminary experiments also suggest that RA and BMP2 have the ability to induce such nuclear receptor coactivators in SCG neurons. Therefore, it is interesting to speculate that the BMP-specific signaling molecule such as Smad1 acts as a coactivator of the RA–RARα transcription complex.
Higher sensitivities of embryonic SCG neurons to BMP2–RA compared with postnatal neurons for the induction of GDNF responsiveness (present study) and NT3 responsiveness (Kobayashi et al., 1998) also highlight the role of RA–BMPs in the development of sympathetic neurons. This view is shared by Wyatt et al. (1999) who reported on the developmental loss of RA sensitivity for the regulation of TrkA/C expression in mouse sympathetic neurons. Recently, it was reported that postnatal development of SCG was completely impaired in mice lacking GFRα-3, whereas embryonic development of SCG was much less affected (Nishino et al., 1999). These results suggest that neurotrophic factors–receptors other than artemin–GFRα-3 are required for the embryonic development of SCG. Although GFRα-3 and GFRα-2 are expressed in both embryonic and mature postnatal SCG neurons, the expression of GFRα-1 is restricted to the embryonic period (Nishino et al., 1999). It is suggested that GDNF and its specific receptor GFRα-1, which is under the control of RA and BMPs, are rather used (or required) for the embryonic development of SCG neurons, consistent with the analysis on GDNF(−/−) mice (Moore et al., 1996). No defects of SCG in GFRα-1(−/−) mice (Cacalano et al., 1998; Enomoto et al., 1998) might have resulted from functional compensation by other ligands and receptors of GDNF–neurotrophin families.
To verify further the role of RA–BMPs in the acquisition of GDNF responsiveness during development, it is helpful to clarify the source of GDNF for developing SCG neurons and characteristics of its regulation. Previously, we showed that the BMP2–RA treatment induces expression of NT3 and its receptor TrkC to form an autocrine loop of NT3 action in SCG neurons (Kobayashi et al., 1998). However, it is unlikely that the BMP2–RA treatment also induced the autocrine loop of GDNF action, because the BMP2–RA-treated SCG neurons surviving without added neurotrophic factors were completely abolished by anti-NT3 antibody (Kobayashi et al., 1998). On the other hand, our present results suggest that RA and BMPs act also on Schwann cells or Schwann cell-like cells (satellite cells) within or in the periphery of sympathetic ganglia to increase the GDNF expression. Thus, it is highly probable that RA and BMPs promote the paracrine loop of the GDNF action by acting on both neuronal and glial components of the sympathetic system during development.
Acquisition of responsiveness to both NT3 and GDNF at the initial stage of development of sympathetic neurons before they become responsive to the target-derived NGF should have profound implications. Under circumstances in which the local supply of either GDNF or NT3 (or any relevant neurotrophic factor) is limited, convergence of the GDNF and NT3 signal transduction pathways should help the survival of the sympathetic neurons until they extend axons to connect peripheral target tissue that supply NGF. Alternatively, convergence of the GDNF and NT3 signal transduction pathways may give rise to unique cellular functions that are mobilized by neither GDNF nor NT3 alone, but are required temporarily for the sympathetic neuron development. Further studies are underway to prove these possibilities.
Footnotes
This work was supported by the Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture, Japan and grants from the Akiyama Foundation, the Cell Science Foundation, and the Terumo Foundation. M.K. was a recipient of fellowship of the Japan Society for the Promotion of Science for Japanese Junior Scientists. We are grateful to Yamanouchi Pharmaceuticals Co. Ltd. (Tokyo, Japan) for the supply of BMP2. We are indebted to Dr. M. Klaus of Hoffmann-La Roche (Basel, Switzerland) for the generous gift of synthetic retinoids. We thank Prof. K. Kurihara (Aomori University, Aomori, Japan) for continuous support and encouragement and S. Bayley (Asahikawa Medical College, Asahikawa, Japan) for correcting our use of language.
Drs. Thang and Kobayashi contributed equally to this work.
Correspondence should be addressed to Dr. Ichiro Matsuoka, Graduate School of Pharmaceutical Sciences, Hokkaido University, Kita-12-Nishi-6, Kita-Ku, Sapporo 060–0812, Japan. E-mail:matsuoka@pharm.hokudai.ac.jp.
REFERENCES
- 1.Apfel C, Bauer F, Crettaz M, Forni L, Kamber M, Kaufmann F, LeMotte P, Pirson W, Klaus M. A retinoic acid receptor alpha antagonist selectively counteracts retinoic acid effects. Proc Natl Acad Sci USA. 1992;89:7129–7133. doi: 10.1073/pnas.89.15.7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arenas E, Trupp M, Akerud P, Ibanez CF. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron. 1995;15:1465–1473. doi: 10.1016/0896-6273(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 3.Baloh RH, Tansey MG, Golden JP, Creedon DJ, Heuckeroth RO, Keck CL, Zimonjic DB, Popescu NC, Johnson EMJ, Milbrandt J. TrnR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron. 1997;18:793–802. doi: 10.1016/s0896-6273(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 4.Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EMJ, Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/s0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- 5.Beck KD, Valverde J, Alexi T, Poulsen K, Moffat B, Vandlen RA, Rosenthal A, Hefti F. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature. 1995;373:339–341. doi: 10.1038/373339a0. [DOI] [PubMed] [Google Scholar]
- 6.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 7.Bunone G, Borrello MG, Picetti R, Bongarzone I, Peverali FA, de Franciscis V, Della Valle G, Pierotti MA. Induction of RET proto-oncogene expression in neuroblastoma cells precedes neuronal differentiation and is not mediated by protein synthesis. Exp Cell Res. 1995;217:92–99. doi: 10.1006/excr.1995.1067. [DOI] [PubMed] [Google Scholar]
- 8.Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran J, Maden M. Nerve growth factor acts via retinoic acid synthesis to stimulate neurite outgrowth. Nat Neurosci. 1999;2:307–308. doi: 10.1038/7214. [DOI] [PubMed] [Google Scholar]
- 12.Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- 13.Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EMJ, Milbrandt J. GFRα1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- 14.Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 15.Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 1996;17:595–606. doi: 10.1016/s0896-6273(00)80193-2. [DOI] [PubMed] [Google Scholar]
- 16.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 17.Henion PD, Weston JA. Retinoic acid selectively promotes the survival and proliferation of neurogenic precursors in cultured neural crest cell populations. Dev Biol. 1994;161:243–250. doi: 10.1006/dbio.1994.1024. [DOI] [PubMed] [Google Scholar]
- 18.Holst A, Lefcort F, Rohrer H. TrkA expression levels of sympathetic neurons correlate with NGF- dependent survival during development and after treatment with retinoic acid. Eur J Neurosci. 1997;9:2169–2177. doi: 10.1111/j.1460-9568.1997.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 19.Ivanchuk SM, Eng C, Cavenee WK, Mulligan LM. The expression of RET and its multiple splice forms in developing human kidney. Oncogene. 1997;14:1811–1818. doi: 10.1038/sj.onc.1201016. [DOI] [PubMed] [Google Scholar]
- 20.Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis JC, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 21.Jing S, Yu Y, Fang M, Hu Z, Holst PL, Boone T, Delaney J, Schultz H, Zhou R, Fox GM. GFRalpha-2 and GFRalpha-3 are two new receptors for ligands of the GDNF family. J Biol Chem. 1997;272:33111–33117. doi: 10.1074/jbc.272.52.33111. [DOI] [PubMed] [Google Scholar]
- 22.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- 24. Klein RD, Sherman D, Ho WH, Stone D, Bennett GL, Moffat B, Vandlen R, Simmons L, Gu Q, Hongo JA, Devaux B, Poulsen K, Armanini M, Nozaki C, Asai N, Goddard A, Phillips H, Henderson CE, Takahashi M, Rosenthal A. A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature 387 1997. 717 721[Erratum (1997) 392:210]. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi M, Fujii M, Kurihara K, Matsuoka I. Bone morphogenetic protein-2 and retinoic acid induce neurotrophin-3 responsiveness in developing rat sympathetic neurons. Brain Res Mol Brain Res. 1998;53:206–217. doi: 10.1016/s0169-328x(97)00291-x. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi M, Kurihara K, Matsuoka I. Retinoic acid induces BDNF responsiveness of sympathetic neurons by alteration of Trk neurotrophin receptor expression. FEBS Lett. 1994;356:60–65. doi: 10.1016/0014-5793(94)01238-5. [DOI] [PubMed] [Google Scholar]
- 27.Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EMJ, Milbrandt J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Cogswell CA, LoTurco JJ. Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci. 1998;18:8853–8862. doi: 10.1523/JNEUROSCI.18-21-08853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 30.Lo L, Anderson DJ. Postmigratory neural crest cells expressing c-RET display restricted developmental and proliferative capacities. Neuron. 1995;15:527–539. doi: 10.1016/0896-6273(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzo MJ, Eng C, Mulligan LM, Stonehouse TJ, Healey CS, Ponder BA, Smith DP. Multiple mRNA isoforms of the human RET proto-oncogene generated by alternate splicing. Oncogene. 1995;10:1377–1383. [PubMed] [Google Scholar]
- 32.Mabie PC, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schtz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin CA, Ziegler LM, Napoli JL. Retinoic acid, dibutyryl-cAMP, and differentiation affect the expression of retinoic acid receptors in F9 cells. Proc Natl Acad Sci USA. 1990;87:4804–4808. doi: 10.1073/pnas.87.12.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuoka I, Meyer M, Thoenen H. Cell-type-specific regulation of nerve growth factor (NGF) synthesis in non-neuronal cells: comparison of Schwann cells with other cell types. J Neurosci. 1991;11:3165–3177. doi: 10.1523/JNEUROSCI.11-10-03165.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehler MF, Mabie PC, Zhang D, Kessler JA. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997;20:309–317. doi: 10.1016/s0166-2236(96)01046-6. [DOI] [PubMed] [Google Scholar]
- 37.Milbrandt J, de Sauvage FJ, Fahrner TJ, Baloh RH, Leitner ML, Tansey MG, Lampe PA, Heuckeroth RO, Kotzbauer PT, Simburger KS, Golden JP, Davies JA, Vejsada R, Kato AC, Hynes M, Sherman D, Nishimura M, Wang LC, Vandlen R, Moffat B, Klein RD, Poulsen K, Gray C, Garces A, Johnson EMJ. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron. 1998;20:245–253. doi: 10.1016/s0896-6273(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 38.Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 39.Niederreither K, McCaffery P, Drager UC, Chambon P, Dolle P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech Dev. 1997;62:67–78. doi: 10.1016/s0925-4773(96)00653-3. [DOI] [PubMed] [Google Scholar]
- 40.Nishino J, Mochida K, Ohfuji Y, Shimazaki T, Meno C, Ohishi S, Matsuda Y, Fujii H, Saijoh Y, Hamada H. GFR alpha3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron. 1999;23:725–736. doi: 10.1016/s0896-6273(01)80031-3. [DOI] [PubMed] [Google Scholar]
- 41.Olson L. The coming of age of the GDNF family and its receptors: gene delivery in a rat Parkinson model may have clinical implications. Trends Neurosci. 1997;20:277–279. doi: 10.1016/s0166-2236(97)01098-9. [DOI] [PubMed] [Google Scholar]
- 42.Patrone G, Puliti A, Bocciardi R, Ravazzolo R, Romeo G. Sequence and characterisation of the RET proto-oncogene 5′ flanking region: analysis of retinoic acid responsiveness at the transcriptional level. FEBS Lett. 1997;419:76–82. doi: 10.1016/s0014-5793(97)01435-x. [DOI] [PubMed] [Google Scholar]
- 43.Reissmann E, Ernsberger U, Francis-West PH, Rueger D, Brickell PM, Rohrer H. Involvement of bone morphogenetic protein-4 and bone morphogenetic protein-7 in the differentiation of the adrenergic phenotype in developing sympathetic neurons. Development. 1996;122:2079–2088. doi: 10.1242/dev.122.7.2079. [DOI] [PubMed] [Google Scholar]
- 44.Robinson M, Adu J, Davies AM. Timing and regulation of trkB and BDNF mRNA expression in placode- derived sensory neurons and their targets. Eur J Neurosci. 1996;8:2399–2406. doi: 10.1111/j.1460-9568.1996.tb01203.x. [DOI] [PubMed] [Google Scholar]
- 45.Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 46.Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell. 1996;85:331–343. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- 47.Tahira T, Ishizaka Y, Itoh F, Nakayasu M, Sugimura T, Nagao M. Expression of the ret proto-oncogene in human neuroblastoma cell lines and its increase during neuronal differentiation induced by retinoic acid. Oncogene. 1991;6:2333–2338. [PubMed] [Google Scholar]
- 48.Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumae U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–788. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 49.Trupp M, Ryden M, Jornvall H, Funakoshi H, Timmusk T, Arenas E, Ibanez CF. Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol. 1995;130:137–148. doi: 10.1083/jcb.130.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuzuki T, Takahashi M, Asai N, Iwashita T, Matsuyama M, Asai J. Spatial and temporal expression of the ret proto-oncogene product in embryonic, infant and adult rat tissues. Oncogene. 1995;10:191–198. [PubMed] [Google Scholar]
- 51.Varley JE, Maxwell GD. BMP-2 and BMP-4, but not BMP-6, increase the number of adrenergic cells which develop in quail trunk neural crest cultures. Exp Neurol. 1996;140:84–94. doi: 10.1006/exnr.1996.0118. [DOI] [PubMed] [Google Scholar]
- 52.Wu TC, Wang L, Wan YJ. Retinoic acid regulates gene expression of retinoic acid receptors α, β and γ in F9 mouse teratocarcinoma cells. Differentiation. 1992;51:219–224. doi: 10.1111/j.1432-0436.1992.tb00699.x. [DOI] [PubMed] [Google Scholar]
- 53.Wyatt S, Davies AM. Regulation of expression of mRNAs encoding the nerve growth factor receptors p75 and trkA in developing sensory neurons. Development. 1993;119:635–648. doi: 10.1242/dev.119.3.635. [DOI] [PubMed] [Google Scholar]
- 54.Wyatt S, Andres R, Rohrer H, Davies AM. Regulation of neurotrophin receptor expression by retinoic acid in mouse sympathetic neuroblasts. J Neurosci. 1999;19:1062–1071. doi: 10.1523/JNEUROSCI.19-03-01062.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa M, Watanabe M, Kashiwagi K, Toriyabe T, Kawabata M, Miyazono K, Kato S. Convergence of transforming growth factor-β and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283:1317–1321. doi: 10.1126/science.283.5406.1317. [DOI] [PubMed] [Google Scholar]
- 56.Zhang D, Mehler MF, Song Q, Kessler JA. Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. J Neurosci. 1998;18:3314–3326. doi: 10.1523/JNEUROSCI.18-09-03314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]