

Abstract
Glycogen synthase kinase-3β (GSK3β) activity is negatively regulated by several signal transduction cascades that protect neurons against apoptosis, including the phosphatidylinositol-3 kinase (PI-3 kinase) pathway. This suggests the interesting possibility that activation of GSK3β may contribute to neuronal apoptosis. Consequently, we evaluated the role of GSK3β in apoptosis in cultured cortical neurons induced by trophic factor withdrawal or by PI-3 kinase inhibition. Neurons were subjected to several apoptotic paradigms, including serum deprivation, serum deprivation combined with exposure to NMDA receptor antagonists, or treatment with PI-3 kinase inhibitors. These treatments all led to stimulation of GSK3β activity in cortical neurons, which preceded the induction of apoptosis. Expression of an inhibitory GSK3β binding protein or a dominant interfering form of GSK3β reduced neuronal apoptosis, suggesting that GSK3β contributes to trophic factor withdrawal-induced apoptosis. Furthermore, overexpression of GSK3β in neurons increased apoptosis, indicating that activation of this enzyme is sufficient to trigger programmed cell death. Although destabilization of β-catenin is an important physiological effect of GSK3β activation, expression of a mutant β-catenin that is not destabilized by GSK3β did not protect against apoptosis. We conclude that inhibition of GSK3β is one of the mechanisms by which PI-3 kinase activation protects neurons from programmed cell death.
Keywords: cortical neurons, glycogen synthase kinase-3β, phosphatidylinositol-3 kinase, Akt, β-catenin, NMDA, glutamate, brain-derived neurotrophic factor, apoptosis, signal transduction
Differentiated cells, including neurons in the CNS, require the presence of survival factors to suppress the intrinsic cell death machinery and thereby avoid apoptosis (Raff et al., 1993; Park et al., 1997). The regulation of apoptosis by survival factors is therefore critical for normal development and proper functioning of multicellular organisms. In addition, abnormal apoptosis in CNS neurons may play a significant role in neurodegenerative diseases (Raff et al., 1993; Stefanis et al., 1997;Estus, 1998).
A number of survival factors for neurons have been identified, including serum, insulin-like growth factor-1, neurotrophins, and NMDA (Levi-Montalcini and Booker, 1960; D'Mello et al., 1993; Koh et al., 1995; Datta and Greenberg, 1998; F. X. Zhang et al., 1998). These factors activate the phosphatidylinositol-3 kinase (PI-3 kinase) pathway, which is one of several signal transduction pathways implicated in the survival of neurons (Yao and Cooper, 1995; Castellino and Chao, 1996; D'Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997; Parrizas et al., 1997; Philpott et al., 1997; Yamada et al., 1997; Chao et al., 1998; Crowder and Freeman, 1998; F. X. Zhang et al., 1998; Hetman et al., 1999). Although effectors downstream from PI-3 kinase that mediate neuron survival have not been completely identified, one likely candidate is protein kinase Akt (also known as PKB or RAC), a Ser/Thr-protein kinase that is activated by PI-3 kinase (Dudek et al., 1997; Franke et al., 1997; Hemmings, 1997; Kauffmann-Zeh et al., 1997; Kulik et al., 1997; Philpott et al., 1997; Crowder and Freeman, 1998; Murga et al., 1998). Akt phosphorylates and inhibits glycogen synthase kinase-3β (GSK3β) (Cross et al., 1995; Moule et al., 1997), although GSK3β may also be regulated by other PI-3 kinase-dependent, but Akt-independent, pathways (Delcommenne et al., 1998; Kobayashi and Cohen, 1999). Because the PI-3 kinase–Akt pathway is neural-protective and negatively regulates GSK3β activity, GSK3β may be an important downstream proapoptotic target that contributes to apoptosis in neurons. Consistent with this hypothesis, Pap and Cooper (1998) demonstrated that GSK3β activity is required for apoptosis induced by inhibition of PI-3 kinase in Rat1 fibroblasts and neuronal-like PC12 cells. However, the role of GSK3β in apoptosis in primary cultured CNS neurons has not been evaluated. Here, we examined the activity of GSK3β after trophic withdrawal and quantitated the effect of inhibiting or stimulating the GSK3β pathway on induction of apoptosis. Our data indicate that this pathway plays an important role in the regulation of neuronal apoptosis.
MATERIALS AND METHODS
Materials. The following plasmids have been described previously: pON260 (Cherrington and Mocarski, 1989), expression constructs for Xenopus GSK3β binding protein (GBP) (Yost et al., 1998), rat GSK3β (Dominguez et al., 1995), and Myc-tagged expression vectors for wild-type and mutantXenopus β-catenin (Pierce and Kimelman, 1995). The polyclonal anti-GSK3β antibody SC8257 used for immunoprecipitation was from Santa Cruz Biotechnology (Santa Cruz, CA); the anti-Akt and the anti-phospho-Ser 473 Akt antibodies were from New England Biolabs (Beverly, MA).
Cell culture and transfection. Cortical neurons were prepared from newborn Sprague Dawley rats as described previously (Xia et al., 1996; Hetman et al., 1999). The culture procedure used results in cell population consisting of at least 90% neurons at 5 daysin vitro (DIV) (Hetman et al., 1999). Cortical neurons were transiently transfected at 3 DIV using a calcium-phosphate coprecipitation protocol (Xia et al., 1996) with modifications (Hetman et al., 1999).
Serum deprivation. Serum deprivation was performed with neurons at 4–6 DIV as described previously (Hetman et al., 1999). Briefly, cells were washed twice with serum-free basal medium Eagle (BME) (Sigma, St. Louis, MO) and incubated in serum-free BME supplemented with 35 mm glucose, 1 mml-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2.5 μmcytosine arabinoside in the presence or absence of NMDA receptor antagonists dizocilpine maleate (MK-801) (10 μm) or 2-amino-5-phosphonovaleric acid (APV) (100 μm). Control cells were washed similarly and then incubated for matched time points in serum-containing conditioned medium with or without 10 μmMK-801.
DNA ladder assay. To examine DNA cleavage, soluble cytoplasmic DNA was isolated from 4 × 106 cells and subjected to 1.8% agarose gel electrophoresis (Hockenbery et al., 1990; Hetman et al., 1999)
Quantitation of apoptosis by nuclear morphological changes.To visualize nuclear morphology, cells were fixed in 4% paraformaldehyde and stained with 2.5 μg/ml the DNA dye Hoechst 33258 (bis-benzimide; Sigma) (Hetman et al., 1999). Apoptosis was quantitated by scoring the percentage of cells with apoptotic nuclear morphology at the single cell level after Hoechst staining. Uniformly stained nuclei were scored as healthy, viable neurons. Condensed or fragmented nuclei were scored as apoptotic. To obtain unbiased counting, slides were coded, and cells were scored blind without knowledge of their previous treatment. Statistical analysis of the data were performed using one- or two-way ANOVA, followed by post hoc tests.
GSK3β kinase assay. GSK3β activity was quantitated using an immune complex kinase assay. Cell extracts were prepared as described previously (Xia et al., 1995); 200 μg of protein extracts from each sample were incubated at 4 °C for 3–4 hr with 0.8 μg of the anti-GSK3β antibody prebound to protein G Sepharose (Sigma). The immunoprecipitates were washed twice with 50 mm Tris, pH 7.5, 0.5 mLiCl, and 1 mm DTT and twice with 50 mm Tris, pH 7.5, and 1 mmDTT. Kinase assays were then performed as described previously (Wang et al., 1994). The phosphorylated peptide KRREILSRRPS(P)YR, with sequence derived from cAMP response element-binding protein, was used as the substrate for the kinase assay. Quantification of kinase activity was achieved by counting the amount of 32P incorporated into the substrate.
Western analysis and immunostaining. Western blot analysis for anti-phospho-Akt and immunostaining were performed as described previously (Xia et al., 1995, 1996; Hetman et al., 1999). Transfected cells were detected by immunostaining with a polyclonal antibody against β-galactosidase and Texas-Red-conjugated goat antibody to rabbit IgG. Cells transfected with the Myc epitope-tagged constructs were also immunostained with a monoclonal antibody to c-Myc (9E10), followed by fluorescein-conjugated goat antibody to mouse IgG.
RESULTS
Cortical neurons undergo apoptosis after serum withdrawal, serum withdrawal with inhibition of NMDA receptors, or after treatment with LY294002
We have shown previously that cultured rat cortical neurons undergo apoptosis after serum withdrawal and that activation of the PI-3 kinase pathway is the dominant mechanism for serum-dependent survival in these neurons (Hetman et al., 1999). The objective of this study was to test the hypothesis that inhibition of GSK3β through the PI-3 kinase–Akt pathway is one of the downstream mechanisms that mediate PI-3 kinase-dependent neuronal survival. Cortical neuron apoptosis was induced by serum deprivation, serum deprivation together with exposure to NMDA receptor antagonists MK-801 or APV, or treatment with LY294002, a pharmacological PI-3 kinase inhibitor (Vlahos et al., 1994). These three apoptotic paradigms are all expected to decrease PI-3 kinase signaling and were chosen to test the generality of our hypothesis.
Apoptosis was measured by monitoring neurite degeneration, cell body shrinkage, nuclei fragmentation or condensation, and DNA cleavage into oligonucleosome fragments manifested as “DNA laddering,” hallmarks of apoptosis (Raff et al., 1993; Stefanis et al., 1997). Consistent with our previous report (Hetman et al., 1999), serum withdrawal or LY294002 treatment induced apoptosis in cortical neurons (Figs.1, 2). Because activation of NMDA receptors promotes survival in cultured cerebellar neurons (F. X. Zhang et al., 1998; Bhave et al., 1999), we also monitored apoptosis when serum withdrawal was combined with NMDA receptor antagonists MK-801 or APV (Watkins and Collingridge, 1989). Although MK-801 or APV treatment alone had little effect on neuronal survival in the presence of serum, they both greatly potentiated serum deprivation-induced apoptosis (Figs. 1, 2). These data suggest that both serum and NMDA receptor activity are required for optimal survival of cortical neurons. Furthermore, serum withdrawal combined with NMDA receptor antagonists should be a useful model to study mechanisms that regulate activity-dependent neuronal survival.
Fig. 1.

Withdrawal of trophic support induces apoptosis in cortical neurons. A, Representative photomicrographs of cortical neurons (6 DIV) treated for 24 hr with control wash (+S), serum deprivation (−S), or serum deprivation together with exposure to the NMDA receptor antagonist MK-801 (−S+MK). The top panels are phase contrast photomicrographs (Phase), and the bottom panels are Hoechst-stained nuclei to visualize nuclei morphology (Hoechst). Arrows indicate healthy cells with uniformly stained nuclei. Arrowheads identify cells with apoptotic morphology, including shrunken cell bodies, fragmented processes, and condensed or fragmented nuclei. B, DNA fragmentation manifested as a DNA ladder. Cortical neurons were treated with serum deprivation with exposure to MK-801 (−S+MK) for 24 hr. Washed cells exposed to serum were used as control (+S). Positions of molecular size markers (MW) are indicated on theleft in base pairs. Similar results were obtained with serum deprivation alone or with LY294002 treatment.
Fig. 2.
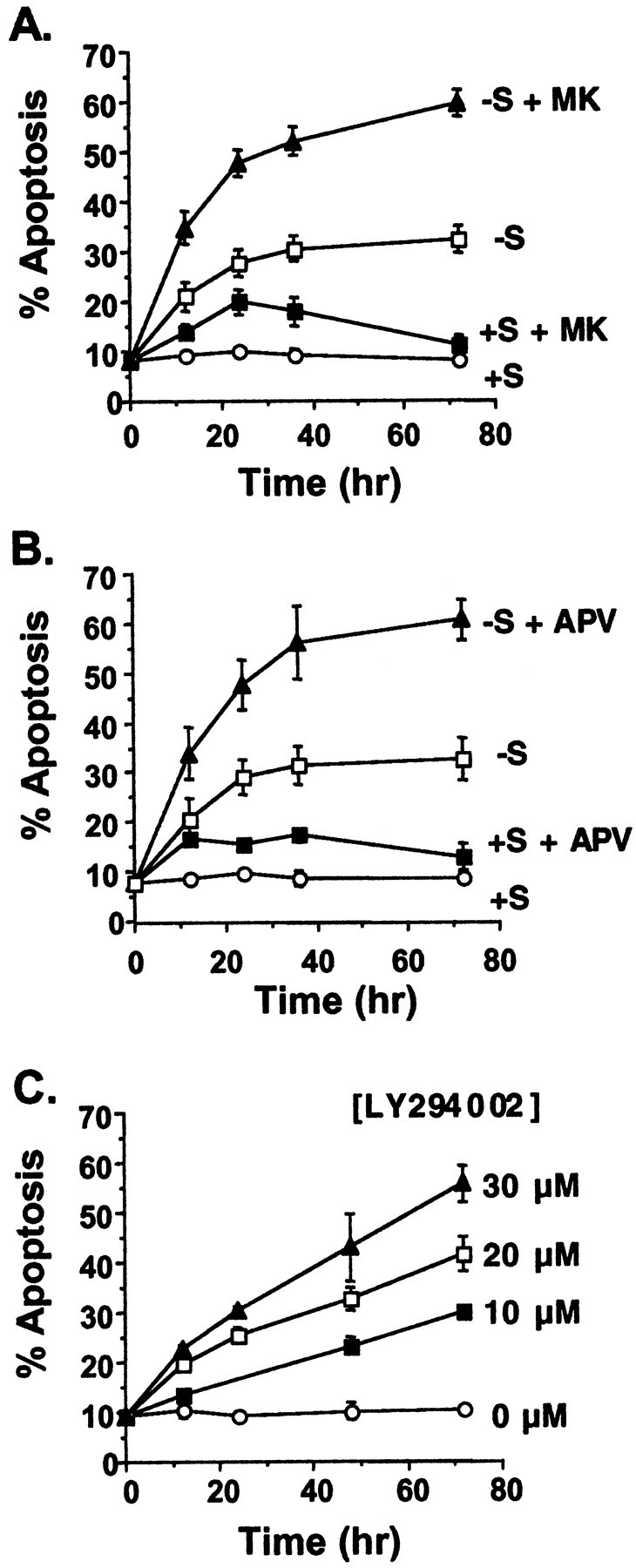
Quantification of apoptosis induced by trophic support withdrawal. Cortical neurons (6 DIV) were treated for 24 hr with control wash (+S) and serum deprivation (−S) with or without exposure to MK-801 (MK) (A) or APV (B), or treated for 24 hr with the PI-3 kinase inhibitor LY294002 in the presence of serum (C). Averages of duplicate determinations in three (A) or two (B, C) independent experiments are shown. At least 1500 cells were scored for each data point in each experiment. Error bars represent SEM. Both MK-801 and APV significantly potentiated apoptotic death after serum withdrawal (−S+MK or −S+APV compared with −S) ( p < 0.0001, ANOVA).
Trophic withdrawal inhibits PI-3 kinase and activates GSK3β
To evaluate the effect of trophic withdrawal on PI-3 kinase, we assayed its activity by Western analysis using an antibody that only recognizes phosphorylated Akt because PI-3 kinase phosphorylates and activates Akt (Franke et al., 1997). Phosphorylation of Akt on Ser-473 is primarily dependent on PI-3 kinase activity (Franke et al., 1997). As anticipated, serum deprivation or serum deprivation plus MK-801 reduced Akt phosphorylation, indicative of PI-3 kinase inactivation (Fig. 3). LY294002 at 30 μm almost completely inhibited PI-3 kinase (Fig. 3).
Fig. 3.
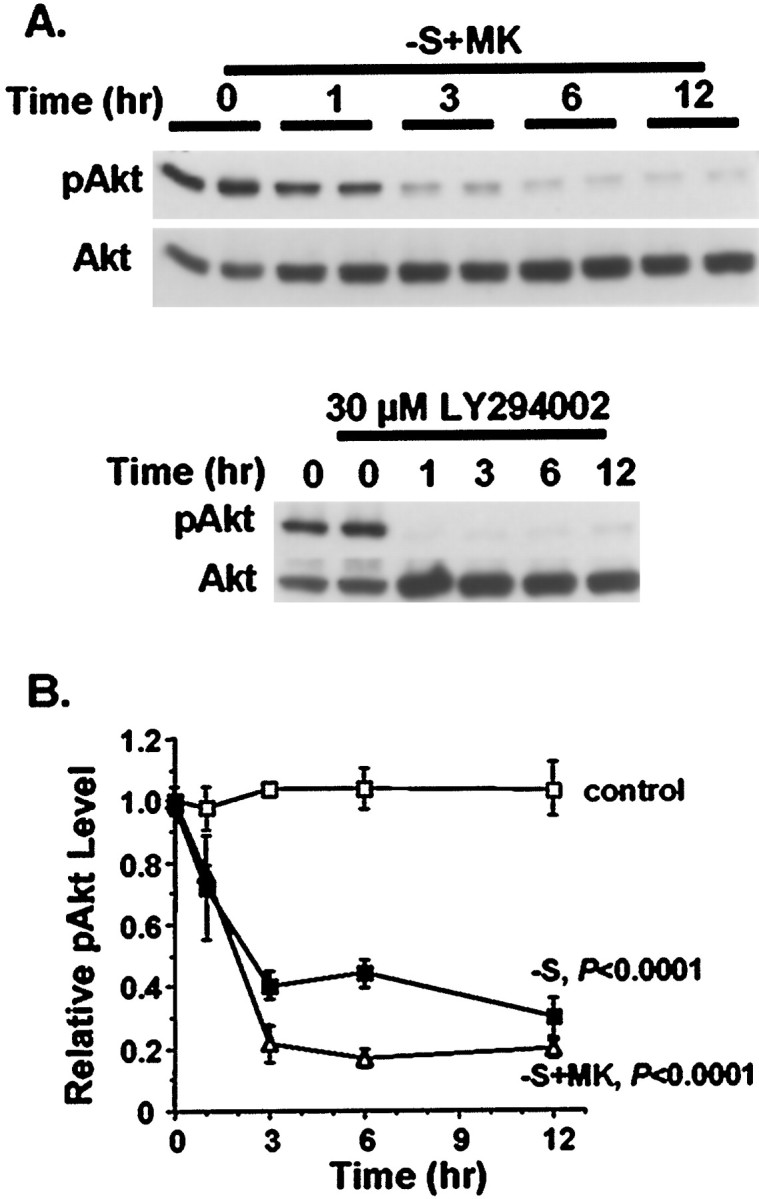
Inhibition of the PI-3 kinase–Akt pathway after trophic support withdrawal. Cortical neurons were treated for the indicated times with 30 μm LY294002, serum deprivation (−S), or serum deprivation with exposure to 10 μm MK-801(−S+MK). Neurons washed similarly but then placed in serum-containing conditioned media were used as controls (control). A, Representative phospho-Akt Western analysis (pAkt) demonstrating reduced Akt phosphorylation at Ser-473, indicative of PI-3 kinase inhibition. The blots were reprobed to show that the total level of Akt (Akt) remained constant. B, Quantitation of Akt phosphorylation by densitometric analysis of pAkt Western blots. Serum withdrawal and serum withdrawal together with MK-801 significantly decreased Akt phosphorylation compared with control washed neurons (p < 0.0001, ANOVA). Data represent averages of duplicate determinations in three independent experiments. Error bars are SEM.
Inhibition of PI-3 kinase leads to activation of GSK3β in PC12 and other non-neuronal cells (Cross et al., 1995; Moule et al., 1997; Pap and Cooper, 1998). To determine whether this is also the case in primary cultured postmitotic neurons, GSK3β activity was measured by an immune complex kinase assay. Serum deprivation or serum deprivation plus MK-801 caused a statistically significant (p < 0.0001) activation of GSK3β compared with control washed neurons (Fig.4A). Similarly, direct inhibition of PI-3 kinase by LY294002 also activated GSK3β in cortical neurons (Fig. 4B). In all three paradigms, GSK3β was activated 75–100% relative to control treated neurons, its activity remained elevated for at least 6 hr after the initial treatment, and GSK3β activation preceded the peak of morphological changes associated with apoptosis (36–72 hr). These data suggest that inhibition of PI-3 kinase and subsequent activation of GSK3β may contribute to neuronal apoptosis induced by trophic withdrawal.
Fig. 4.
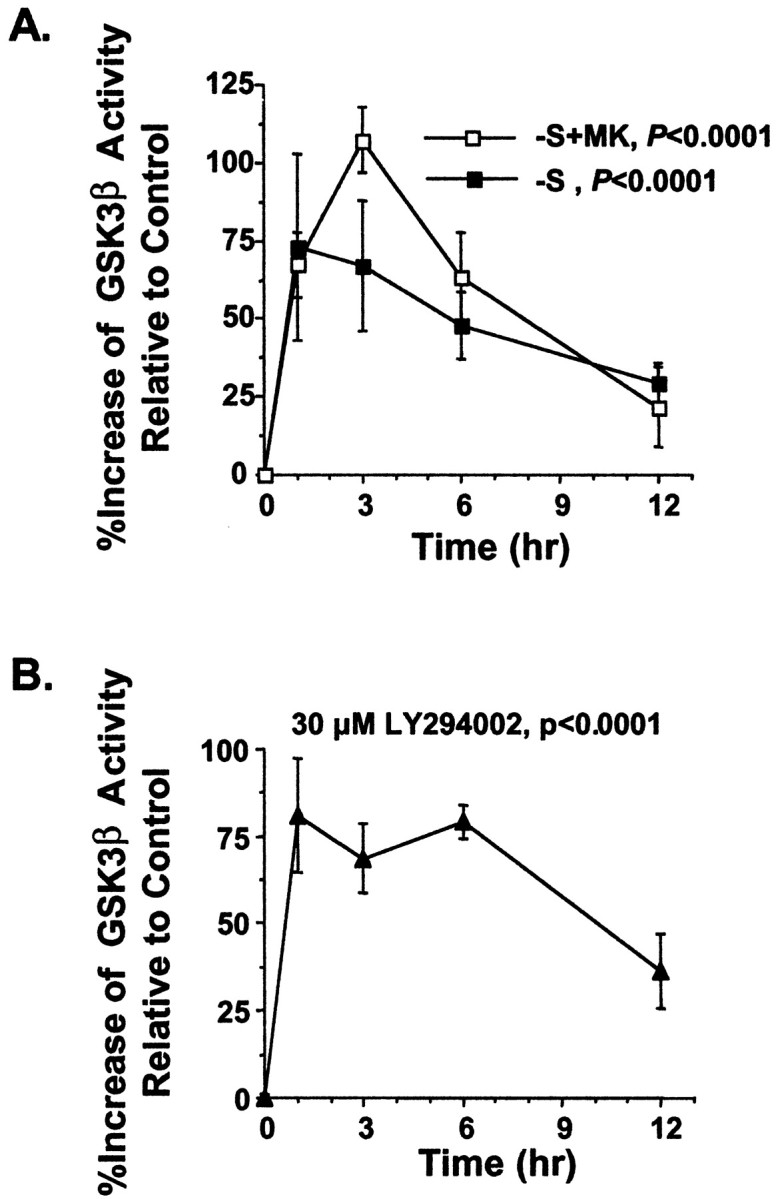
Activation of GSK3β in cortical neurons after trophic deprivation. Cortical neurons were treated as described in Figure 3. A, Serum withdrawal in the absence (−S) or presence of MK-801 (−S+MK) significantly activated GSK3β compared with control washed neurons (p < 0.0001, ANOVA). B, GSK3β was activated in neurons by treatment with LY294002 (p < 0.0001, ANOVA). Results in all panels are averages of duplicate determinations in three independent experiments. Error bars are SEM.
BDNF suppresses GSK3β activation and protects cortical neurons from apoptosis after trophic withdrawal
If trophic withdrawal-induced apoptosis is mediated by inhibition of PI-3 kinase and subsequent activation of GSK3β, then factors that activate PI-3 kinase might prevent GSK3β activation and apoptosis. We reported earlier that brain-derived neurotrophic factor (BDNF) activates PI-3 kinase in cultured cortical neurons and protects these neurons from serum withdrawal-induced apoptosis (Hetman et al., 1999). Here, we tested whether BDNF can also block cortical neuron apoptosis after serum deprivation plus MK-801 and whether the neuroprotective effect of BDNF correlated with activation of PI-3 kinase and inhibition of GSK3β.
Cortical neurons were treated with serum deprivation plus 10 μm MK-801 to induce apoptosis (Fig.5A). Approximately 50% of cells underwent apoptosis 24 hr after the treatment. However, only 20% of the cells were apoptotic in the presence of BDNF; the neural protection afforded by BDNF was primarily reversed by cotreatment with LY294002, suggesting a role for the PI-3 kinase in BDNF protection against apoptosis induced by serum deprivation plus MK-801 (Fig.5A). Moreover, BDNF increased Akt phosphorylation (Fig.5B,C) and prevented GSK3β activation (Fig. 5D) after serum deprivation plus MK-801 treatment. These data are consistent with the hypothesis that inhibition of GSK3β activity may be one of the anti-apoptotic mechanisms used by the PI-3 kinase pathway.
Fig. 5.

BDNF inhibits apoptosis, which correlates with activation of PI-3 kinase and inhibition of GSK3β in trophic-deprived neurons. Cortical neurons were treated with serum deprivation plus 10 μm MK-801(−S+MK) in the presence or absence of 10 ng/ml BDNF. A, BDNF protected cortical neurons from apoptosis compared with vehicle treated controls (Veh) (**p < 0.0001, ANOVA). Apoptosis was scored 24 hr after the treatment. Addition of 30 μm LY294002 (BDNF+LY) reversed the protective effect of BDNF, suggesting a requirement for PI-3 kinase activity in BDNF protection. B, BDNF induced Akt phosphorylation at Ser-473, indicative of PI-3 kinase activation. Akt phosphorylation (pAkt) was examined by Western analysis at indicated times after BDNF treatment. The blots were reprobed to show that the total level of Akt (Akt) remained constant.C, Quantitation of Akt phosphorylation by densitometric analysis of pAkt Western blots. D, BDNF significantly inhibited GSK3β activity in neurons deprived of trophic support (p < 0.001, ANOVA). Results in allpanels are averages of triplicate determinations in at least two independent experiments. Error bars are SEM.
Direct inhibition of GSK3β protects cortical neurons from trophic withdrawal-induced apoptosis
To determine whether GSK3β activation is necessary for trophic withdrawal-induced apoptosis, we transiently transfected cortical neurons with an inhibitory GSK3β binding protein (GBPwt) (Yost et al., 1998) and examined the effect of blocking GSK3β activation on neuronal apoptosis (Fig. 6). In control experiments, neurons were transfected with an inactive GBP mutant (GBPmt), which does not bind or inhibit GSK3β (Yost et al., 1998). Expression of the wild-type or mutant GBP was confirmed by immunocytochemistry (data not shown). Expression of the wild-type or mutant GBP did not significantly affect the rate of basal cell death without apoptotic treatment (Fig. 6). However, transfection of neurons with GBPwt significantly protected them from apoptosis induced by serum deprivation, serum deprivation together with MK-801 treatment, or treatment with LY294002 (Fig.6B).
Fig. 6.
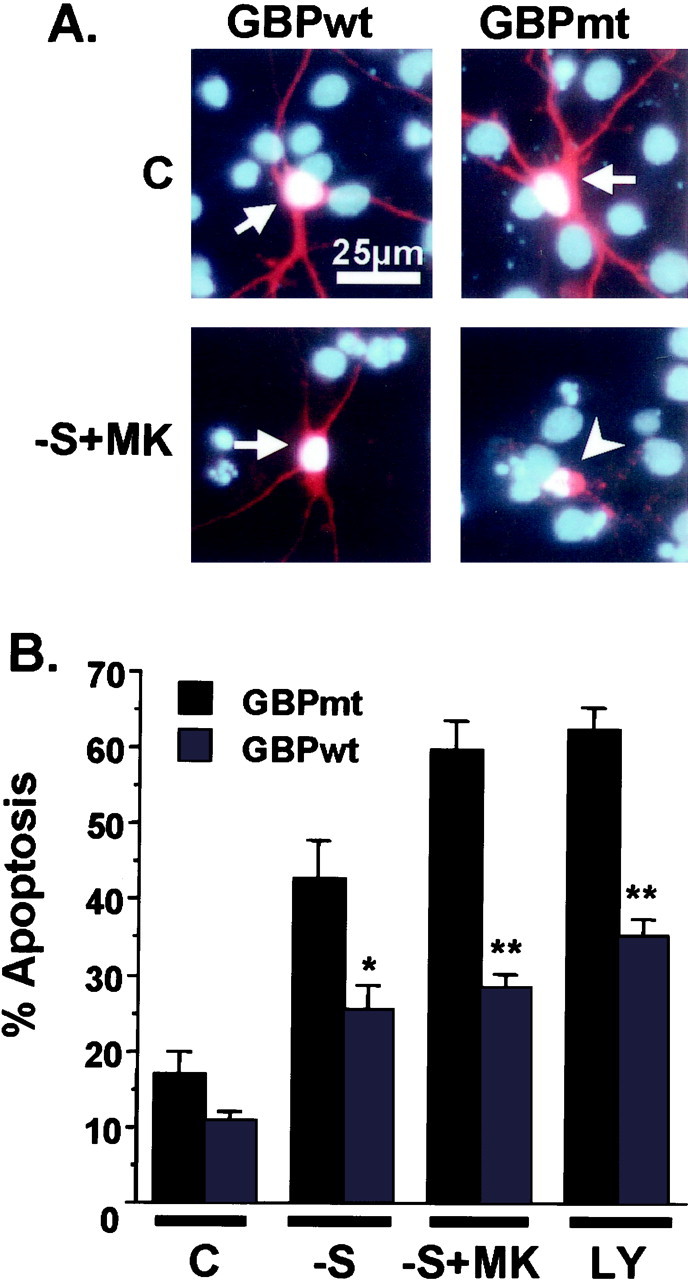
Expression of GBP, a GSK3β inhibitor, protects trophic-deprived neurons from apoptosis. Cortical neurons were transfected with 4 μg of plasmid DNA encoding GBPwt or GBPmt, which is unable to bind or inhibit GSK3β. Cells were also cotransfected with an expression vector encoding β-galactosidase (2 μg) as a marker for transfection. Three days after transfection, neurons were either fixed directly (C) or treated with serum deprivation (−S), serum deprivation with 10 μm MK-801 (−S+MK), or 30 μm LY294002 (LY) for 24 hr.A, Representative immunofluorescence photomicrographs of cortical neurons. Transfected cells were identified by β-galactosidase immunostaining (red cells). To reveal nuclear morphology, cells were counter-stained with Hoechst 33258 (blue). Arrows indicate transfected cells with healthy morphology. The arrowhead identifies a transfected cell with apoptotic morphology after serum deprivation combined with MK-801 treatment. B, Expression of the wild-type but not the mutant GBP inhibited apoptosis. Apoptosis in the transfected cell population was quantitated. Data are averages of duplicate determinations in three independent experiments. At least 1000 transfected cells were scored for each data point. Error bars are SEM. Statistically significant differences are indicated (*p < 0.01; **p < 0.0001, one-way ANOVA, factor-transfected plasmid).
We also transiently transfected cortical neurons with a kinase dead dominant negative form of GSK3β (GSK3βdn) (Dominguez et al., 1995) and examined its effect on neuronal apoptosis (Fig.7). In control experiments, neurons were transfected with the cloning vector pEF1α. Transfection with GSK3βdn or the vector had no effect on basal cell death without any treatment. In contrast, expression of GSK3βdn caused a statistically significant reduction in apoptosis triggered by serum deprivation, serum deprivation plus MK-801 treatment, or treatment with LY294002 (*p < 0.01; **p < 0.0001). These data suggest that activation of GSK3β contributes to apoptosis induced by trophic deprivation.
Fig. 7.

Expression of a dominant negative mutant form of GSK3β protects trophic-deprived neurons from apoptosis. Cortical neurons were transfected with 4 μg of plasmid DNA encoding a dominant negative form of rat GSK3β (GSKdn). The empty cloning vector pEF1α was used as control (vector). Cells were also cotransfected with an expression vector encoding β-galactosidase (2 μg) as a marker for transfection. Three days after transfection, neurons were either fixed directly (C) or treated with serum deprivation (−S), serum deprivation with 10 μm MK-801 (−S+MK), or 30 μm LY294002 (LY) for 24 hr. Apoptosis in the transfected cell population was quantitated. Expression of the dominant negative GSK3β inhibited apoptosis. Data are averages of duplicate determinations in three independent experiments. At least 1000 transfected cells were scored for each data point. Error bars are SEM. Statistically significant differences are indicated (*p < 0.01; **p < 0.0001, one-way ANOVA, factor-transfected plasmid).
Activation of GSK3β is sufficient to induce cortical neuron apoptosis
If trophic withdrawal-induced apoptosis in cortical neurons is mediated by activation of GSK3β, then direct and selective activation of GSK3β might be sufficient to induce apoptosis. Therefore, cortical neurons were transfected with plasmids encoding either the wild-type or a kinase dead dominant negative mutant of GSK3β (Dominguez et al., 1995). The corresponding cloning vector (pEF1α) was used as a control. Transfection of neurons with the wild-type but not the kinase dead mutant form of GSK3β significantly increased apoptosis (Fig.8). Furthermore, apoptosis induced by expression of GSK3β was inhibited by BDNF in a PI3 kinase-dependent manner (data not shown). Thus, direct and selective activation of GSK3β is sufficient to induce apoptosis in cortical neurons.
Fig. 8.

Activation of GSK3β is sufficient to induce neuronal apoptosis. Cortical neurons were transfected with 1 or 4 μg of plasmid DNA encoding either wild-type (wt) or a dominant negative mutant of GSK3β (dn). Cells were also cotransfected with 2 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. The cloning vector pEF1α was used as a control (vector) and to supplement the total DNA to 6 μg in each case. Three days after transfection, cells were fixed and immunostained. Apoptosis in transfected cell population (β-galactosidase-positive) was scored. Data are averages of duplicate determinations in three independent experiments. At least 1000 transfected cells were scored for each data point. Error bars are SEM. Statistical analysis was performed as described in Figure 7(**p < 0.0001).
Degradation of β-catenin is not critical for cortical neuron apoptosis induced by withdrawal of trophic support
Activated GSK3β leads to phosphorylation and subsequent degradation of β-catenin (Miller and Moon, 1996). This is one of the best characterized biochemical events downstream of GSK3β activation (Miller and Moon, 1996). To determine whether β-catenin degradation is important for GSK3β-induced apoptosis, we transiently transfected cortical neurons with either wild-type or a mutant form of β-catenin in which all four GSK3β-targeted serine residues were mutated to alanines (Miller and Moon, 1996). This mutant β-catenin is very stable and is resistant to GSK3β-induced degradation (Miller and Moon, 1996). Our results confirmed this observation. Expression of the wild-type or mutant β-catenin before apoptotic treatment was demonstrated by immunocytochemistry (Fig.9A). Furthermore, the mutant β-catenin protein was indeed stable, whereas the wild-type β-catenin protein was undetectable in these neurons after any of the three apoptotic treatments (data not shown). These results are consistent with the notion that apoptotic treatments cause activation of endogenous GSK3β and subsequent degradation of the wild-type but not mutant β-catenin. If β-catenin degradation is important for GSK3β-induced apoptosis, then overexpression of the stable β-catenin mutant might protect neurons from apoptosis. However, expression of either the wild-type or the mutant β-catenin had no significant effect on cortical neuron apoptosis after serum deprivation, serum deprivation together with MK-801 treatment, or treatment with LY294002 (Fig. 9B). Therefore, stabilization of β-catenin is ineffective in blocking GSK3β-mediated apoptosis, suggesting that β-catenin is not the critical substrate by which GSK3β triggers neuron death.
Fig. 9.
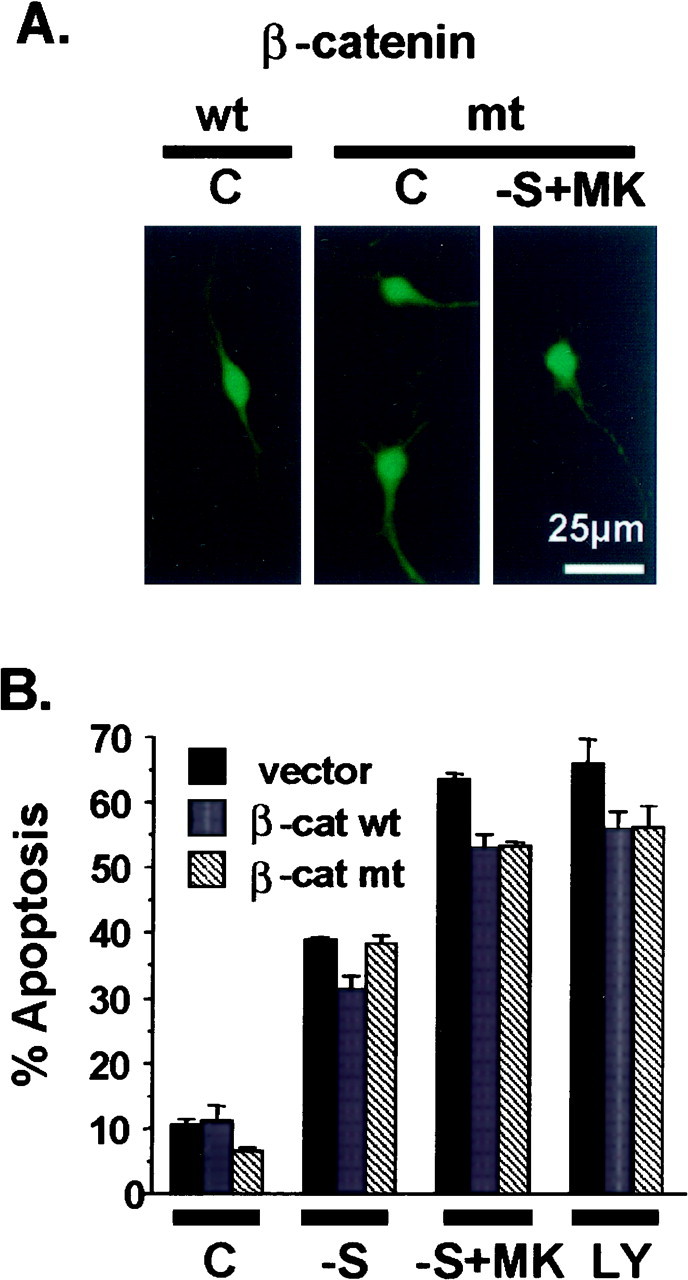
Expression of β-catenin does not protect cortical neurons from apoptosis induced by trophic withdrawal. Cortical neurons were transfected with 4 μg of plasmid DNA encoding the wild-type (β-cat wt) or a mutant form (β-cat mt) of β-catenin. The cloning vector was used as a control (vector). All four of the serine residues in β-catenin, which are targeted by GSK3β, were mutated to alanines in the mutant form of β-catenin, which is stable and not subject to GSK3β-induced degradation. Cells were also cotransfected with an expression vector encoding β-galactosidase (2 μg) as a marker for transfection. Two days after transfection, neurons were either fixed directly (C) or treated with serum deprivation (−S), serum deprivation with 10 μm MK-801 (−S+MK), or 30 μmLY294002 (LY) for 24 hr. A, Representative photomicrographs depicting immunostaining with 9E10 antibody to detect transfected Myc-tagged β-catenin.B, Apoptosis in the transfected cell population (β-galactosidase-stained cells) was quantitated. Data are averages of duplicate determinations. Similar results were obtained in two independent experiments. At least 350 transfected cells were scored for each condition. Error bars are SEM.
DISCUSSION
The goal of this study was to test the hypothesis that inhibition of GSK3β through PI-3 kinase pathway is one of the downstream mechanisms that mediate PI-3 kinase-dependent neuronal survival. Using cultured cortical neurons, we identified three apoptotic paradigms in which PI-3 kinase activity was suppressed while GSK3β was activated. These include serum deprivation, serum deprivation with inhibition of NMDA receptors, or treatment with a PI-3 kinase inhibitor LY294002. Our data demonstrate that apoptosis induced by all three paradigms was accompanied by inhibition of PI-3 kinase, as well as activation of GSK3β. Furthermore, direct inhibition of GSK3β by expression of either GBP, a specific and potent inhibitor of GSK3β (Yost et al., 1998), or a dominant negative mutant form of GSK3β (Dominguez et al., 1995) partially prevented cell death induced by all three treatments. In contrast, expression of wild-type GSK3β alone was sufficient to induce cortical neuron apoptosis, even in the presence of serum. Moreover, BDNF-mediated protection against trophic deprivation required PI-3 kinase activity and correlated with inhibition of GSK3β. GSK3β is the substrate of Akt (Cross et al., 1995; Moule et al., 1997), although it might also be inhibited by other PI-3 kinase-dependent pathways (Delcommenne et al., 1998; Kobayashi and Cohen, 1999). Although we cannot distinguish between these two possibilities, our results strongly suggest that GSK3β is an important inducer of neuronal apoptosis and that PI-3 kinase-promoted neuronal survival may involve negative regulation of GSK3β.
Pap and Cooper (1998) reported that nerve growth factor leads to GSK3β inactivation via the PI-3 kinase pathway. Furthermore, inhibition of PI-3 kinase by either LY294002 treatment or expression of dominant negative mutants interfering with PI-3 kinase–Akt pathway induces apoptosis in Rat1 fibroblasts and neuronal-like PC12 cells, whereas inhibition of GSK3β prevented apoptosis (Pap and Cooper, 1998). Together with our findings, these data suggest that inhibition of GSK3β activity by the PI-3 kinase signaling pathway may be a general mechanism for survival of neurons and non-neuronal cells. Activation of the PI-3 kinase–Akt signal transduction system may promote survival through several mechanisms, including inhibition of GSK3β, phosphorylation of proapoptotic proteins Bad, FKHRL1 (Datta et al., 1997; del Peso et al., 1997; Brunet et al., 1999), and phosphorylation of caspase-9 (Cardone et al., 1998). This suggests that multiple mechanisms may work in parallel downstream from PI-3 kinase to suppress apoptosis.
The mechanism for induction of apoptosis by GSK3β remains undefined. GSK3β phosphorylates four serine residues at the N-terminal region of β-catenin and causes β-catenin degradation (Miller and Moon, 1996; Yost et al., 1996). It has been proposed that destabilization of β-catenin potentiates neuronal apoptosis induced by β-amyloid peptide (Z. Zhang et al., 1998). This suggested the interesting hypothesis that GSK3β-induced apoptosis involves degradation of β-catenin. However, our data do not support this general hypothesis because overexpression of wild-type or a stable mutant form of β-catenin did not rescue cortical neuron from trophic deprivation-induced apoptosis.
In addition to β-catenin and glycogen synthase, several other substrates for GSK3β have been identified, some of which are worth consideration as candidate mediators of GSK3β-induced cell death. For example, mitochondrial pyruvate dehydrogenase is phosphorylated and inhibited by GSK3β; the subsequent metabolic failure might cause neuron death (Hoshi et al., 1996). GSK3β also phosphorylates insulin receptor substrate 1 (IRS-1) and converts IRS-1 into an inhibitor of insulin receptor tyrosine kinase activity (Eldar-Finkelman and Krebs, 1997). Because IRS-1 is critical in the signaling of insulin and insulin-like growth factor and both factors promote neuronal survival, phosphorylation of IRS-1 by GSK3β may contribute to GSK3β-induced cell death. Furthermore, GSK3β phosphorylates microtubule-associated protein tau into Alzheimer's disease-like forms (PH-tau) found in tangles (Hanger et al., 1992; Mandelkow et al., 1992; Ishiguro et al., 1993; Mulot et al., 1994). The appearance of PH-tau is associated with early alterations in neurites associated with Alzheimer's disease (Goedert et al., 1995). Tau phosphorylation by GSK3β may cause axonal dysfunction and trigger neuronal apoptosis. Regardless of the downstream target, our data strongly indicate that inhibition of GSK3β by PI-3 kinase is an important mechanism for neuronal survival.
Our finding that serum withdrawal-induced apoptosis of cortical neurons is potentiated by NMDA antagonists suggests that optimal cortical neuron survival requires both trophic factors and NMDA receptor activity. The role of NMDA receptor activity in supporting neuronal survival has also been demonstrated in cultured cerebellar granule neurons (F. X. Zhang et al., 1998; Bhave et al., 1999) and in forebrain cortical neurons in vivo (Ikonomidou et al., 1999). Collectively, these findings support the notion that neuronal survival depends on both the availability of peptide trophic factors and neuronal activity.
In summary, we have discovered that cortical neuron apoptosis induced by trophic withdrawal is mediated in part by GSK3β activation. Our findings add GSK3β to the list of potential drug targets for pharmacotherapy of neurodegenerative disorders and suggest that GSK3β may play a critical role in neuronal apoptosis.
Footnotes
This work was supported by Pilot Grant 1810 from the Royalty Research Fund at the University of Washington (Z.X.), and National Institute of Neurological Disorders and Stroke Grants NS37359 (Z.X.) and HD27262 (D.K). M.H. completed part of this work while on tenure of a fellowship award from the American Heart Association, Washington Affiliate. J.E.C. was supported by National Institutes of Health, Genetic Approaches to Aging Postdoctoral Training Grant 2 T32 AG00057-21. We thank Dr. J. R. Miller for helpful discussion and Drs. M. Pap and G. M. Cooper for providing GSK3β constructs.
Correspondence should be addressed to Zhengui Xia, Department of Environmental Health, Box 357234, University of Washington, Seattle, WA 98195. E-mail: zxia@u.washington.edu.
REFERENCES
- 1.Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 3.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 4.Castellino AM, Chao MV. Trans-signaling by cytokine and growth factor receptors. Cytokine Growth Factor Rev. 1996;7:297–302. doi: 10.1016/s1359-6101(96)00038-x. [DOI] [PubMed] [Google Scholar]
- 5.Chao M, Casaccia-Bonnefil P, Carter B, Chittka A, Kong H, Yoon SO. - Neurotrophin receptors: mediators of life and death. Brain Res Brain Res Rev. 1998;26:295–301. doi: 10.1016/s0165-0173(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 6.Cherrington JM, Mocarski ES. Human cytomegalovirus iel transactivates the α promoter-enhancer via an 18-base-pair repeat element. J Virol. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 8.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta SR, Dudek H, Tao X, Masters S, Fu HA, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 12.Datta SR, Greenberg ME. Molecular mechanisms of neuronal survival and apoptosis. In: O'Malley B, editor. Hormones signaling. Academic; San Diego: 1998. pp. 257–306. [Google Scholar]
- 13.del Peso L, GonzalezGarcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 14.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominguez I, Itoh K, Sokol SY. Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc Natl Acad Sci USA. 1995;92:8498–8502. doi: 10.1073/pnas.92.18.8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao RJ, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 17.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Estus S. Gene induction and neuronal apoptosis. In: Mattson MP, editor. Neuroprotective signal transduction. Humana; Totowa, NJ: 1998. pp. 83–94. [Google Scholar]
- 19.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 20.Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Gotz J, Burki K, Cohen P. Molecular dissection of the paired helical filament. Neurobiol Aging. 1995;16:325–334. doi: 10.1016/0197-4580(95)00017-9. [DOI] [PubMed] [Google Scholar]
- 21.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- 22.Hemmings BA. Signal transduction–Akt signaling: linking membrane events to life and death decisions. Science. 1997;275:628–630. doi: 10.1126/science.275.5300.628. [DOI] [PubMed] [Google Scholar]
- 23.Hetman M, Kanning K, Smith-Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular-signal-regulated kinase and phosphatidylinositol-3 kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 24.Hockenbery D, Nuñez G, Millman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 25.Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci USA. 1996;93:2719–2723. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 27.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 28.Kauffmann-Zeh A, RodriguezViciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3- phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339:319–328. [PMC free article] [PubMed] [Google Scholar]
- 30.Koh J-Y, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 31.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levi-Montalcini R, Booker B. Destruction of sympathetic ganglia in mammals by an antisera to nerve growth factor protein. Proc Natl Acad Sci USA. 1960;46:384–391. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 34.Miller JR, Moon RT. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- 35.Miller TM, Tansey MG, Johnson EM, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 36.Moule SK, Welsh GI, Edgell NJ, Foulstone EJ, Proud CG, Denton RM. Regulation of protein kinase B and glycogen synthase kinase-3 by insulin and beta-adrenergic agonists in rat epididymal fat cells. Activation of protein kinase B by wortmannin-sensitive and -insensitive mechanisms. J Biol Chem. 1997;272:7713–7719. doi: 10.1074/jbc.272.12.7713. [DOI] [PubMed] [Google Scholar]
- 37.Mulot SF, Hughes K, Woodgett JR, Anderton BH, Hanger DP. PHF-tau from Alzheimer's brain comprises four species on SDS-PAGE which can be mimicked by in vitro phosphorylation of human brain tau by glycogen synthase kinase-3 beta. FEBS Lett. 1994;349:359–364. doi: 10.1016/0014-5793(94)00702-0. [DOI] [PubMed] [Google Scholar]
- 38.Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinase gamma. J Biol Chem. 1998;273:19080–19085. doi: 10.1074/jbc.273.30.19080. [DOI] [PubMed] [Google Scholar]
- 39.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 40.Park DS, Stefanis L, Greene LA. Ordering the multiple pathways of apoptosis. Trends Cardiovasc Med. 1997;7:294–301. doi: 10.1016/S1050-1738(97)00090-X. [DOI] [PubMed] [Google Scholar]
- 41.Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 42.Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce SB, Kimelman D. Regulation of Spemann organizer formation by the intracellular kinase Xgsk-3. Development. 1995;121:755–765. doi: 10.1242/dev.121.3.755. [DOI] [PubMed] [Google Scholar]
- 44.Raff MC, Barres BA, Burne J, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 45.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 46.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 47.Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- 48.Watkins JC, Collingridge GL. The NMDA receptor. Oxford UP; Oxford: 1989. [Google Scholar]
- 49.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 50.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamada M, Ohnishi H, Sano S, Nakatani A, Ikeuchi T, Hatanaka H. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chem. 1997;272:30334–30339. doi: 10.1074/jbc.272.48.30334. [DOI] [PubMed] [Google Scholar]
- 52.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 53.Yost C, Farr GHR, Pierce SB, Ferkey DM, Chen MM, Kimelman D. GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell. 1998;93:1031–1041. doi: 10.1016/s0092-8674(00)81208-8. [DOI] [PubMed] [Google Scholar]
- 54.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 55.Zhang FX, Rubin R, Rooney TA. N-Methyl-d-aspartate inhibits apoptosis through activation of phosphatidylinositol 3-kinase in cerebellar granule neurons. A role for insulin receptor substrate-1 in the neurotrophic action of N-methyl-d- aspartate and its inhibition by ethanol. J Biol Chem. 1998;273:26596–26602. doi: 10.1074/jbc.273.41.26596. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]