

Abstract
The localization of cannabinoid (CB) receptors to GABAergic interneurons in the hippocampus indicates that CBs may modulate GABAergic function and thereby mediate some of the disruptive effects of marijuana on spatial memory and sensory processing. To investigate the possible mechanisms through which CB receptors may modulate GABAergic neurotransmission in the hippocampus, whole-cell voltage-clamp recordings were performed on CA1 pyramidal neurons in rat brain slices. Stimulus-evoked GABAA receptor-mediated IPSCs were reduced in a concentration-dependent manner by the CB receptor agonist WIN 55,212–2 (EC50 of 138 nm). This effect was blocked by the CB1 receptor antagonist SR141716A (1 μm) but not by the opioid antagonist naloxone. In contrast, evoked GABAB-mediated IPSCs were insensitive to the CB agonist. WIN 55,212–2 also reduced the frequency of spontaneous, action potential-dependent IPSCs (sIPSCs), without altering action potential-independent miniature IPSCs (mIPSCs), measured while sodium channels were blocked by tetrodotoxin (TTX). Blockade of voltage-dependent calcium channels (VDCCs) by cadmium also eliminated the effect of WIN 55,212–2 on sIPSCs. Depolarization of inhibitory terminals with elevated extracellular potassium caused a large increase in the frequency of mIPSCs that was inhibited by both cadmium and WIN 55,212–2. The presynaptic effect of WIN 55,212–2 was also investigated using the potassium channel blockers barium and 4-aminopyridine. Neither of these agents significantly altered the effect of WIN 55,212–2 on evoked IPSCs. Together, these data suggest that presynaptic CB1 receptors reduce GABAA- but not GABAB-mediated synaptic inhibition of CA1 pyramidal neurons by inhibiting VDCCs located on inhibitory nerve terminals.
Keywords: brain slice, calcium channels, cannabis, electrophysiology, GABAA receptors, GABABreceptors, hippocampal, marijuana, potassium channels, presynaptic, ruthenium red
The pharmacological actions of marijuana within the mammalian CNS are attributable to specific interactions between the active constituents of the drug, collectively known as cannabinoids (CBs) and their receptors. Two subtypes of CB receptor, known as CB1 and CB2, have been identified. The CB1 receptor is expressed in high concentrations throughout the CNS, whereas the CB2 receptor is expressed primarily in immune cells (Pertwee, 1997; Axelrod and Felder, 1998). Cannabinoid receptors interact with G-proteins to alter the activities of enzymes, such as adenylyl cyclase, and to modulate ion channels (Matsuda, 1997; Pertwee, 1997). Studies of CB1 receptor localization within the CNS have revealed moderate to high densities throughout several cortical areas, including the hippocampus (Herkenham et al., 1990; Howlett et al., 1990; Pettit et al., 1998;Tsou et al., 1998; Katona et al., 1999). Given the well established role of the hippocampus in learning and memory processes, it is likely that the adverse effects of marijuana on spatial learning tasks, short-term memory, and attention are attributable to its actions within this brain region (Miller and Branconnier, 1983; Murray, 1986;Deadwyler et al., 1990; Hampson and Deadwyler, 1998).
Pyramidal neurons in area CA1 of the hippocampus receive both excitatory and inhibitory inputs from intrinsic and extrinsic sources, and they communicate with various cortical and limbic regions (Knowles, 1992). Several recent studies have addressed the role of CB1 receptors in modulating excitatory synaptic transmission in the hippocampus. Thus, presynaptic inhibition of glutamate release onto CA1 pyramidal neurons by CBs has been described previously (Misner and Sullivan, 1999), and it has been suggested that this occurs through the inhibition of voltage-dependent Ca2+channels (VDCCs) of the N and P/Q classes (Twitchell et al., 1997; Shen and Thayer, 1998; Sullivan, 1999). In contrast, although CB1-mediated inhibition of GABAergic synaptic transmission has been demonstrated in the basal ganglia (Chan et al., 1998; Szabo et al., 1998) and medulla (Vaughan et al., 1999), similar studies have not been performed in the hippocampus. The largest GABAergic input to CA1 pyramidal neurons is derived from a diverse network of intrinsic interneurons (Freund and Buzsáki, 1996; Buzsáki, 1997). Although these interneurons represent only a small fraction (∼10%) of the total hippocampal neuronal population, each interneuron forms multiple synapses onto its cellular targets. In this way, the release of GABA by interneurons provides a means to coordinate pyramidal cell activity and hippocampal output (Cobb et al., 1995; Buzsáki, 1997). The transmitter released from the interneurons onto pyramidal cells can interact with either GABAA or GABABreceptors, generating fast IPSCs mediated by the activation of Cl− channels or slow IPSCs mediated by the activation of K+ channels, respectively (Alger and Nicoll, 1982; Solis and Nicoll, 1992; Ling and Benardo, 1994). Recently, Katona and colleagues (1999) demonstrated that CB1 receptors were located on the axon terminals of a specific subpopulation of cholecystokinin-immunoreactive interneurons and that CB1 receptor activation reduced [3H]GABA release. However, this study did not determine whether CB1 receptors inhibited synaptic GABA release, nor did it identify the mechanism(s) involved in this modulation. Several potential mechanisms may mediate CB modulation of GABAergic transmission. For example, the inhibition of VDCCs (Shen and Thayer, 1998; Sullivan, 1999), the activation of voltage-dependent K+ channels (VDKCs) and voltage-independent K+ channels (Deadwyler et al., 1995; Mackie et al., 1995), the inhibition of GABA uptake (Maneuf et al., 1996), or the activation of endogenous opioid pathways (Chen et al., 1990) have all been proposed (Tanda et al., 1997). In the present study, we compare the effects of CB1 receptor activation on GABAA- and GABAB-mediated synaptic transmission in the hippocampus, and we examine the potential mechanisms involved in this modulation. We demonstrate that CB1 receptor activation inhibits GABAA- but not GABAB-mediated IPSCs through a presynaptic mechanism that likely involves the inhibition of VDCCs.
MATERIALS AND METHODS
Slice preparation. All protocols were performed under National Institutes of Health Guidelines and were approved by the Institutional Animal Care and Use Committee (National Institute on Drug Abuse, Intramural Research Program, Baltimore, MD). Male Sprague Dawley rats (Charles River Laboratories, Raleigh, NC), 14- to 30-d-old, were killed by decapitation, and their brains rapidly removed and placed in ice-cold oxygenated artificial CSF (aCSF) (see below). The brain was then blocked in a coronal plane ∼2 mm anterior and 5 mm posterior to bregma using a razor blade. The posterior end of the tissue block was then glued to the stage of a vibrating tissue slicer (Technical Products International, St. Louis, MO) using cyanoacrylate. A midsagittal cut was then made with a scalpel blade to separate the two hemispheres, and brain slices were cut at 300 μm nominal thickness. The slices were then transferred to a beaker containing aCSF and aerated with 95% O2–5% CO2 at room temperature in which they were stored for at least 90 min before they were transferred to the recording chamber. During recordings, slices were continuously superfused with aCSF at a rate of 2 ml/min. All recordings were performed at room temperature (∼ 22° C). Control aCSF consisted of (in mm): NaCl 126, KCl 3.0, MgCl2 1.5, CaCl2 2.4, NaH2PO4 1.2, glucose 11.0, and NaHCO3, 26, saturated with 95% O2 and 5% CO2. In a few experiments, Ca2+ and Mg2+ were omitted from the buffer and were instead applied at selected concentrations by superfusion using a calibrated syringe pump (Razel Scientific Instruments Inc., Stamford, CT).
Pyramidal neuron recording. Whole-cell patch-clamp recordings of spontaneous IPSCs (sIPSCs) and tetrodotoxin (TTX)-resistant miniature IPSCs (mIPSCs) from pyramidal cells were obtained using methods described previously (Lupica, 1995; Miller et al., 1997). Briefly, recordings were performed using an Axoclamp-2A or an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA) and electrodes pulled from borosilicate thick-walled capillary tubing (inner diameter of 0.75 mm, outer diameter of 1.5 mm; Sutter Instrument Co., Novato, CA). Cells were voltage clamped at −60 to −90 mV using whole-cell electrodes containing (in mm): CsCl 125.0, HEPES 10.0, EGTA 1.0, CaCl2 0.1, Mg2+-ATP 2.0, Na+-GTP 0.2, and the quaternary lidocaine derivative QX-314 2, pH 7.2–7.4. Series resistance was monitored continuously using small (10 mV), hyperpolarizing voltage steps (200 msec). Only cells demonstrating <20 MΩ series resistance were used in these experiments. In most cases, the series resistance did not change appreciably during the recording period. However, in cases in which the series resistance increased, there was a noticeable decrease in whole-cell conductance and a sudden and sustained decrease in the holding current. When this occurred, the cell was not used in further analyses. The glutamate receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX) (10 μm) and d-(−)-2-amino-5-phosphonopentanoic acid (APV) (40 μm) were continuously present in the aCSF to block EPSPs and to isolate the presynaptic interneurons from excitatory afferent input. Spontaneous and mIPSCs were amplified fivefold to 100-fold, filtered at 1–3 kHz, and recorded to videotape for later analysis. Epochs of 1–3 min of data were digitized at 4–10 kHz using a National Instruments (Austin, TX) Lab PC 1200 analog-to-digital converter and the Strathclyde electrophysiology software package (courtesy of Dr. John Dempster, Strathclyde University, Glasgow, UK) and then analyzed using a personal computer-based program (Mini Analysis 4.3; Synaptosoft, Leonia, NJ). Averaged mIPSCs were generated by aligning individual events by rise time, and a peak to decay single exponential fit was applied (before and during drug application) using the formula y =A1 * exp(−x/τ) + Baseline, where A1is the peak amplitude, and τ is the time constant for decay.
Evoked GABAA-receptor-mediated IPSCs (evIPSCs) were generated in the presence of DNQX and APV using a bipolar tungsten stimulating electrode placed near (<100 μm) the recording electrode, within stratum radiatum. Evoked GABAB-mediated IPSCs were isolated by including picrotoxin (100 μm) in the superfusion buffer and by using stimulation intensities that were twofold to 2.5-fold greater than those necessary to evoke GABAA-mediated IPSCs (see below). To monitor whole-cell access, a constant hyperpolarizing step pulse (10–20 mV, 200 msec) was delivered after each stimulus using a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Stimulation (0.1 msec duration) was delivered at 30 sec intervals using a constant current unit (A.M.P.I.) and the pulse generator. Current output was adjusted to evoke a submaximal response in each experiment (<200 μA). In those cells in which sIPSCs or mIPSCs were not analyzed and in all cases in which GABAB-receptor mediated responses were measured, recordings of evIPSCs were performed using K-gluconate-filled electrodes. These whole-cell electrodes had resistances of 7–10 MΩ when filled with the following solution (in mm): K+-gluconate 125.0, KCl 10.0, HEPES 10.0, EGTA 1.0, CaCl2 0.1, Mg2+-ATP 2.0, and Na+-GTP 0.2, adjusted to pH 7.2–7.4 with 1 m KOH and brought to 270–280 mOsm with deionized water. Analyses of predrug and postdrug effects on evIPSCs were performed using personal computer-based software (Neuropro SCOPE; R.C. Electronics, Goleta, CA).
Chemicals. Drugs were obtained from the following sources. TTX, DNQX, picrotoxin, ruthenium red, bicuculline methiodide, DAMGO [Tyr-D-Ala2,N-CH3-Phe4,Gly-ol-enkephalin], CdCl2, 4-aminopyridine, naloxone, and BaCl2 were from Sigma (St. Louis, MO). APV and WIN 55,212–2 were from Tocris Cookson (Ballwin, MO). SR141716A was obtained from the National Institute on Drug Abuse drug supply system. CGP 35348 was a generous gift from Drs. D. Scholer and H. Kaufmann (CIBA-Geigy Ltd., Basel, Switzerland). WIN 55,212–2 and SR141716A were prepared as concentrated (10–100 mm) stock solutions in DMSO. Final (bath) concentrations were <0.01% DMSO. All drugs were made up at either 50 or 100 times the desired final concentration in deionized water and then added to the flow of the superfusion medium using a calibrated syringe pump (Razel Scientific Instruments Inc.).
Statistical analysis. Group data are presented as the mean ± SEM in all cases. Drug-induced changes in cumulative sIPSC and mIPSC amplitude and interevent interval distributions were analyzed for statistical significance using the Kolmogorov–Smirnov (K–S) test (Mini Analysis 4.3) and a conservative critical probability level of p < 0.01. All other statistical tests were performed using a critical probability of p< 0.05 (Prism version 2.01; GraphPad Software, San Diego CA).Post hoc analysis was performed only when an ANOVA yielded a significant (p < 0.05) main effect.
RESULTS
Effect of WIN 55,212–2 on evoked GABAA IPSCs in pyramidal neurons
In cells clamped at −80 mV, using a CsCl-based internal solution, a single stimulus evoked a fast inward current that was abolished by application of either the GABAA receptor antagonist bicuculline methiodide (20 μm) or the Cl− channel blocker picrotoxin (100 μm). After a 3–5 min period to allow for stabilization of the baseline response, the CB receptor agonist WIN 55,212–2 was applied via the aCSF. As shown in Figure1, application of WIN 55,212–2 (5 μm) resulted in a slow, time-dependent decrease in the evoked GABAA receptor-mediated IPSC. Maximal inhibition of the response generally occurred within 5–7 min of drug application, and the peak inhibition of the response was 47 ± 4% (n = 12) with 5 μm WIN 55,212–2. Because of the lipid soluble nature of WIN 55,212–2, reversal of the drug effect by washout was not possible within the temporal parameters of the recording session. Therefore, we attempted to block the effect using the selective CB1 antagonist SR141716A (Rinaldi-Carmona et al., 1994). Because the antagonist is also highly lipid soluble, we found it necessary to preincubate the slices with it for 10–15 min before application of WIN 55,212–2. Alone, SR141716A (1 μm) had no significant effect on the evIPSC (115 ± 17% of control; n = 9) (Fig.1A). However, as shown in Figure 1, SR141716A significantly antagonized the effect of WIN 55,212–2 on the evIPSC (120 ± 11% of control; n = 9).
Fig. 1.
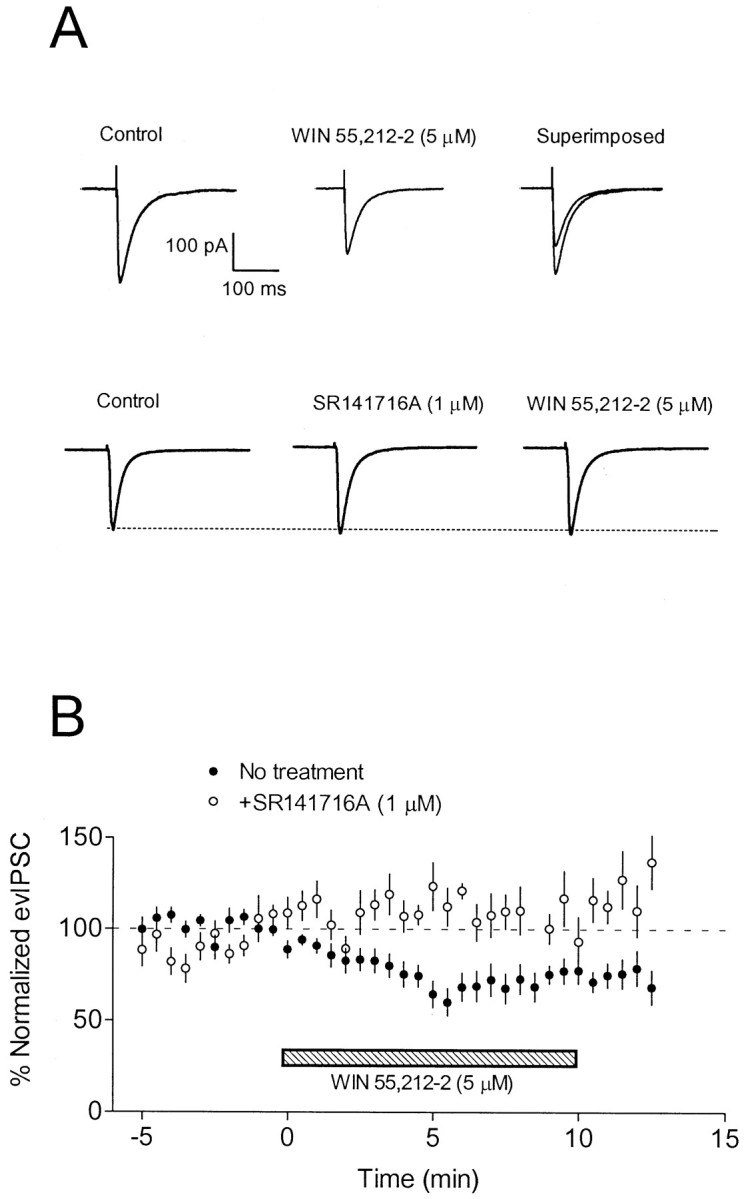
Effect of WIN 55,212–2 on stimulus-evoked GABAA receptor-mediated IPSCs in CA1 pyramidal neurons. Whole-cell recordings were performed using CsCl-based electrode solution at a holding potential of −80 mV. Currenttraces represent an average of 5–10 sweeps.A, In the top series oftraces, application of 5 μm WIN 55,212–2 reduced the GABAA IPSC. Control and drugtraces are superimposed for clarity. In a different cell shown in the bottom series of traces, application of SR141716A (1 μm; 10 min) did not alter the IPSC. In the continued presence of SR141716A, WIN 55,212–2 had no effect on the IPSC. B, Summary of the effect of WIN 55–212, plotted as a percentage change (mean ± SEM) from control. Closed circles represent the effect of WIN 55,212–2 alone (n = 12), and open circles represent the effect of WIN 55,212–2 after pretreatment with SR141716A (n = 9).
We also performed a concentration–response analysis using the sequential cumulative administration of increasing concentrations (2–4 concentrations per slice) of WIN 55,212–2. For these experiments, IPSCs were measured using K+- gluconate-filled electrodes because of the increased stability of these recordings over long periods of time. As shown in Figure2, WIN 55,212–2 inhibited the evoked IPSCs in a concentration-dependent manner, with an estimated EC50 of 138 nm. In these experiments, stable, peak effects of the lower concentrations of WIN 55,212–2 occurred more slowly (10–15 min) than at higher concentrations of the drug (5–7 min). However, we ensured response stability by measuring 3–5 similar consecutive responses at a given drug concentration. Also, when the effect of the 1 μm concentration of WIN 55,212–2 (n = 3), observed at the end of the cumulative administration paradigm, was compared with the effect at the same concentration in WIN 55,212–2-naïve neurons (n = 3), no significant differences were observed (p < 0.05; two-tailed Student's ttest). This suggests that the effects of WIN 55,212–2 were concentration-dependent and that CB1 receptor desensitization did not occur during the cumulative administration of the drug.
Fig. 2.

Concentration-dependent effect of WIN 55,212–2 on evoked GABAA IPSCs in CA1 pyramidal neurons.A, Recording from a single neuron using a K+-gluconate-based electrode solution at a holding potential of −55 mV. Control and WIN 55,212–2 concentrations are labeled for each trace. Traces represents an average of 5–10 sweeps taken at the peak of a stable drug response.B, Concentration–response curve for WIN 55,212–2. Each data point represents the mean ± SEM of the maximal inhibition of the evoked IPSC (n = 3–12 cells). The EC50 estimated from the fitted curve is 138 nm.
Several studies have demonstrated that CB-mediated actions in the CNS could be explained by the stimulation of opioid receptors (Chen et al., 1990; Tanda et al., 1997). Because of these observations and because opioid receptors are known to inhibit GABA release in the hippocampus (Siggins and Zieglgansberger, 1981; Cohen et al., 1992; Lupica, 1995), we examined the possibility that the effects of WIN 55,212–2 might be mediated via activation of an endogenous opioid pathway. Slices were pretreated with the opioid antagonist naloxone (5 μm), and the effects of WIN 55,212–2 (1 μm) were tested on GABAA evIPSCs in the presence of naloxone. The reduction in evIPSC amplitude in the presence of naloxone (64 ± 3% of control; n = 10) was not significantly different than the reduction of evIPSCs by 1 μm WIN 55,212–2 alone (56 ± 7% of control; n = 6;p = 0.29; unpaired t test). This suggests that the inhibition of GABAA-mediated IPSCs by WIN 55,212–2 was not attributable to activation of opioid receptors by either the agonist itself or endogenous opioid peptides.
Effect of WIN 55,212–2 on GABAB IPSCs
The results described above demonstrate that CB1 receptor activation reduces fast, GABAA-mediated IPSCs in CA1 pyramidal neurons. However, CA1 neurons also demonstrate a slow IPSC, mediated by activation of GABAB receptors and the opening of inwardly rectifying G-protein-coupled K+ channels (GIRK) (Alger and Nicoll, 1982; Solis and Nicoll, 1992; Ling and Benardo, 1994). In addition, it has been proposed that GABAB-mediated IPSCs occur in response to GABA release from afferent fibers that are distinct from those mediating GABAA IPSCs (Nurse and Lacaille, 1997). Therefore, to determine whether GABAB IPSCs were also sensitive to modulation by CB1 receptors and whether this subset of GABAergic fibers was sensitive to presynaptic inhibition by CB1 receptors, we measured GABAB IPSCs in CA1 pyramidal neurons. Stimulus-evoked GABAB IPSCs were isolated in neurons voltage clamped at −55 to −65 mV in the presence of DNQX (10 μm), APV (40 μm), and picrotoxin (100 μm). The IPSCs observed under these conditions were significantly reduced (p < 0.01; n = 6) by application of the GABAB receptor antagonist CGP 35348 (100 μm) (Fig. 3), and they reversed polarity at membrane potentials between −90 and −110 mV, suggesting that they were mediated by K+ currents (Ek of −96 mV with [K+]o of 3.0 mm and [K+]i of 135.0 at 20° C, using the Nernst equation). In contrast to its effect on the GABAA-mediated IPSC, WIN 55,212–2 (1 μm) did not significantly alter the amplitude of the GABAB-mediated response (p > 0.05; n = 7; one-way ANOVA) (Fig. 3). However, in agreement with a previous study (Lupica et al., 1992), these GABAB responses were significantly inhibited presynaptically by the μ-opioid agonist DAMGO (1 μm; p < 0.05;n = 4) (Fig. 3B).
Fig. 3.

Effect of WIN 55,212–2 on GABABreceptor-mediated IPSCs. A, Recording from a single neuron clamped at −55 mV using K+-gluconate-based internal solution. APV (40 μm), DNQX (10 μm), and picrotoxin (100 μm) were included in the superfusion medium. Traces represent an average of 8–10 sweeps. Application of the GABAB antagonist CGP 35348 (100 μm) reduced the IPSC amplitude, and this effect reversed within ∼10 min. Subsequent application of WIN 55,212–2 (1 μm; 15 min) did not decrease the IPSC.B, Summary of the effects of WIN 55,212–2 (1 μm; n = 7) (WIN), CGP 35348 (100 μm;n = 6) (CGP), and the μ-opioid agonist DAMGO (1 μm; n = 4) on the GABAB response (*p < 0.05, **p < 0.01 vs control; one-way ANOVA, followed by Tukey–Kramer post hoc analysis). For comparison, the effect of 1 μm WIN 55,212–2 on the GABAAreceptor-mediated IPSC is shown (solid bar) (n = 6; **p < 0.01 vs WIN effect on GABAB IPSC; unpaired Student's ttest).
Effect of WIN 55,212–2 on spontaneous GABAA IPSCs
The inhibition of GABAA-mediated evoked IPSCs by WIN 55,212–2 could involve either presynaptic or postsynaptic mechanisms. To distinguish between these possibilities, we conducted studies examining the effects of WIN 55,212–2 on spontaneously occurring IPSCs. Action potential-dependent sIPSCs were measured in pyramidal neurons under voltage clamp at −80 mV (n = 6). Treatment with WIN 55,212–2 (5 μm) significantly reduced the mean amplitude of the sIPSCs from −16.7 ± 3.7 to −11.9 ± 1.4 pA (p < 0.05; Wilcoxon signed rank test). In addition, the mean frequency of sIPSCs was significantly reduced by WIN 55,212–2 from 2.0 ± 0.3 Hz in control to 1.2 ± 0.1 Hz (p < 0.05; Wilcoxon signed rank test). The effects of WIN 55,212–2 on sIPSC amplitude and frequency are shown in Figure4. Significant differences in both the cumulative amplitude and cumulative interevent interval distributions (p < 0.001; Kolmogorov–Smirnov test) were observed in five of six cells tested during WIN 55,212–2 application.
Fig. 4.

Effect of WIN 55,212–2 on spontaneous, action potential-dependent IPSCs in a CA1 pyramidal neuron. Recording was performed using a CsCl-based electrode solution at a holding potential of −80 mV. A, Traces represent portions of 2 min epochs recorded before (Control) and during the peak drug effect (∼7 min after 5 μm WIN 55,212–2 application). B, Cumulative interevent interval distribution shown for the same cell, revealing a significant increase in the interevent interval (i.e., decreased frequency;p < 0.001; K–S test) during WIN 55,212–2 application. C, Cumulative amplitude distribution obtained from the same cell reveals a significant decrease in sIPSC amplitude (p < 0.001; K–S test) in the presence of WIN 55,212–2. The mean sIPSC amplitude in this cell was decreased from −29.5 pA (n = 392 events) to −18.4 pA (n = 145 events). D, Summary of the effect of 5 μm WIN 55,212–2 (WIN) on the amplitude and frequency of sIPSCs (mean ± SEM; n = 5). Significant reductions were observed in both amplitude and frequency (*p< 0.05; Wilcoxon signed rank test). Calibration: 50 pA, 500 msec.
Effects of WIN 55,212–2 on miniature IPSCs
To further confirm a presynaptic mechanism for the actions of WIN 55,212–2 on GABA release and to assess the role of voltage-dependent ion channels in this effect, we measured action potential-independent mIPSCs in the presence of the voltage-dependent Na+ channel blocker TTX (1 μm). We reasoned that any action of WIN 55,212–2 on postsynaptic sensitivity to GABA or on GABA uptake processes should be manifested as a change in mIPSC amplitude or mIPSC kinetics. In all cells, the efficacy of the TTX block of Na+ channels was monitored by observing the disappearance of the evIPSC during maximal electrical stimulation. A complete elimination of the evoked response usually occurred within 2 min after beginning the TTX application. As shown in Figure5, application of TTX alone reduced both the frequency and amplitude of sIPSCs. However, the mIPSCs remaining after TTX application were completely insensitive to WIN 55,212–2 (1–5 μm). Thus, WIN 55,212–2 did not produce a shift in either the cumulative amplitude or cumulative interevent interval mIPSC distributions in any of these cells (p > 0.05; Kolmogorov–Smirnov test; n = 6). Mean mIPSC amplitudes were −9.2 ± 3.3 pA in TTX (control) versus −9.5 ± 0.5 pA during WIN 55,212–2 application (p > 0.05; Wilcoxon signed rank test). The average frequency of mIPSCs was 0.68 ± 0.18 Hz in TTX versus 0.61 ± 0.15 Hz during WIN 55,212–2 (p > 0.05; Wilcoxon signed rank test).
Fig. 5.

Effect of WIN 55,212–2 on action potential-independent (TTX-insensitive) mIPSCs in a single CA1 pyramidal neuron. Holding potential, −80 mV. A,Traces represent portions of 2 min epochs recorded before TTX application (Control), 10 min into the TTX (500 nm) application, and 10 min into the WIN 55,212–2 (5 μm) application. B, Cumulative interevent interval distributions for each treatment condition in the same cell. A significant decrease in the frequency of events was observed during TTX (p < 0.001; K–S test). During WIN 55,212–2 application, no further change (p > 0.05; K–S test) in the distribution was observed. C, Cumulative amplitude distribution for the same cell demonstrating a decrease in amplitude during TTX (p < 0.001; K–S test). The mean amplitude decreased from −12.6 pA (n = 518 events) to −8.8 pA (n = 43 events) in TTX. However, no further change in the mean amplitude (−9.3 pA; n = 56 events) was observed during WIN 55,212–2 treatment. D, Summary of mIPSC amplitude and frequency (mean ± SEM;n = 6) before TTX (Con, open bars), during TTX (filled bars) (*p < 0.05 vs control; Wilcoxon signed rank test), and during WIN 55,212–2 (5 μm) (hatched bars). Calibration: 25 pA, 500 msec.
In a further attempt to identify any postsynaptic effects of WIN 55,212–2 on the sensitivity to GABA or on GABA uptake, mIPSCs were averaged before and during WIN 55,212–2 application, and single exponential decay time constants (τ) were fit to these waveforms (see Materials and Methods). During TTX alone, τ was 28.8 ± 2.2 msec, whereas during WIN 55,212–2 application, τ was 28.9 ± 3.1 msec. These values were not significantly different (n = 6; p = 0.98; paired Student'st test). Similarly, the 10–90% rise times of mIPSCs were not significantly affected by WIN 55,212–2 (control, 2.9 ± 0.2 msec; WIN, 2.3 ± 0.3 msec; p = 0.07; paired Student's t test; n = 6). Thus, because WIN 55,212–2 did not alter the amplitudes or kinetics of mIPSCs , it is unlikely that postsynaptic sensitivity to GABA was altered or that GABA uptake was disrupted in these neurons.
Because the frequency of mIPSCs was significantly slower than sIPSC frequency, we considered the possibility that our inability to observe modulation of mIPSCs might be because of the small number of events available to analyze in the presence of TTX. Therefore, to increase the relative frequency of mIPSCs in the presence of TTX, we used the polyvalent cation ruthenium red, which blocks VDCCs and enhances mIPSC frequency via a Ca2+-independent mechanism (Trudeau et al., 1996; Sciancalepore et al., 1998; Cibulsky and Sather, 1999). Ruthenium red (200 μm) increased mIPSC frequency threefold to fivefold in every cell tested, which, on average, was similar to the frequency of action potential-dependent sIPSCs (sIPSCs, 2.0 ± 0.3 Hz; TTX, 0.60 ± 0.11 Hz; ruthenium red, 2.3 ± 0.78 Hz; n = 9; data not shown). However, ruthenium red had no effect on mIPSC amplitude (TTX, −8.6 ± 0.6 pA; ruthenium red, −8.8 ± 0.9 pA; n = 9). Similar to the lack of effects of WIN 55,212–2 on mIPSCs, a 1 μm concentration of this agonist had no effect on mIPSC frequency (WIN 55,212–2, 2.3 ± 0.8 Hz) or amplitude (WIN 55,212–2, −8.5 ± 1.1 pA) in the presence of ruthenium red. Together, the above data suggest that WIN 55,212–2 altered neither the postsynaptic sensitivity to GABA nor its rate of clearance and that the effects of the CB1 agonist on sIPSCs were presynaptic. Also, the absence of CB1 receptor-mediated effects on mIPSCs was not attributable to the smaller number of synaptic currents available for the analysis.
Effects of WIN 55,212–2 on sIPSCs in the presence of cadmium
The experiments involving TTX demonstrated that, when voltage-dependent Na+ channels were blocked, the effects of WIN 55,212–2 on GABA release were eliminated. This could indicate that the presynaptic actions of WIN 55,212–2 were attributable to either a direct action on voltage-dependent Na+ channels or on another voltage-dependent current activated by Na+channel-induced depolarization. To distinguish between these possibilities, we examined the actions of WIN 55,212–2 on spontaneous IPSCs during the blockade of VDCCs by CdCl2. Bath application of CdCl2 (200 μm) completely eliminated the evIPSC and reduced the amplitude and the frequency of sIPSCs (Fig. 6). However, as shown in Figure 6, application of WIN 55,212–2 (1–5 μm;n = 6) in the presence of CdCl2did not produce an additional shift in either the cumulative amplitude sIPSC distribution or the interevent interval distribution in any cell (p > 0.05; Kolmogorov–Smirnov test). The mean sIPSC amplitudes in these experiments were −11.8 ± 1.0 pA in CdCl2 and −11.5 ± 1.5 pA during WIN 55,212–2 (p > 0.05; Wilcoxon signed rank test). A slight increase in the mean frequency was observed during WIN 55,212–2 treatment in these experiments (CdCl2, 0.49 ± 0.08 Hz; WIN 55,212–2, 0.63 ± 0.13 Hz), but it was not statistically significant (p > 0.05; Wilcoxon signed rank test). These data demonstrate that the blockade of VDCCs, like the blockade of VD Na+channels, eliminated the CB1 receptor-mediated inhibition of GABA release.
Fig. 6.

Effect of WIN 55,212–2 on spontaneous IPSCs in a CA1 pyramidal neuron during CdCl2 application. Holding potential, −80 mV. A, Traces represent portions of 2 min epochs obtained before treatment (Control), 10 min into CdCl2 (200 μm) application, and 13 min into WIN 55,212–2 (1 μm) application. B, Cumulative interevent interval distribution for the same cell. CdCl2 produced a significant increase in the interevent interval (p < 0.001; K–S test). During WIN 55,212–2 treatment in the presence of CdCl2, there was no further change in the distribution (p> 0.05; K–S test). C, Cumulative amplitude distribution for the same cell reveals that CdCl2 treatment significantly (p < 0.001; K–S test) reduced the amplitude from control. The mean amplitude decreased from −17.9 pA (n = 241 events) in control to −13.0 pA (n = 126 events) during CdCl2. A significant change in the amplitude distribution was not observed during WIN 55,212–2 application in the presence of CdCl2(p > 0.05; K–S test; mean amplitude, −10.7 pA; n = 92 events). D, Summary of mean ± SEM amplitude and frequency changes of sIPSCs (n = 6) during CdCl2(Cd) (*p < 0.05 vs control; Wilcoxon signed rank test) and WIN 55,212–2 in the presence of CdCl2 (WIN). Calibration: 25 pA, 500 msec.
Effect of WIN 55,212–2 on Ca2+-dependent mIPSCs during depolarization with KCl
The experiments described above indicated that the presynaptic modulation of GABA release by CB1 receptors could be occluded by blockade of either Na+ channels by TTX or VDCCs by CdCl2. Because the activation of VDCCs and the release of GABA are dependent on the depolarization initiated by Na+ action potentials, we hypothesized that TTX acted indirectly to inhibit VDCC activity and thus occlude the effect of the CB1 agonist, whereas CdCl2 acted directly on VDCCs to occlude this effect. However, although these experiments supported the hypothesis that CBs inhibit GABA release by modulating VDCC activity, they did not permit us to dismiss the possibility of direct modulation of VD Na+channels by WIN 55,212–2. In an effort to dissociate VDCC from VD Na+ channel activity, we increased the contribution of presynaptic VDCCs by directly depolarizing the inhibitory terminals with elevated [K+]o in the presence of TTX.
In the presence of TTX, the elevation of extracellular K+ from 3 to 15 mm produced a significant increase in both the amplitude (TTX, −11.8 ± 3.6 pA; high K+, −21.6 ± 8.2 pA;n = 6) and the frequency (TTX, 0.5 ± 0.7 Hz; high K+, 13.7 ± 5.6 Hz; n= 6) of mIPSCs in every cell tested (p < 0.01; K–S test; n = 6), although the absolute magnitude of the increase varied considerably from cell to cell. To confirm that larger and more frequent mIPSCs observed in high [K+]o were caused by increased VDCC activity, we applied CdCl2 (200 μm). In each neuron, CdCl2 reduced the frequency (mean, 2.5 ± 0.98 Hz; n = 6) and amplitude (−18.1 ± 5.3 pA) of the mIPSCs, suggesting that the elevated [K+]o increased GABA release as a result of increased VDCC activity (Fig.7). Under conditions of elevated [K+]o, WIN 55,212–2 (1 μm) significantly reduced the frequency of mIPSCs to 59 ± 9% of the baseline recorded during application of TTX and high [K+]o (Fig. 7). In addition, mIPSC amplitude was significantly reduced by WIN 55,212–2 in four of six neurons (p < 0.01; K–S test). Thus, the CB1 receptor-mediated inhibition of GABA release was restored under conditions that favored VDCC activity, during the blockade of VD Na+ channels.
Fig. 7.

Effect of WIN 55,212–2 on mIPSCs during depolarization of inhibitory terminals with elevated [K+]o. Holding potential, −80 mV using a CsCl-based internal solution. A,Traces representing portions of 2 min epochs acquired during the sequential administration of TTX (500 nm; in standard 3 mm[K+]o), elevated [K+]o (KCl) (15 mm), WIN 55,212–2 (1 μm), and CdCl2 (200 μm). B, Cumulative interevent interval distribution for the same cell. WIN 55,212–2 produced a significant (p < 0.001; K–S test) increase in the interevent interval distribution during application of high [K+]o and TTX, indicating a decrease in frequency. Subsequent application of CdCl2 also resulted in a significant (p < 0.001; K–S test) shift in the interevent interval distribution, indicating that the direct activation of VDCCs contributed to the enhancement of mIPSC frequency in elevated [K+]o. C, Cumulative amplitude distribution for the same cell. Both WIN 55,212–2 and the subsequent application of CdCl2 produced a significant (p < 0.001; K–S test) decrease in the cumulative amplitude distribution relative to high [K+]o. The mean amplitudes were as follows: high [K+], −10.6 pA (n = 637 events); WIN 55,212–2, −8.4 pA (n = 367 events); and CdCl2, −7.9 pA (n = 264 events). D, Summary of the effects of WIN 55,212–2 (WIN) (1 μm; n = 6) and CdCl2(Cd) (200 μm;n = 6) on the mean frequency and amplitude. Data represent the mean ± SEM and are shown as a percentage of the baseline obtained in high [K+]o (*p < 0.05 vs baseline; Student's two-tailed t test). Although the mean amplitude was not significantly affected, a significant (p < 0.001; K–S test) shift in the cumulative amplitude distribution was observed in four of six cells. Calibration: 25 pA, 500 msec.
Effects of K+ channel blockade on the inhibition of evIPSCs by WIN 55,212–2
The experiments described above demonstrated that CB1 receptor activation presynaptically reduced GABA release onto CA1 pyramidal neurons and suggested that the inhibition of VDCCs was involved. However, because CBs have also been shown to modulate voltage-dependent K+ channels (IA) in hippocampal pyramidal neurons (Deadwyler et al., 1995) and voltage-independent K+ channels in cellular expression systems (Mackie et al., 1995; Jin et al., 1999), we sought to assess the potential role of presynaptic K+ channels in the CB1 receptor-mediated inhibition of GABA release. Recordings were performed on neurons voltage clamped at −45 to −55 mV using a K+-gluconate-based internal solution (see Materials and Methods), and K+ channels were blocked by either BaCl2 (300 μm) or 4-AP (100 μm). Evoked IPSC amplitudes and durations were increased by both BaCl2 (amplitudes, 138 ± 10% of control;n = 9) and 4-AP (amplitudes, 148 ± 15% of control; n = 9) (Fig. 8). Application of WIN 55,212–2 (1 μm) failed to reduce the evIPSC in the presence of either of the K+ channel blockers (Fig. 8). However, because the blockade of presynaptic K+channels is known to delay repolarization of the synaptic terminal and thereby prolong the presynaptic action potential (for discussion, seeLupica and Dunwiddie, 1993), it was likely that Ca2+ influx into the terminal was increased and that the GABA release process was saturated. Because this shift in the Ca2+ dependence of GABA release might confound our ability to observe modulation by the CB1 receptor agonist, we sought to “normalize” this action by lowering [Ca2+]o to generate evIPSCs that were similar in amplitude to those observed before K+ channel blockade. Thus, after BaCl2 (n = 3) or 4-AP (n = 4) application, the Ca2+ concentration was adjusted to between 1.5 and 2.0 mm (from 2.4 mm, with corresponding Mg2+ levels increased to maintain osmolarity). This manipulation reduced the evIPSC to 70–100% of control levels. As shown in Figure 8, in the presence of BaCl2 or 4-AP and under conditions of lowered Ca2+, the ability of WIN 55,212–2 (1 μm) to reduce evIPSCs was not changed compared with its effect in the absence of these manipulations (p > 0.05 vs 1 μm WIN 55,212–2 alone; one-way ANOVA). This suggests that CB1 receptor activation did not inhibit IPSCs through the modulation of 4-AP- and BaCl2-sensitive K+channels.
Fig. 8.
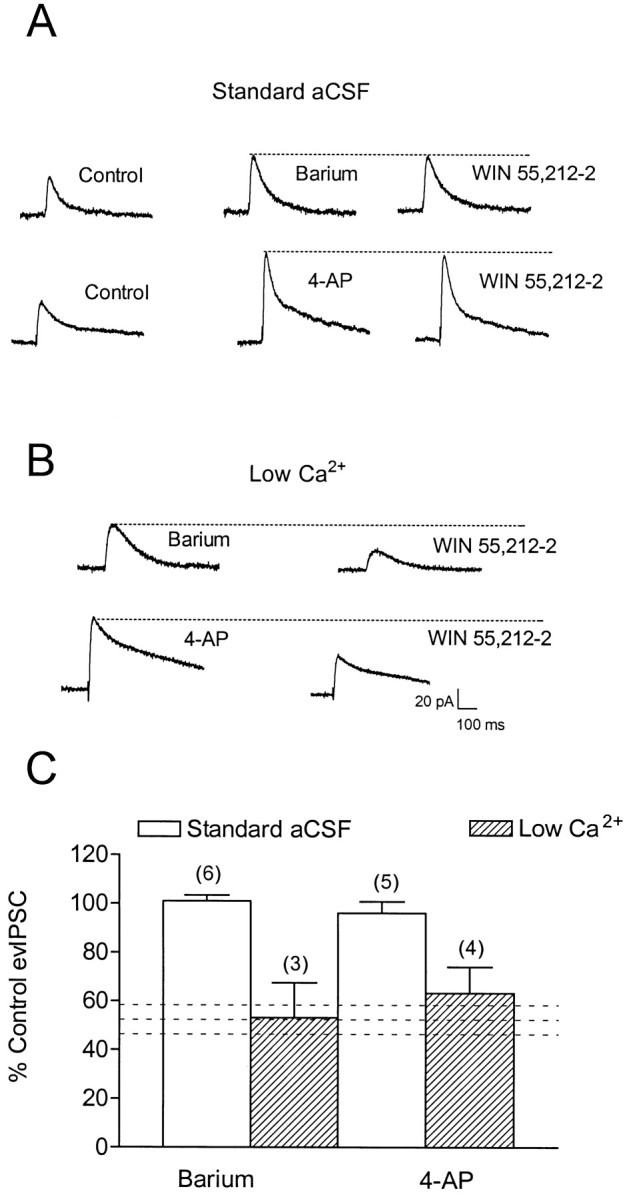
Effect of K+ channel blockade on WIN 55,212–2-mediated inhibition of evoked GABAA-mediated IPSCs in CA1 pyramidal neurons. Recordings were performed using a K+-gluconate-based electrode solution at a holding potential of −45 to −55 mV. A, Under standard conditions (2.4 mmCaCl2), BaCl2 (300 μm) (top traces) and 4-AP (100 μm) (bottom traces) produced a large increase in the IPSC. Application of WIN 55,212–2 (1 μm) in the presence of BaCl2 or 4-AP failed to cause a significant reduction in the IPSC. B, Under conditions in which the extracellular Ca2+ was reduced to 1.2–2.0 mm after BaCl2 (top trace) or 4-AP (bottom trace) application, WIN 55,212–2 reduced the IPSC.C, Summary of the effects of WIN 55,212–2 (1 μm) in the presence of BaCl2 (300 μm) or 4-AP (100 μm) under either standard (open bars) or low Ca2+(hatched bars) aCSF. Data represent the mean ± SEM of the number of cells given in parentheses. The horizontal dashed lines represent the effect of 1 μm WIN 55,212–2 on GABAA IPSCs in standard aCSF without BaCl2 or 4-AP (mean ± SEM; n = 4).
DISCUSSION
The present study demonstrates that the CB1 agonist WIN 55,212–2 acted presynaptically to inhibit GABA release onto GABAA receptors in the CA1 region of the hippocampus. This was in contrast to the actions of this CB1 agonist on GABAB-mediated synaptic transmission, which was not affected at a concentration of WIN 55,212–2 that maximally inhibited GABAA-mediated IPSCs. In addition, this electrophysiological analysis permitted the exclusion of possible postsynaptic effects of WIN 55,212–2 on the postsynaptic sensitivity to GABA, GABA uptake (Maneuf et al., 1996), and the indirect modulation of GABA release via the activation of opioid receptors (Tanda et al., 1997). We also provide evidence that the CB1 receptor inhibition of GABAA-mediated synaptic transmission likely occurs through the inhibition of VDCCs and probably not through alterations in Na+ or K+ channel activity. These results thereby extend those demonstrating that CB1 receptors were located on the inhibitory terminals of hippocampal interneurons and that K+-stimulated [3H]GABA release was modulated by WIN 55,212–2 (Katona et al., 1999).
Although the adverse affects of marijuana on memory and cognitive function have long been ascribed to its actions in the hippocampus (Drew and Miller, 1974; Miller and Branconnier, 1983; Essman, 1984;Hampson and Deadwyler, 1998), the description of CB binding sites in this brain region has provided strong corroborative evidence in favor of this hypothesis (Herkenham et al., 1990; Howlett et al., 1990;Breivogel and Childers, 1998). Furthermore, the development of potent ligands for the CB1 receptor has provided a means to directly assess the effects of CBs in the CNS (Pertwee, 1997). One of the hallmarks of CNS CB function that has emerged from these studies is that activation of CB1 receptors can presynaptically inhibit fast synaptic transmission in a variety of brain regions. Thus, CB1 agonists can inhibit glutamatergic transmission in the cerebellum (Levenes et al., 1998) and the hippocampus (Shen et al., 1996; Misner and Sullivan, 1999;Sullivan, 1999), whereas GABAergic synaptic transmission is inhibited by CBs in the substantia nigra (Chan et al., 1998), the striatum (Szabo et al., 1998), the medulla (Vaughan et al., 1999), and the hippocampus (present study).
CB1 receptors modulate GABAA- but not GABAB-mediated synaptic responses
The present study demonstrated that WIN 55,212–2 inhibited evoked GABAA receptor-mediated IPSCs in a concentration-dependent manner, without affecting GABAB-mediated IPSCs. The differential modulation of GABAA and GABAB synapses by monoamine receptor agonists has been shown previously in midbrain dopamine neurons (Johnson et al., 1992; Cameron and Williams, 1993;Shoji et al., 1999). However, it is not clear whether GABAA and GABAB synaptic responses arise from different populations of inhibitory terminals in the hippocampus (Nurse and Lacaille, 1997). Our results support the hypothesis that the innervation of GABAA and GABAB receptors arises from distinct inhibitory terminals and are consistent with the idea that CB1 receptors are found on inhibitory basket cell terminals (Katona et al., 1999) that constitute a major source of GABAA-mediated synaptic input onto CA1 pyramidal neuron somata (Buhl et al., 1995). This differential targeting of inhibitory terminals by CB1 receptors is in contrast to μ-opioid receptors, which inhibit both GABAA- and GABAB-mediated synaptic transmission in the hippocampus (Lupica et al., 1992).
Our estimated EC50 value for WIN 55,212–2-mediated inhibition of GABAA IPSCs (138 nm) is in reasonable agreement with the values of 41 nm for inhibition of [3H]GABA release (Katona et al., 1999) and 30 nm for inhibition of [3H]acetylcholine release in hippocampal slices (Gifford and Ashby, 1996). In agreement with previous data (Collins et al., 1995; Katona et al., 1999), the effect of WIN 55,212–2 was completely blocked by the CB1 antagonist SR141716A. However, the lack of an effect of SR141716A alone on evoked IPSCs suggests that endogenous CBs do not modulate ongoing GABAergic synaptic transmission under basal conditions in hippocampal slices (Katona et al., 1999). In addition, the inability of naloxone to significantly alter the effect of WIN 55,212–2 indicates that CB1 receptors did not reduce GABA release through either the direct activation of opioid receptors or the stimulation of endogenous opioid release in the hippocampus. This is in contrast to studies showing that such a mechanism exists for the CB-induced increase in dopamine release in the nucleus accumbens (Chen et al., 1990; Tanda et al., 1997).
The effects of CB1 receptors on GABAergic transmission are presynaptic
Several experiments were performed to determine whether the effect of WIN 55,212–2 on GABAergic neurotransmission was presynaptic. First, we compared the effects of WIN 55,212–2 on action potential-dependent sIPSCs with its effects on action potential-independent mIPSCs. These results showed that sIPSCs were inhibited by WIN 55,212–2, and that mIPSCs were completely unaffected. Thus, postsynaptic changes in the sensitivity to GABA or a change in mIPSC kinetics caused by a slowing of the rate of uptake of GABA were not observed (Maneuf et al., 1996). Second, the elimination of the effects of WIN 55,212–2 on spontaneous IPSCs by the VDCC blocker CdCl2 also indicated that the effects of the agonist were presynaptic. Changes in mIPSC frequency are often used to demonstrate the modulation of transmitter release through direct actions on nerve terminals (Cohen et al., 1992;Thompson et al., 1993; Lupica, 1995; Manzoni and Williams, 1999). However, dissociation of the effects of neuromodulators on sIPSCs and mIPSCs can also provide evidence for a presynaptic mechanism of action, because the loss of a drug effect in the presence of TTX may indicate that some action potential-dependent process in the presynaptic neuron was required to observe the modulation (Miller et al., 1997).
Mechanism of presynaptic inhibition of GABA release by CB1 receptors
Despite providing evidence in support of a presynaptic mechanism for WIN 55,212–2, the analysis of sIPSCs and mIPSCs did not distinguish among the several possible ion channel targets of CB1 receptors. Because somatic VDCCs (Shen and Thayer, 1998; Sullivan, 1999) and VDKCs (Deadwyler et al., 1995) have been implicated in the effects of the CBs, we hypothesized that at least one of these classes of ion channels was modulated by WIN 55,212–2. We reasoned that the block of the effect of WIN 55,212–2 by TTX indicated that CB1 receptors inhibited Na+ channels directly or that CB1 receptors inhibited VDCCs or VDKCs that were activated by the Na+ channel-dependent depolarization. Our hypothesis that the inhibition of GABA release occurred as a result of the inhibition of VDCCs was derived from the observations that (1) the inhibitory effect of WIN 55,212–2 on sIPSCs was also eliminated when VDCCs “downstream” of Na+channels were blocked by CdCl2, and (2) the effect of WIN 55,212–2 on sIPSCs was restored when inhibitory terminals were depolarized and VDCCs activated directly by elevated [K+]o during Na+ channel blockade with TTX. However, because distinct classes of VDCCs are known to differentially inactivate according to the level of sustained depolarization, the VDCCs that were recruited by elevated [K+]o may represent only a subset of the VDCCs activated by a brief depolarization, such as that initiated by an action potential (Doze et al., 1995). Nevertheless, our results clearly demonstrate that elevating [K+]orecruited VDCCs that could support GABA release and that these channels were sensitive to inhibition by both CdCl2 and WIN 55,212–2. Thus, although these data do not conclusively establish which VDCCs were involved in this process, they do support the hypothesis that GABA release is inhibited by CB1 receptor modulation of VDCCs. In this way, the modulation of presynaptic VDCCs in the inhibition of GABA release is similar to that described for the CB1-mediated inhibition of glutamate release in hippocampal cultures (Twitchell et al., 1997; Sullivan, 1999).
Although the inhibition of VDCCs represents a likely mechanism for the CB1-mediated inhibition of synaptic GABA release, these experiments did not eliminate the possibility that WIN 55,212–2 may also act on presynaptic K+ channels. This mechanism was important to evaluate because K+channels have been implicated as targets in the actions of other presynaptic modulators of neurotransmitter release (Simmons and Chavkin, 1996; Vaughan et al., 1997). Furthermore, the CB1 receptor is known to activate voltage-dependent K+channels (IA) in hippocampal neurons (Deadwyler et al., 1993, 1995) and voltage-independent, GIRKs in cellular expression systems (Mackie et al., 1995; Matsuda, 1997; Garcia et al., 1998; Jin et al., 1999). Therefore, we assessed the effect of blockade of the IA-like channel with 4-AP and the GIRK channel with BaCl2 on the modulation of evIPSCs by WIN 55,212–2. Although both BaCl2 and 4-AP appeared to block the inhibition of evIPSCs by WIN 55,212–2 at physiological concentrations of Ca2+ (2.4 mm), an important caveat must be considered. Because blockade of presynaptic K+ channels prolongs the depolarization of the nerve terminal, Ca2+ influx is enhanced (Fig. 8). This increase in intraterminal Ca2+ can then “saturate” the neurotransmitter release process (Lupica and Dunwiddie, 1993), confounding the observation of the inhibition of evoked IPSCs, particularly if VDCCs are indeed modulated. In support of this hypothesis, the inhibitory effect of WIN 55,212–2 on evIPSCs was restored when [Ca2+]o was lowered (1.5–2.0 mm) during BaCl2 or 4-AP treatment. Thus, we conclude that neither 4-AP nor BaCl2 blocked the modulation of evIPSCs by WIN 55,212–2. In this respect, our data are similar to previous studies demonstrating that the ability of 4-AP to reduce presynaptic inhibition by adenosine could be reversed by decreasing [Ca2+]o (Klapstein and Colmers, 1992) or by increasing [Mg2+]o (Lupica and Dunwiddie, 1993).
Conclusions
The GABAergic interneurons of the hippocampus play a critical role in the synchronization of pyramidal cell activity and thereby contribute to oscillatory patterns, such as theta rhythm, that are important in the encoding of spatial and sensory information (O'Keefe, 1993; Cobb et al., 1995; Buzsáki, 1997; Paulsen and Moser, 1998). Presynaptic inhibition of GABA release by CBs would therefore be expected to interfere with this synchronization, perhaps explaining the disruptive effects of marijuana on spatial memory and learning tasks (Heyser et al., 1993; Ameri, 1999). Our observation that CBs differentially modulate GABAergic synapses, together with the fact that CBs also inhibit glutamatergic transmission in the hippocampus (Shen et al., 1996; Misner and Sullivan, 1999), suggests that the activation of CB1 receptors is likely to have complex effects on hippocampal circuitry. It remains to be determined whether inhibitory or excitatory synapses display differential sensitivity to CBs or whether the specific Ca2+ channels differ between these populations of synapses. Despite these unresolved issues, it is apparent that the adverse effects of marijuana on cognitive processes are attributable in part to actions on fast synaptic transmission in the hippocampus.
Footnotes
This work was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health. We thank Dr. James A. Bell for his assistance in analyzing the data and in helpful discussions on this manuscript.
Correspondence should be addressed to Dr. Carl R. Lupica, National Institute on Drug Abuse, Intramural Research Program, Building C, Room 267, 5500 Nathan Shock Drive, Baltimore, MD 21224. E-mail:clupica@intra.nida.nih.gov.
REFERENCES
- 1.Alger BE, Nicoll RA. Pharmacological evidence for two kinds of GABA receptor on rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:125–41. doi: 10.1113/jphysiol.1982.sp014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 3.Axelrod J, Felder CC. Cannabinoid receptors and their endogenous agonist, anandamide. Neurochem Res. 1998;23:575–581. doi: 10.1023/a:1022418217479. [DOI] [PubMed] [Google Scholar]
- 4.Breivogel CS, Childers SR. The functional neuroanatomy of brain cannabinoid receptors. Neurobiol Dis. 1998;5:417–431. doi: 10.1006/nbdi.1998.0229. [DOI] [PubMed] [Google Scholar]
- 5.Buhl EH, Cobb SR, Halasy K, Somogyi P. Properties of unitary IPSPs evoked by anatomically identified basket cells in the rat hippocampus. Eur J Neurosci. 1995;7:1989–2004. doi: 10.1111/j.1460-9568.1995.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 6.Buzsáki G. Functions for interneuronal nets in the hippocampus. Can J Physiol Pharmacol. 1997;75:508–515. [PubMed] [Google Scholar]
- 7.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 8.Chan PK, Chan SC, Yung WH. Presynaptic inhibition of GABAergic inputs to rat substantia nigra pars reticulata neurones by a cannabinoid agonist. NeuroReport. 1998;9:671–675. doi: 10.1097/00001756-199803090-00020. [DOI] [PubMed] [Google Scholar]
- 9.Chen JP, Paredes W, Li J, Smith D, Lowinson J, Gardner EL. Delta 9-tetrahydrocannabinol produces naloxone-blockable enhancement of presynaptic basal dopamine efflux in nucleus accumbens of conscious, freely-moving rats as measured by intracerebral microdialysis. Psychopharmacology (Berl) 1990;102:156–162. doi: 10.1007/BF02245916. [DOI] [PubMed] [Google Scholar]
- 10.Cibulsky SM, Sather WA. Block by ruthenium red of cloned neuronal voltage-gated calcium channels. J Pharmacol Exp Ther. 1999;289:1447–1453. [PubMed] [Google Scholar]
- 11.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 12.Cohen GA, Doze VA, Madison DV. Opioid inhibition of GABA release from presynaptic terminals of rat hippocampal interneurons. Neuron. 1992;9:325–335. doi: 10.1016/0896-6273(92)90171-9. [DOI] [PubMed] [Google Scholar]
- 13.Collins DR, Pertwee RG, Davies SN. Prevention by the cannabinoid antagonist, SR141716A, of cannabinoid- mediated blockade of long-term potentiation in the rat hippocampal slice. Br J Pharmacol. 1995;115:869–870. doi: 10.1111/j.1476-5381.1995.tb15889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deadwyler SA, Heyser CJ, Michaelis RC, Hampson RE. The effects of delta-9-THC on mechanisms of learning and memory. NIDA Res Monogr. 1990;97:79–93. [PubMed] [Google Scholar]
- 15.Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- 16.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 17.Doze VA, Cohen GA, Madison DV. Calcium channel involvement in GABAB receptor-mediated inhibition of GABA release in area CA1 of the rat hippocampus. J Neurophysiol. 1995;74:43–53. doi: 10.1152/jn.1995.74.1.43. [DOI] [PubMed] [Google Scholar]
- 18.Drew WG, Miller LL. Cannabis: neural mechanisms and behavior—a theoretical review. Pharmacology. 1974;11:12–32. doi: 10.1159/000136463. [DOI] [PubMed] [Google Scholar]
- 19.Essman EJ. Marijuana intoxication in rats: interruption of recent memory and effect on brain concentration of delta 9-tetrahydrocannabinol. Psychol Rep. 1984;55:563–567. doi: 10.2466/pr0.1984.55.2.563. [DOI] [PubMed] [Google Scholar]
- 20.Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 21.Garcia DE, Brown S, Hille B, Mackie K. Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J Neurosci. 1998;18:2834–2841. doi: 10.1523/JNEUROSCI.18-08-02834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gifford AN, Ashby CRJ. Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212–2, and is potentiated by the cannabinoid antagonist, SR 141716A. J Pharmacol Exp Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- 23.Hampson RE, Deadwyler SA. Role of cannabinoid receptors in memory storage. Neurobiol Dis. 1998;5:474–482. doi: 10.1006/nbdi.1998.0223. [DOI] [PubMed] [Google Scholar]
- 24.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heyser CJ, Hampson RE, Deadwyler SA. Effects of delta-9-tetrahydrocannabinol on delayed match to sample performance in rats: alterations in short-term memory associated with changes in task specific firing of hippocampal cells. J Pharmacol Exp Ther. 1993;264:294–307. [PubMed] [Google Scholar]
- 26.Howlett AC, Bidaut-Russell M, Devane WA, Melvin LS, Johnson MR, Herkenham M. The cannabinoid receptor: biochemical, anatomical and behavioral characterization. Trends Neurosci. 1990;13:420–423. doi: 10.1016/0166-2236(90)90124-s. [DOI] [PubMed] [Google Scholar]
- 27.Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, Mackie K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson SW, Mercuri NB, North RA. 5-Hydroxytryptamine1B receptors block the GABAB synaptic potential in rat dopamine neurons. J Neurosci. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klapstein GJ, Colmers WF. 4-Aminopyridine and low Ca2+ differentiate presynaptic inhibition mediated by neuropeptide Y, baclofen and 2-chloroadenosine in rat hippocampal CA1 in vitro. Br J Pharmacol. 1992;105:470–474. doi: 10.1111/j.1476-5381.1992.tb14277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knowles WD. Normal anatomy and neurophysiology of the hippocampal formation. J Clin Neurophysiol. 1992;9:252–263. [PubMed] [Google Scholar]
- 32.Levenes C, Daniel H, Soubrie P, Crepel F. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol (Lond) 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling DS, Benardo LS. Properties of isolated GABAB-mediated inhibitory postsynaptic currents in hippocampal pyramidal cells. Neuroscience. 1994;63:937–944. doi: 10.1016/0306-4522(94)90561-4. [DOI] [PubMed] [Google Scholar]
- 34.Lupica CR. Delta and mu enkephalins inhibit spontaneous GABA-mediated IPSCs via a cyclic AMP-independent mechanism in the rat hippocampus. J Neurosci. 1995;15:737–749. doi: 10.1523/JNEUROSCI.15-01-00737.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupica CR, Dunwiddie TV. Adenosine modulation of glutamate-mediated synaptic transmission in the hippocampus. In: Dunwiddie TV, Lovinger DM, editors. Presynaptic receptors in the mammalian brain. Birkhauser; Boston: 1993. pp. 104–126. [Google Scholar]
- 36.Lupica CR, Proctor WR, Dunwiddie TV. Dissociation of mu and delta opioid receptor-mediated reductions in evoked and spontaneous synaptic inhibition in the rat hippocampus in vitro. Brain Res. 1992;593:226–238. doi: 10.1016/0006-8993(92)91312-3. [DOI] [PubMed] [Google Scholar]
- 37.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maneuf YP, Crossman AR, Brotchie JM. Modulation of GABAergic transmission in the globus pallidus by the synthetic cannabinoid WIN 55,212–2. Synapse. 1996;22:382–385. doi: 10.1002/(SICI)1098-2396(199604)22:4<382::AID-SYN9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 39.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda LA. Molecular aspects of cannabinoid receptors. Crit Rev Neurobiol. 1997;11:143–166. doi: 10.1615/critrevneurobiol.v11.i2-3.30. [DOI] [PubMed] [Google Scholar]
- 41.Miller KK, Hoffer A, Svoboda KR, Lupica CR. Cholecystokinin increases GABA release by inhibiting a resting K+ conductance in hippocampal interneurons. J Neurosci. 1997;17:4994–5003. doi: 10.1523/JNEUROSCI.17-13-04994.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller LL, Branconnier RJ. Cannabis: effects on memory and the cholinergic limbic system. Psychol Bull. 1983;93:441–456. [PubMed] [Google Scholar]
- 43.Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–6805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray JB. Marijuana's effects on human cognitive functions, psychomotor functions, and personality. J Gen Psychol. 1986;113:23–55. doi: 10.1080/00221309.1986.9710540. [DOI] [PubMed] [Google Scholar]
- 45.Nurse S, Lacaille JC. Do GABAA and GABAB inhibitory postsynaptic responses originate from distinct interneurons in the hippocampus? Can J Physiol Pharmacol. 1997;75:520–525. [PubMed] [Google Scholar]
- 46.O'Keefe J. Hippocampus, theta, and spatial memory. Curr Opin Neurobiol. 1993;3:917–924. doi: 10.1016/0959-4388(93)90163-s. [DOI] [PubMed] [Google Scholar]
- 47.Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998;21:273–278. doi: 10.1016/s0166-2236(97)01205-8. [DOI] [PubMed] [Google Scholar]
- 48.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 49.Pettit DA, Harrison MP, Olson JM, Spencer RF, Cabral GA. Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J Neurosci Res. 1998;51:391–402. doi: 10.1002/(SICI)1097-4547(19980201)51:3<391::AID-JNR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 50.Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 51.Sciancalepore M, Savic N, Gyori J, Cherubini E. Facilitation of miniature GABAergic currents by ruthenium red in neonatal rat hippocampal neurons. J Neurophysiol. 1998;80:2316–2322. doi: 10.1152/jn.1998.80.5.2316. [DOI] [PubMed] [Google Scholar]
- 52.Shen M, Thayer SA. The cannabinoid agonist Win55,212–2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- 53.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shoji Y, Delfs J, Williams JT. Presynaptic inhibition of GABAB-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:2347–2355. doi: 10.1523/JNEUROSCI.19-06-02347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siggins GR, Zieglgansberger W. Morphine and opioid peptides reduce inhibitory synaptic potentials in hippocampal pyramidal cells in vitro without alteration of membrane potential. Proc Natl Acad Sci USA. 1981;78:5235–5239. doi: 10.1073/pnas.78.8.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simmons ML, Chavkin C. κ-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol. 1996;50:80–85. [PubMed] [Google Scholar]
- 57.Solis JM, Nicoll RA. Pharmacological characterization of GABAB-mediated responses in the CA1 region of the rat hippocampal slice. J Neurosci. 1992;12:3466–3472. doi: 10.1523/JNEUROSCI.12-09-03466.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 59.Szabo B, Dorner L, Pfreundtner C, Norenberg W, Starke K. Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rat corpus striatum. Neuroscience. 1998;85:395–403. doi: 10.1016/s0306-4522(97)00597-6. [DOI] [PubMed] [Google Scholar]
- 60.Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common μ1 opioid receptor mechanism. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- 61.Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- 62.Trudeau LE, Doyle RT, Emery DG, Haydon PG. Calcium-independent activation of the secretory apparatus by ruthenium red in hippocampal neurons: a new tool to assess modulation of presynaptic function. J Neurosci. 1996;16:46–54. doi: 10.1523/JNEUROSCI.16-01-00046.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 64.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 65.Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- 66.Vaughan CW, McGregor IS, Christie MJ. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. Br J Pharmacol. 1999;127:935–940. doi: 10.1038/sj.bjp.0702636. [DOI] [PMC free article] [PubMed] [Google Scholar]