

Abstract
Growth cone motility is regulated by changes in actin dynamics. Actin depolymerizing factor (ADF) is an important regulator of actin dynamics, and extracellular signal-induced changes in ADF activity may influence growth cone motility and neurite extension. To determine this directly, we overexpressed ADF in primary neurons and analyzed neurite lengths. Recombinant adenoviruses were constructed that express wild-type Xenopus ADF/cofilin [XAC(wt)], as well as two mutant forms of XAC, the active but nonphosphorylatable XAC(A3) and the less active, pseudophosphorylated XAC(E3). XAC expression was detectable on Western blots 24 hr after infection and peaked at 3 d in cultured rat cortical neurons. Peak expression was ∼75% that of endogenous ADF. XAC(wt) expression caused a slight increase in growth cone area and filopodia but decreased filopodia numbers on neurite shafts. At maximal XAC levels, neurite lengths increased >50% compared with controls infected with a green fluorescent protein-expressing adenovirus. Increased neurite extension was directly related to the expression of active XAC. Expression of the XAC(E3) mutant did not increase neurite extension, whereas expression of the XAC(A3) mutant increased neurite extension but to a lesser extent than XAC(wt), which was partially phosphorylated. XAC expression had minimal, if any, impact on F-actin levels and did not result in compensatory changes in the expression of endogenous ADF or actin. However, F-actin turnover appeared to increase based on F-actin loss after treatment with drugs that block actin polymerization. These results provide direct evidence that increased ADF activity promotes process extension and neurite outgrowth.
Keywords: actin depolymerizing factor, cofilin, growth cones, actin dynamics, phosphorylation, cortical neurons
The regulated assembly and disassembly of F-actin is essential for the formation of appropriate connections during neuronal development, as well as for regenerative growth after injury, because growth cone advance and navigation relies on local alterations of the actin cytoskeleton in response to external cues (Bentley and Toroian-Raymond, 1986; Forscher and Smith, 1988; Fan et al., 1993; Lin and Forscher, 1993; O'Connor and Bentley, 1993). Actin depolymerizing factor (ADF) and cofilin are ∼19 kDa actin-monomer-sequestering, F-actin-depolymerizing proteins that play a substantial role in regulating actin dynamics (Theriot, 1997; Bamburg, 1999). ADF and cofilin are closely related in sequence and have similar activities. Proteins of the ADF/cofilin family are ubiquitously expressed in eukaryotes, with activities conserved across phylogeny. For example, ADF/cofilin mutations in yeast are lethal but rescued by addition of mammalian ADF (Iida et al., 1993). The activity of all vertebrate ADF/cofilin proteins is regulated by phosphorylation. Phosphorylation at a single site (ser3) inhibits its binding to actin monomers and its actin-depolymerizing activity (Morgan et al., 1993;Agnew et al., 1995). ADF is a substrate for LIM kinase (Arber et al., 1998; Yang et al., 1998). In vivo studies underscore the importance of ADF/cofilin phosphorylation. Injections intoXenopus blastomeres of ADF/cofilin mutants that cannot be phosphorylated block cytokinesis (Abe et al., 1996), whereas injections of phosphorylatable ADF have no effect (Abe et al., 1995).
Growth cone motility is regulated by the assembly of actin filaments at the leading edge and disassembly at the central domain (Lin and Forscher, 1995). Several lines of evidence suggest that ADF/cofilins are important regulators of this actin treadmilling and may regulate growth cone motility: (1) in vitro ADF increases actin monomer addition at the barbed ends and loss at the pointed ends of F-actin (Carlier et al., 1997); (2) ADF is essential for actin-based motility of Listeria (Carlier et al., 1997; Rosenblatt et al., 1997); (3) overexpression of cofilin increasesDictyostelium cell motility (Aizawa et al., 1996); (4) ADF binds to actin filaments at the leading edge of fibroblast lamellipodia, especially in labile regions in which actin filament disassembly occurs (Svitkina and Borisy, 1999); (5) ADF and cofilin are abundant in neuronal growth cones (Bamburg and Bray, 1987; Jensen et al., 1993); and (6) signals that influence growth cone motility alter the phosphorylation of ADF (Meberg et al., 1998).
Much of ADF/cofilin in cells is in the phosphorylated, inactive form. Circumstantial evidence from many cell types suggests that signal-induced ADF/cofilin dephosphorylation is important for regulating actin dynamics and altering cell morphology. For example, dephosphorylation occurs in response to nerve growth factor stimulation of PC12 cells, and increases in both intracellular calcium and cAMP increase dephosphorylation in neurons (Meberg et al., 1998). In general, signals that promote process extension decrease ADF/cofilin phosphorylation (Meberg et al., 1998). However, to date no direct evidence exists that ADF/cofilin regulates actin dynamics or process extension in neurons. To directly test this, recombinant adenoviruses were constructed to overexpress ADF/cofilin in primary neurons and assess effects on neurite extension.
MATERIALS AND METHODS
Recombinant adenovirus construction and infection.The cDNAs containing the coding regions for wild-typeXenopus ADF/cofilin [XAC(wt)], the mutant less active, pseudophosphorylated form of XAC(wt) [XAC(E3)], the mutant active, nonphosphorylatable form of XAC(wt) [XAC(A3)], enhanced green fluorescent protein (GFP) (Clontech, Palo Alto, CA), and XAC(E3)-GFP were subcloned into the shuttle plasmid pACCMVpLpA (Gómez-Foix et al., 1992) between the cytomegalovirus (CMV) promoter and the SV40 polyadenylation sequence. Replication-defective recombinant adenoviruses were prepared by homologous recombination between the pJM17 plasmid (Microbix Biosystems Inc., Toronto, Canada) (McGrory et al., 1988) and the left-end shuttle plasmid pACCMVpLpA in 293 cells, using methods similar to those described by others (Gómez-Foix et al., 1992). The resultant recombinant adenoviruses were plaque-purified three times, expanded, and titered. Recombinant adenoviruses were screened for expression of the introduced genes by fluorescent microscopy and/or Western blot analysis.
For infection, recombinant adenoviruses were added to the culture medium, and 16–20 hr later, approximately half of the medium was replaced with fresh medium. For primary neuronal cultures, infection was performed 4 hr after plating. For cell lines, infection was typically performed 24 hr after the cells were split.
Protein isolation and Western blot analysis. Cells were rinsed four times with ice-cold PBS before addition of SDS lysis buffer (Morgan et al., 1993). Cell lysates were then scraped from the dishes, heated in a boiling water bath for 5 min, and sonicated, and the proteins were precipitated after chloroform–methanol extraction of lipids (Wessel and Flügge, 1984). Proteins were resuspended in the appropriate buffer for subsequent gel electrophoresis, and the protein concentration was determined using a filter paper dye-binding assay (Minamide and Bamburg, 1990). SDS-PAGE on 15% polyacrylamide gels, Western blotting, and densitometry were performed as described previously (Bamburg et al., 1991; Morgan et al., 1993). For two-dimensional gels, nonequilibrium pH gradient electrophoresis was used in the first dimension.
For determination of soluble versus insoluble actin in cells, a Triton X-100 extraction procedure was used (Minamide et al., 1997), and then the proteins were processed as described above. Briefly, 60 mm dishes were washed four times with PBS then extracted 1 min in 450 μl of 50 mm MES, pH 6.5, 1 mm EGTA, 50 mm KCl, 1 mm MgCl2, 1 mm PMSF, 10 mm NaF, 0.5% Triton X-100, and 0.5% protease inhibitor cocktail (Minamide et al., 1997). The Triton extraction buffer (the soluble fraction) was then removed and placed in a tube containing 20% SDS. After removal of the extraction buffer, the Triton-insoluble cytoskeletal fraction was obtained by scraping the dish in SDS lysis buffer.
Proteins were electroblotted onto polyvinylidene difluoride membranes (Gelman Sciences, Ann Arbor, MI) and then immunostained with one of the following primary antibodies: (1) rabbit antiserum to chick ADF, which recognizes both phosphorylated (pADF) and unphosphorylated forms of ADF (Morgan et al., 1993); (2) mouse monoclonal antibody to cofilin (mAb-22), which recognizes both phosphorylated and unphosphorylated forms of cofilin (Abe et al., 1989); (3) rabbit antiserum to pAC, an antibody specific for the phosphorylated forms of ADF, cofilin, and XAC (Meberg et al., 1998); (4) rabbit antiserum to XAC, which recognizes XAC but not ADF or cofilin on Western blots (Fig.1); and (5) monoclonal antibody C4 to actin (ICN, Costa Mesa, CA). The secondary antibodies used were alkaline phosphatase-conjugated goat anti-rabbit or goat anti-mouse IgGs (Sigma, St. Louis, MO). Chemiluminescent detection with CDP-Star (Tropix) was followed by staining with nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphatep-toluidine salt (Life Technologies, Gaithersburg, MD). Quantification of spot or band densities from blots was performed as described previously (Meberg et al., 1998). Protein concentrations were determined by comparison with recombinant ADF and XAC standards run on each gel.
Fig. 1.
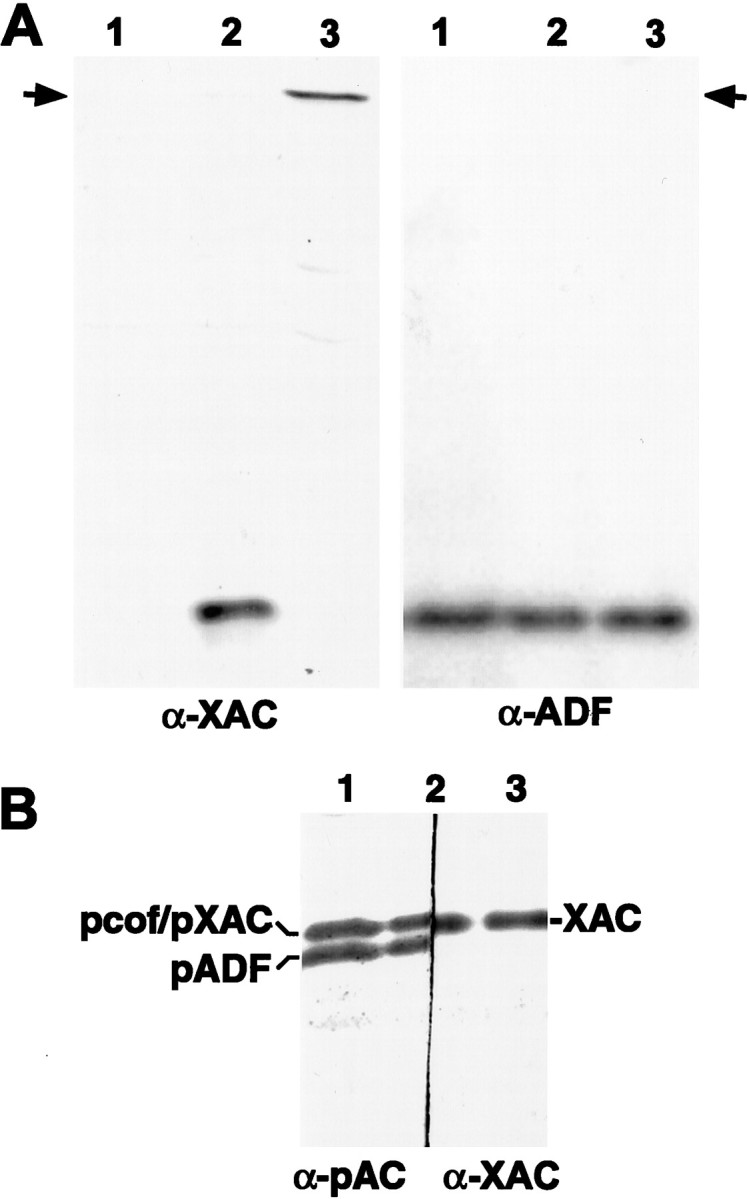
Specificity of ADF and XAC antibodies. Swiss 3T3 cells were infected with recombinant adenoviruses at an MOI of 100 pfu/cell, and cell extracts were harvested 2 d later.A, Duplicate protein blots were immunostained with the XAC (α-XAC) or the ADF (α-ADF) antibody. Uninfected control cultures (lane 1) exhibited no immunoreactivity to the XAC antibody (left panel) but strong immunoreactivity to the ADF antibody (right panel), indicating that the XAC antibody does not recognize rodent ADF. Cells infected with adXAC(wt) (lane 2) or adXAC(E3)-GFP (lane 3) did exhibit XAC immunoreactivity (left panel), indicating that XAC was expressed by the recombinant adenoviruses. Duplicate samples from these adenovirus-infected cells exhibited ADF immunoreactivity at levels equivalent to uninfected controls (right panel). Therefore, the ADF antibody did not appear to recognize XAC, because no immunoreactivity was apparent at the location of the XAC(E3)-GFP protein (indicated by arrow) and only a single band appeared in extracts from cells expressing XAC(wt). The XAC(E3)-GFP chimera migrated with an apparent mass of 45 kDa. B, The relative electrophoretic mobility of XAC compared with rodent ADF/cofilin was determined by cutting a protein blot in half and then using XAC (right side) or pAC (left side) antibodies on the respective halves. The same sample from adXAC(wt)-infected cells was run in all three lanes, and the blot cut down the middle of lane 2. The pAC antibody recognizes only the phosphorylated forms of ADF (pADF) and cofilin (pcof), which are two separate bands (Meberg et al., 1998). XAC had an electrophoretic mobility similar to pcofilin and slower than pADF. Therefore, the pAC antibody cannot discriminate between phosphorylated XAC and phosphorylated cofilin on immunoblots from one-dimensional gels. Again, the XAC antibody did not cross-react with ADF, or a doublet would have appeared. A total of 8 μg of protein was loaded in each lane on all of these blots.
Cell culture. Primary cultures of cortical neurons were prepared from embryonic day 18 fetal rats. Cortices were minced and then incubated for 15 min in HBSS containing 10 mm HEPES, 0.2% trypsin, and 1 mg/ml DNase I (Sigma). After three rinses in DMEM–10% FBS, the tissue was triturated with a fire-polished Pasteur pipette. Cell aliquots were slowly frozen in DMEM–10% FBS containing 8% DMSO and stored in liquid nitrogen for later use. Thawed cells were diluted in DMEM–10% FBS and plated on poly-d-lysine-coated Petri dishes or glass coverslips. After 2–4 hr, the medium was replaced with Neurobasal medium containing B27 supplements (Life Technologies).
Primary cultures of spinal cord neurons were removed from 6 or 7 d chick embryos and incubated for 10 min in F-12 media containing 0.2% trypsin with EDTA (Sigma). The medium was then replaced with DMEM–10% FBS, and the tissue was triturated with a fire-polished Pasteur pipette. Cells were preplated for 2 hr to remove non-neuronal cells and then plated onto poly-d-lysine-coated culture dishes in DMEM–10% FBS containing uridine (300 μm; Sigma) and 5′-fluoro-2′-deoxyuridine (120 μm; Sigma) as an antimitotic agent.
Quantitative analyses of neuronal morphology and phalloidin labeling. Cells were typically fixed 15–30 min with 4% paraformaldehyde–0.1% glutaraldehyde in PBS at room temperature. For better preservation of filopodia and growth cone morphology, some cultures were fixed in 2% glutaraldehyde in PBS. Quantitative analysis of neuronal morphology and fluorescence intensity was performed using Metamorph (Universal Imaging, West Chester, PA) or Olympix 2000 (Olympus America, Melville, NY) software on digitally captured images. Isolated neurons were selected for analysis so that individual neurites could be traced without overlap or fasciculation with other neuronal processes. Morphology measures were obtained by analyzing >100 neurons in two to three different culture dishes, unless otherwise noted. Cultures were not analyzed if cell densities were reduced in control cultures because of toxic effects of the adenovirus.
Phalloidin was used to label F-actin in growth cones in fixed cultures. After fixation, cultures were permeabilized in 0.1% Triton–PBS, incubated for 15 min with 1 U of Texas Red-phalloidin (Molecular Probes, Eugene, OR), and then washed with PBS before mounting in Prolong Anti-fade (Molecular Probes). Maximum fluorescence in growth cones was measured as the brightest four-pixel region within the growth cone. For tubulin localization, fixed and permeabilized cultures were blocked 60 min with 2% goat serum and 1% bovine serum albumin in PBS before a 60 min incubation with a combination of monoclonal antibodies to β-tubulin acquired from Sigma (clone TUB 2.1) and Amersham Pharmacia Biotech (Arlington Heights, IL). Cultures were then incubated with an Alexa 488-conjugated goat anti-mouse IgG secondary antibody (Molecular Probes) to enable colocalization studies of tubulin with F-actin.
Determination of F-actin stability. For some assays of F-actin turnover–stability, cultures were treated with latrunculin A (LatA) (Molecular Probes) or cytochalasin D (Sigma) and then extracted for 3 min in Triton-extraction buffer (1% Triton X-100, 100 mm PIPES, pH 6.9, 1 mmMgCl2, and 1 mm EGTA) containing 0.5 μm Texas Red-phalloidin to stabilize actin filaments, as described by Svitkina and Borisy (1999). Briefly, a ½ vol of medium containing a 3× concentration of drug was added to cultures for 45 sec to 10 min, and then the medium was removed and replaced with Triton extraction buffer. Cultures were then washed twice in the same buffer without Triton X-100 before a 12–15 min fixation in 4% paraformaldehyde–1% glutaraldehyde. For drug treatments of ≤1 min duration, 37°C medium containing the drug was added to the culture dish, and the dish was left at room temperature until fixation or extraction. For longer drug incubations, culture dishes were returned to the 37°C incubator.
RESULTS
To overexpress ADF/cofilin in neurons, recombinant adenoviruses were constructed containing the coding regions for XenopusADF/cofilin (XAC) cDNAs preceded by a CMV promoter. XAC constructs used were as follows: (1) XAC(wt); (2) the active XAC(A3) mutant (serine3 replaced by alanine), which cannot be regulated by phosphorylation; and (3) the minimally active XAC(E3) mutant (serine3 replaced by glutamate), which is a phosphorylation mimic. Adenoviral-mediated expression of XAC rather than chick ADF allows for expression levels of the introduced ADF/cofilin construct in chick and rodent cells to be assessed independently from endogenous ADF and cofilin, because antibodies that recognize chick and rodent ADF do not cross-react with XAC (Fig. 1). A green fluorescent protein-expressing recombinant adenovirus (adGFP) and an adenovirus containing an inactive XAC(E3)-GFP chimera served as controls. The binding of XAC(E3)-GFP to actin is nearly undetectable when compared with XAC(wt), as determined by DNase I affinity chromatography of extracts from recombinant adenovirus-infected Swiss 3T3 cells (data not shown).
Chick spinal cord neurons infected with GFP- or XAC(wt)-expressing adenoviruses survived and appeared healthy at least 4 d after infection, even when the multiplicity of infection (MOI) was as high as 300 pfu/cell. Rat cortical cultures also remained viable at least 4 d after infection, but only if the MOI was no greater than 150 pfu/cell. At higher concentrations of virus (200–300 pfu/cell), cell death was readily apparent by decreases in cell density 2 d after infection. Adenoviral toxicity was similar for both the GFP- and XAC(wt)-expressing adenoviruses, so it is unlikely that the expression of XAC(wt) was itself responsible for the cell death.
The general morphology of neurons in adXAC(wt)-infected cultures was similar to uninfected or adGFP-infected controls. No obvious changes in cell size, neurite number or thickness, or growth cone morphology were observed (Fig. 2). The number of long neurites in culture did, however, appear to be greater in adXAC(wt)-infected cultures (see below). When growth cones from neurons in adXAC(wt)-infected cultures (150 pfu/cell) were viewed live, they had highly motile filopodia and lamellipodia that could not be qualitatively differentiated from those of controls. Filopodia and lamellipodia were highly dynamic, exhibiting rapid extension and disappearance. Tracking the turnover of individual filopodia was unfeasible, because filopodia often detached from the surface and went out of the plane of focus. Growth rates were not assayed on live growth cones because the average rate of extension was <2 μm/hr.
Fig. 2.
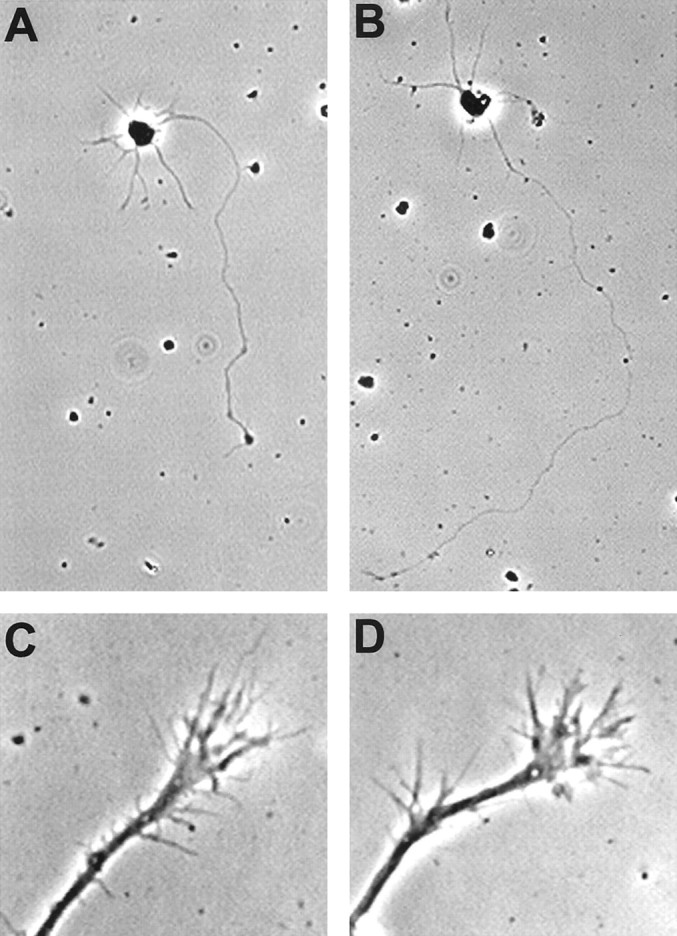
Morphology of rat cortical neurons expressing XAC(wt). Typical isolated neurons are shown, which were grown in culture for 3 d and were either not infected (A) or infected with the XAC(wt)-expressing recombinant adenovirus at 150 pfu/cell (B). Neurons shown were from fixed, low-density cultures. Growth cones of neurons infected with the recombinant adenoviruses had motile growth cones with dynamic lamellipodia and numerous labile filopodia (C, D). Growth cones are shown from presumptive axons of pyramidal neurons 3 d after infection with the GFP-expressing (C) or the XAC(wt)-expressing (D) adenoviruses. Images were taken of growth cones from live neurons.
To better assay possible effects of XAC(wt) expression on growth cone morphology, neurons were fixed, and filopodia number and growth cone area were analyzed. Although changes were relatively small, expression of XAC(wt) consistently increased growth cone area compared with controls (+14.1 ± 2.6%; mean of three independent experiments;p < 0.05; paired t test), as well as the number of growth cone filopodia (+11.6 ± 2.5%; p< 0.01). In contrast to the increase in filopodia number in growth cones, there was a consistent decrease in filopodia number on the neurite shaft within 30 μm of the growth cone (−11.1 ± 4.2% compared with controls; p < 0.05).
Expression levels of XAC constructs and ADF in cultured cells
The amount of XAC(wt) expressed in neuronal cultures increased with the amount of adenovirus added (Fig.3). At 3 d after infection, XAC(wt) expression in chick spinal cord neurons was barely detectable at 50 pfu/cell but increased with higher MOI (Fig. 3A). At 200 pfu/cell, XAC(wt) expression reached 38% of the level of endogenous ADF in the same sample (nearly 0.02% of total protein). XAC(wt) expression was, however, much higher in rat cortical neurons. At 3 d after infection, XAC(wt) expression was clearly evident at 50 pfu/cell and increased with higher MOI (Fig. 3B). At 150 pfu/cell, the protein concentration of XAC(wt) was nearly 0.04% of total protein, with levels 76% that of endogenous ADF (Fig.3B). Because adenoviral-mediated expression of XAC(wt) was much higher in rat cortical neurons than in chick spinal cord neurons, rat cortical neurons were the primary cell type used for further experimentation.
Fig. 3.

Levels of adenoviral-mediated XAC(wt) expression compared with endogenous ADF levels. XAC(wt) expression increased with the addition of increasing amounts of the recombinant adenovirus in both chick spinal cord neurons (A) and rat cortical neurons (B). However, relative XAC(wt) expression in rat cortical neurons was higher than in chick spinal cord neurons infected with the equivalent amount of adenovirus. Theinset in A is a representative Western blot from which the graphed data were obtained. Data shown are from duplicate cultures harvested 3 d after infection (error bars indicate range), except for a single sample from cortical cultures infected with 150 pfu/cell (B). The time course of adenoviral-mediated XAC expression is shown in rat cortical neurons infected with 100 pfu/ml recombinant adenoviruses expressing either XAC(wt) (C) or XAC(A3) (D). Duplicate culture dishes were harvested at all time points (error bars indicate range). The inset Western blot (C) shows XAC(wt) expression from 1–4 d after infection. Control samples (con) infected with the GFP-expressing adenovirus exhibit no XAC immunoreactivity. A graphical representation of the Western blot is shown in C, together with endogenous ADF levels from the same samples.D, The time course of XAC(A3) and endogenous ADF expression is shown.
XAC(wt) expression was low, but evident, 1 d after infection with 100 pfu/cell of adXAC(wt) (Fig. 3C). Expression increased and peaked at 3 d after infection and then declined by 4 d after infection (Fig. 3C). A similar time course was found for XAC(A3) expression, with peak levels at 3 d after infection and then a decline (Fig. 3D). Expression of endogenous ADF did not change with days in culture and was unaffected by changes in expression of the XAC constructs over time (Fig.3C,D), indicating that compensatory changes in ADF expression are not induced by XAC expression in neurons.
Expression levels of the mutant XAC constructs were similar to those of XAC(wt) when rat cortical neurons were infected with the same MOI (Fig.4A). Expression of all constructs was ∼75% that of endogenous ADF. It was a surprise, however, to find that the expression of the XAC(E3)-GFP chimera doubled that of the XAC constructs (Fig. 4A). This higher expression of the GFP chimera was also observed when chick spinal cord neurons were infected (data not shown) and might be attributable to a slowed rate of protein degradation. Expression of XAC(wt), XAC(A3), or XAC(E3) did not affect expression levels of endogenous ADF (Fig.4B) or cofilin (data not shown).
Fig. 4.
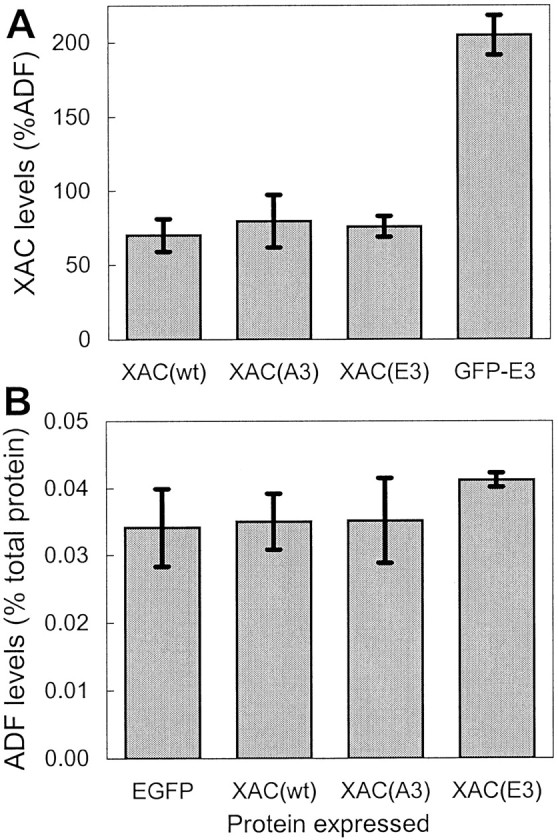
Expression of XAC mutants and effects on endogenous ADF levels. Rat cortical neurons were infected with 100 pfu/cell of the respective recombinant adenoviruses and then harvested 3 d after infection. A, The levels of the XAC(A3) and XAC(E3) mutants were similar to that of XAC(wt). However, the GFP-XAC(E3) chimera was expressed at higher levels than the nonfusion proteins based on relative XAC immunoreactivity. B, The expression of the XAC constructs did not affect endogenous ADF levels, because endogenous ADF levels were similar to those of neurons infected with the control GFP-expressing adenovirus. Samples from duplicate cultures are shown (error bars indicate range).
Effects of ADF/cofilin overexpression on neurite extension
To determine whether increased expression–activity of ADF/cofilin increases neurite outgrowth, low-density cultures of rat cortical neurons were infected with the XAC(wt)-expressing adenovirus 4 hr after plating. After 3 d in culture, the neurons were fixed, and the length of the longest neurite was measured on isolated neurons. Ectopic expression of XAC(wt) increased the length of the longest neurite, with increasing amounts of adenovirus causing a greater increase in length (Fig. 5A). This increase was specific to XAC(wt) expression, because infection with the inactive XAC(E3)-GFP-expressing adenovirus at the same MOI did not increase lengths compared with uninfected controls (Fig.5B).
Fig. 5.
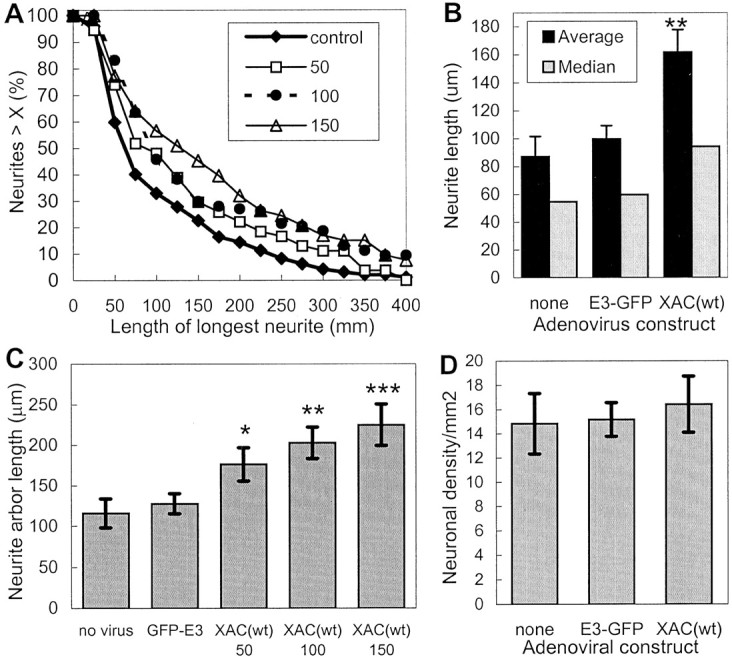
Increased neurite extension induced by the expression of XAC(wt). The length of the longest neurite was measured for rat cortical neurons. All analyses were performed blind of the experimental treatment and taken from a single experiment.A, The length distribution is shown from neurons infected with adGFP-XAC(E3) at 100 pfu/cell (control) or increasing multiplicities of infection with adXAC(wt) (shown in the legend as plaque-forming units per cell). Infection at 50 pfu/cell increased neurite lengths, as shown by a shift to the right of the length distribution curve. Infection at 150 pfu/cell increased lengths even further, with the length distribution curve from neurons infected at 100 pfu/cell falling in between or overlapping that of cultures infected at 50 or 150 pfu/cell. B, Average and median neurite lengths are shown for uninfected cultures and cultures infected with GFP-XAC(E3) and XAC(wt)-expressing adenoviruses at 100 pfu/cell. C, The total arbor length, measured by tracing the length of all branches on the longest neurite, is shown for cultures infected with control adenoviruses (100 pfu/cell) or varying MOIs of adXAC(wt). Statistically significant differences between GFP-E3 and XAC-infected cultures are indicated (*p < 0.05; **p < 0.01; ***p < 0.01; t tests).D, Infection with the adenoviruses did not affect cell densities. Measures were taken from eight randomly selected 1.2 mm2 areas/treatment. Error bars indicate SEM.
ADF/cofilin overexpression could indirectly increase neurite length if neurite branching was reduced. To determine this, the entire neurite arbor was measured by adding up the length of all of the branches on the neurite. Ectopic expression of XAC(wt) resulted in an increase in total arbor length (Fig. 5C), indicating that overexpression of ADF/cofilin increases neurite extension rather than simply decreasing branching. The total number of neurites emanating from each neuron was not altered with XAC(wt) expression (data not shown). Such alterations in the number of neurites would not be expected because neurite initiation occurs before significant expression of XAC(wt), which requires at least 1 d in culture (Fig. 3).
Differences in cell density could also potentially influence length measures, because longer neurites would be more likely to cross other neurites in higher density cultures and therefore not be selected for measurement. However, neuronal densities were similar among uninfected cultures and cultures infected with different adenoviruses (Fig.5D), indicating that measured differences in lengths were not attributable to differences in cell density.
Although the previous length results are from a single experiment, similar results were obtained in several experiments. Ectopic expression of XAC(wt) consistently increased average and median neurite length. However, the effects of XAC(wt) expression on neurite length were more similar and reproducible when median rather than average lengths were compared, because the median length distribution is less affected by experimental differences in neuronal density or outgrowth than average lengths. Median neurite lengths were increased an average of 42 ± 5% (±SEM; four experiments) when neurons were infected with adXAC(wt) at 100 pfu/cell and 62 ± 16% (±SEM; four experiments) when infected at 150 pfu/cell. Overall, the greater the expression of XAC(wt), as determined by a higher multiplicity of infection (Fig. 3), the greater the increase in length. Median neurite length also increased in chick spinal cord neurons infected with adXAC(wt) but only by 13%, again perhaps because of the lower expression level of XAC(wt).
Although the increases in median neurite length in rat cortical neurons were substantial, they may underestimate the effect of ADF/cofilin overexpression on neurite outgrowth. Up to 1 d after infection, XAC(wt) is likely expressed at levels too low to influence outgrowth (Fig. 3C). As expected, at 1 d after infection, neurites of adXAC(wt)-expressing neurons were similar in length to neurites of adGFP-expressing neurons (Fig.6A). However, by 3 d after infection, the median neurite length of adXAC(wt)-expressing neurons was nearly 50% greater than that of adGFP-expressing controls. If the median lengths of neurons at 1 d after infection are subtracted from that at 3 d after infection, then the increase in neurite extension propelled by XAC(wt) expression would have been nearly 80% between 1 and 3 d after infection.
Fig. 6.

Comparison of neurite lengths at different times after adenovirus infection and effects of XAC phosphorylation site mutants. A, Median neurite lengths are shown 1 and 3 d after infection with XAC(wt)- and GFP-expressing adenoviruses at 100 pfu/cell. At 1 d after infection, adXAC(wt)-infected neurons were of similar length to that of adGFP-infected controls. At this time, XAC(wt) levels are still very low in the neurons (see Fig.4). By 3 d after infection, XAC(wt)-expressing neurons have much longer neurites than that of GFP-expressing controls (p < 0.05; t test).B, C, Adenoviral-mediated expression of the constitutively active XAC(A3) mutant increased neurite length but to a lesser extent than XAC(wt). Expression of the inactive XAC(E3) mutant did not affect neurite length. Rat cortical cultures were infected at 150 pfu/ml. A, Results shown are the averages from two independent experiments (error bars indicate range). Median lengths were normalized so that control cultures infected with GFP were at 100%. B, The length distribution shown is from a single experiment. Expression of XAC(wt) increased neurite lengths, as shown by a shift to the right of the length distribution curve compared with neurons expressing XAC(E3). XAC(A3) expression increased the numbers of neurites of intermediate length, but unlike XAC(wt), did not appreciably increase the frequency of longer neurites. At least 150 neurons were analyzed per adenovirus per experiment.
Only active forms of XAC increased neurite length when expressed in cortical neurons. Neurite lengths in neurons expressing the inactive XAC(E3) mutant were the same as in cultures infected with adGFP (Fig.6B,C). Expression of constitutively active XAC(A3) increased median neurite length by 17%, but this was less than observed for XAC(wt) (Fig.6B,C).
Phosphorylation of Xenopus ADF/cofilin in rat neurons
Constitutively active XAC(A3) had less effect on neurite outgrowth than XAC(wt), although XAC(wt) can be inhibited by phosphorylation and the expression level of the two proteins were similar (Fig. 4). To determine whether XAC(wt) is phosphorylated in rat neurons, two-dimensional immunoblots were performed. In uninfected and adGFP-infected cultures, ∼20% of endogenous ADF was phosphorylated (Fig. 7A). When neurons expressed XAC(wt), endogenous ADF phosphorylation increased slightly, but the extent of phosphorylation remained much less than that of XAC(wt) (Fig. 7). Approximately 75% of XAC(wt) was phosphorylated at either 2 or 3 d after infection.
Fig. 7.

Phosphorylation of XAC in rat cortical neurons. Protein blots from two-dimensional gels were immunostained simultaneously with both the XAC and ADF antibodies. Control cultures infected with adGFP exhibit two immunoreactive spots corresponding to phosphorylated (pADF) and unphosphorylated ADF (top). The more acidic phosphoprotein is shown on the left. XAC(wt) expressed in these cells is phosphorylated, as shown by the two additional immunoreactive spots appearing in extracts from adXAC(wt)-infected cells. Theleft spot corresponds to the phosphorylated form of XAC, and the right to the dephosphorylated form (Abe et al., 1996). Extracts were taken from cells harvested 2 d after infection.
Effects of ADF/cofilin overexpression on F-actin levels and F-actin stability
Because ADF/cofilins regulate actin dynamics, the greater total ADF/cofilin expression in neurons expressing XAC likely influences neurite outgrowth through changes in actin polymerization in the growth cones. Therefore, F-actin levels in growth cones were analyzed by labeling with Texas Red-phalloidin. Growth cones from neurons infected with adXAC(wt) exhibited a 17% decrease in maximal phalloidin labeling when compared with neurons from control cultures infected with adGFP (Figs. 8A,9). The average intensity of phalloidin fluorescence in growth cones was also lower in adXAC(wt)-infected neurons (data not shown). However, because ADF/cofilin binding to F-actin blocks phalloidin binding (McGough et al., 1997), the decrease in labeling may simply be a reflection of higher levels of active ADF/cofilin present because of XAC expression. Therefore, an alternative method for assaying F-actin levels was also used. Triton X-100 extraction of cultured neurons was used to determine levels of actin in the Triton-soluble versus the Triton-insoluble cytoskeletal fraction containing F-actin. Expression of XAC(wt) decreased the amount of actin in the Triton-insoluble fraction by <10% (Fig.8B). Therefore, XAC(wt) expression may decrease F-actin levels but to a limited extent. Decreases in F-actin levels could be attributable to changes in actin polymerization or simply reflect a decrease in total actin levels. Total actin levels did not decline after adXAC(wt) infection; at 3 d after infection, actin levels were at 102.8 ± 6.5% of controls (average ± SEM;n = 6, pooled from three experiments).
Fig. 8.
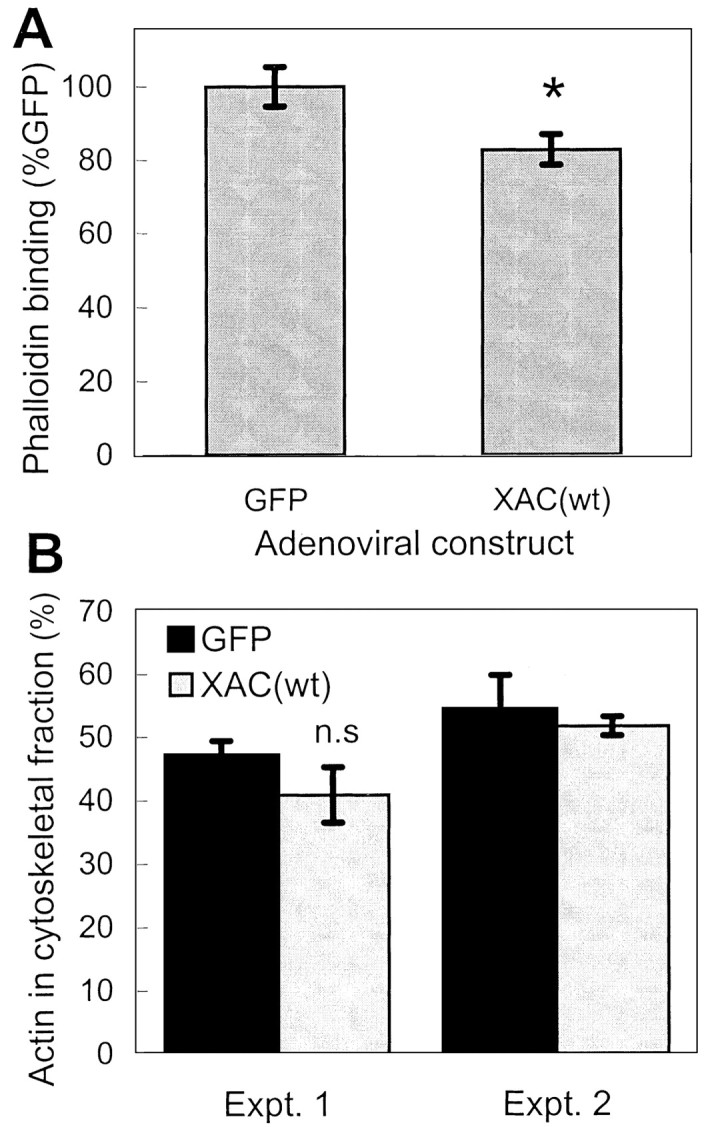
Changes in F-actin levels in neurons expressing XAC(wt). A, Three days after recombinant adenovirus infection, rat cortical cultures were fixed and F-actin was stained with Texas Red-phalloidin. Peak fluorescence intensity measured in growth cones indicated a decrease in phalloidin binding in adXAC(wt)-infected neurons compared with adGFP-infected controls (n > 30; *p < 0.05).B, A second method to determine F-actin levels was to use Triton X-100 to extract soluble actin and compare this with levels of actin in the Triton-insoluble, F-actin-containing fraction. Results from two separate experiments are shown (Expt. 1,n = 4 per group; Expt. 2,n = 2 per group). Differences were not statistically significant (n.s.) in experiment 1.
Fig. 9.
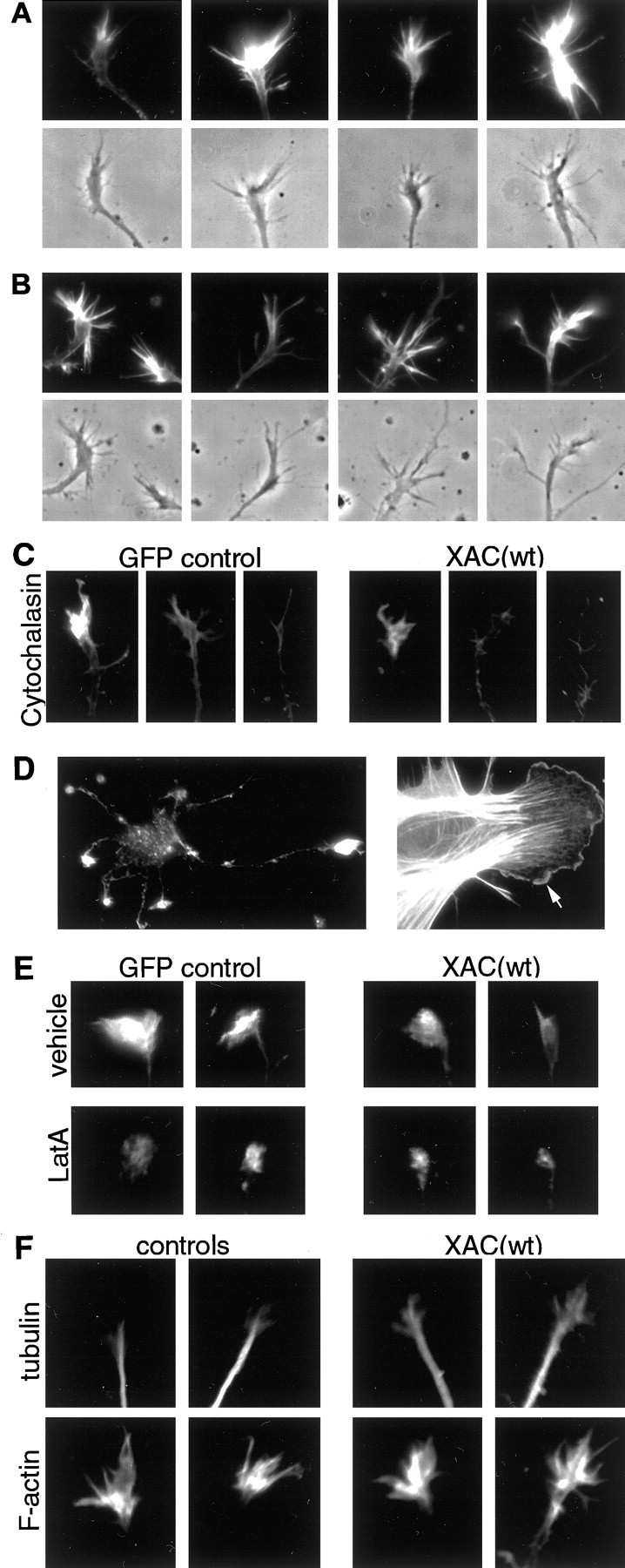
Effects of XAC(wt) expression on the cytoskeleton in growth cones. Representative growth cones shown were treated and fixed 3 d after infection. A, B, Texas Red-phalloidin labeling of growth cones from adenovirus-infected neurons. Growth cones are shown from rat cortical neurons infected with the GFP-expressing control adenovirus (A) or the XAC(wt)-expressing adenovirus (B). No obvious changes in growth cone morphology were evident when XAC(wt) was expressed, although slight decreases in fluorescence were observed (see Fig. 8A). Fluorescence and phase-contrast images are shown for each growth cone. C, Growth cones are shown from neurons treated with 1.0 μm cytochalasin D for 45 sec and then fixed and labeled with Texas Red-phalloidin. Loss of F-actin from growth cones was greater in XAC(wt)-expressing neurons (right panels) than in GFP-expressing controls (left panels). Although greatly reduced in intensity and number, some neurons retained diminished phalloidin staining and filopodia, whereas others lost nearly all phalloidin staining and/or had completely collapsed growth cones. Examples of growth cones within each category are shown.D, Photomicrographs of cells extracted with Triton X-100 in the presence of rhodamine-phalloidin (see Materials and Methods). The Triton-extraction procedure preserved the general morphology of neurons, as well as phalloidin staining in growth cones, although growth cone morphology was compromised. In contrast, fibroblast-like cells retained excellent lamellipodia morphology, as well as enriched F-actin labeling at the leading edge (see arrow).E, Photomicrographs are shown of growth cones from Triton-extracted neurons labeled with rhodamine-phalloidin. Phalloidin staining was reduced in neurons expressing XAC(wt) (right panels) when compared with GFP-expressing controls (left panels). Treatment with 1.0 μm LatA for 3 min before Triton extraction (bottom panels) reduced total phalloidin labeling compared with vehicle-treated controls (top panels). LatA reduced both the intensity and the area of F-actin labeling, with the majority of XAC(wt)-expressing neurons exhibiting no remaining phalloidin labeling (see Fig.10B). F, Neurons were double-labeled with an antibody against tubulin (top panels) and Texas Red-phalloidin (bottom panels). Tubulin immunoreactivity was greater within growth cones of neurons expressing XAC(wt) (right panels) compared with uninfected controls (left panels).
Although XAC(wt) expression did not significantly affect total F-actin levels, it might still increase F-actin turnover or instability in growth cones. To determine this, cultures were treated with agents that block F-actin polymerization. The tip of the longest neurite on each neuron was then scored for the presence of an intact growth cone based on the presence of F-actin. Growth cones were considered “intact” if the neurite tip was wider than the neurite shaft and phalloidin fluorescence was at least three times the intensity found in the neurite shaft. Cyochalasin D caps the plus ends of actin filaments, which leads to F-actin depolymerization and growth cone collapse. When treated with 1 μm cytochalasin D for 45 sec, cortical cultures expressing XAC(wt) had a greater loss of intact growth cones than those expressing GFP (Figs. 9C,10A).
Fig. 10.
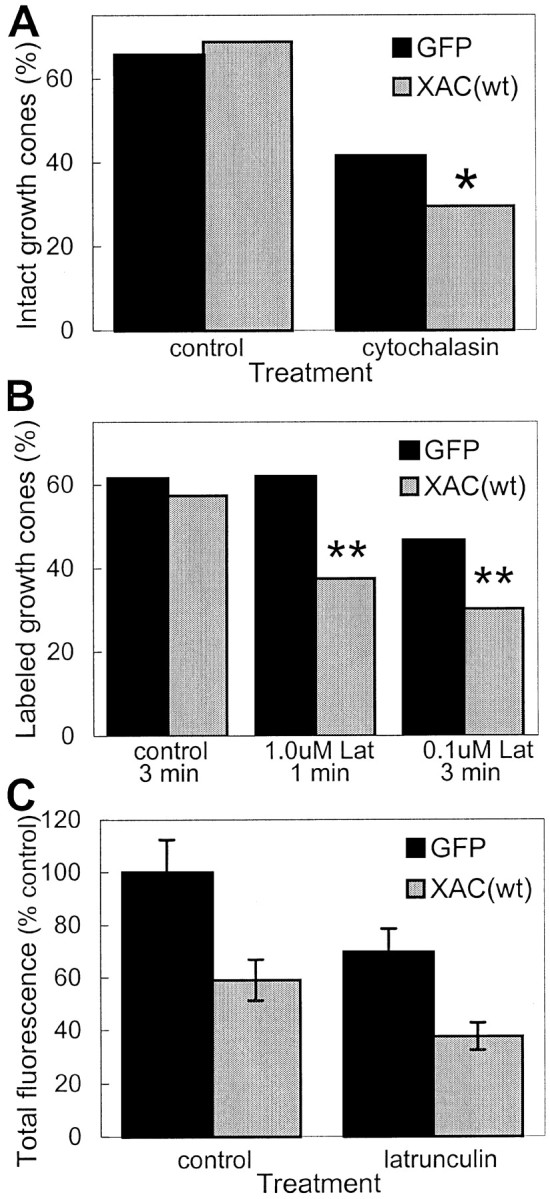
Decreased stability of actin filaments in growth cones of XAC(wt)-expressing neurons. A, Treatment of rat cortical neurons with 1.0 μm cytochalasin D for 45 sec caused a greater loss of intact growth cones in neurons infected with adXAC(wt) than those infected with adGFP (*p < 0.05; χ2 test). The percentage of intact growth cones was similar in vehicle-treated control cultures not exposed to cytochalasin D. Growth cones were considered intact if the neurite tip was wider than the neurite shaft and phalloidin fluorescence was at least three times the intensity found in the neurite shaft. The longest neurite on each neuron was assayed. B, Neurons expressing XAC(wt) were more susceptible to losing identifiable, phalloidin-labeled growth cones than GFP-expressing controls after LatA exposure. Cells were exposed to LatA or control medium and then Triton-extracted in the presence of phalloidin (see Materials and Methods). Treatment of GFP-expressing neurons with 1.0 μmLat A for 1 min had almost no effect on the percentage of labeled growth cones but decreased labeled growth cones by more than a third in XAC(wt)-expressing neurons (**p < 0.01; χ2 test). Treatment with LatA for a longer duration (3 min), even at lower concentration (0.1 μm), caused a greater decline in phalloidin labeling, with the effect again being more pronounced on XAC(wt)-expressing neurons when compared with GFP-expressing ones (**p < 0.01; χ2test). C, Relative fluorescence in growth cones after the Triton-extraction procedure is shown. Averages are expressed relative to total fluorescence in adGFP-infected cultures not exposed to LatA. Because phalloidin fluorescence of growth cones could only be detected in a fraction of XAC(wt)-expressing neurons, only a similar fraction of growth cones from GFP-expressing neurons were selected, corresponding to the most highly labeled growth cones (if 100 XAC(wt)-infected neurons were analyzed and only 30 had identifiable phalloidin-labeled growth cones, then only the brightest-labeled 30 growth cones were selected for comparison from the GFP-infected neurons).
Treatment with LatA leads to F-actin depolymerization by sequestering actin monomers and has therefore been used for studies of F-actin turnover in the lamellipodia of non-neuronal cells (Svitkina and Borisy, 1999). If the overexpression of ADF/cofilin increases actin filament treadmilling in growth cones, then neurons expressing XAC(wt) should exhibit an increased rate of loss of phalloidin labeling after LatA treatment. Treatment of cortical neurons with 2.0 μmLatA for 1 min or 0.3 μm for 10 min did not decrease total fluorescence in phalloidin-labeled growth cones, although treatments of >1 min caused a loss of filopodia and lamellipodia and resulted in brightly fluorescent punctae (data not shown). To avoid the formation of the punctae, we used Triton X-100 extraction methods described by Svitkina and Borisy (1999) to assay actin filament turnover in the lamellipodia of individual non-neuronal cells (see Materials and Methods). Using Triton extraction of untreated neurons, phalloidin-labeled growth cones were identifiable on the majority of neurons (Fig. 10B). However, the typical growth cone morphology was often lost, with filopodia seldom apparent and fluorescence often more bulbous than in conventionally fixed growth cones (Fig. 9D,E). Interestingly, there were a small number of fibroblast-like cells in the cultures, and these retained intact lamellipodia with strong phalloidin labeling at the leading edge, even after the Triton extraction protocol (Fig.9D).
Treatment of control neurons with 0.1 μm LatA for >5 min resulted in an almost complete loss of F-actin from growth cones treated with Triton extraction buffer under standard conditions. With briefer exposures to LatA, phalloidin-labeled growth cones could be identified for the majority of neurons. However, after LatA treatment, a smaller percentage of labeled growth cones were identifiable in XAC(wt)-expressing neurons than in GFP controls (Fig.10B). In addition, adXAC(wt)-infected neurons had less phalloidin labeling of growth cones both with and without LatA treatment (Figs. 9E, 10C). This could indicate either an increased turnover of actin filaments during the extraction procedure itself or an increased binding of ADF/cofilin that blocks phalloidin labeling.
If ADF/cofilin overexpression increases F-actin turnover in growth cones, this could lead to an increased invasion of microtubules into the growth cones, because the actin filament network may block microtubule extension (for review, see Tanaka and Sabry, 1995;Challacombe et al., 1996). To determine this, neurons were double-labeled using tubulin antibodies and phalloidin (Fig.9F). Total tubulin immunofluorescence in growth cones was 46 ± 10.1% greater in adXAC(wt)-infected neurons than in uninfected controls (mean ± range of two experiments;p < 0.05 for both experiments; t test). The increased immunofluorescence was attributable to not only larger growth cones (121.3 ± 7.6% of controls) but also brighter immunofluorescence (121.7 ± 15.9% of controls).
DISCUSSION
Stimulatory effects of XAC expression on neurite outgrowth depended on active forms of ADF/cofilin. Infection of neurons with a GFP-expressing adenovirus did not increase neurite lengths nor did expression of the inactive XAC(E3) mutant. In addition, higher levels of XAC expression resulted in correspondingly greater increases in neurite length. The length effect was not confined to a single neuronal type, because lengths in both rat cortical neurons and chick spinal cord neurons were increased. Length increases were smaller in chick spinal cord neurons, but these neurons also expressed lower levels of XAC. Expression of the constitutively active XAC(A3) mutant also increased length but to a lesser extent than XAC(wt).
Although we expected that increased ADF/cofilin activity would increase neurite growth, this was based on evidence that process extension is inversely correlated with the extent of ADF/cofilin phosphorylation after treatment with growth factors or drugs (Meberg et al., 1998). The current results offer more direct evidence that increases in ADF/cofilin activity, through increased expression of active forms, increase neurite outgrowth. It is surprising that a single actin-binding protein can have such a large influence on neurite outgrowth, because we know of no other protein having such a specific function that can increase neurite outgrowth from primary neurons. Known stimulators of neurite outgrowth are typically signal molecules that regulate multiple substrates or signal transduction pathways. Because ADF/cofilin phosphorylation is influenced by multiple signaling pathways, it is likely that a wide variety of growth-regulating signals exert their effects in part by changing ADF activity. As such, ADF/cofilin may be a major intracellular target of signals that influence neuronal morphology. The spatially regulated activity of ADF in response to discrete extracellular cues might influence growth cone navigation through asymmetric activation of ADF in portions of the growth cone distal and proximal to the cues.
The XAC(A3) mutant did not increase outgrowth to the same extent as XAC(wt). This may seem surprising because XAC(A3) cannot be inactivated by phosphorylation, whereas a large fraction of XAC(wt) was phosphorylated and therefore in an inactive form. However, chick ADF(A3) mutants have only half of the binding affinity of the wild-type protein (Agnew, 1995), and the same difference hold true for XAC (J. Sneider, H. Chen, and J. R. Bamburg, unpublished observations). Even then, XAC(A3) activity should have been at least equal to that of XAC(wt). This suggests that the regulation of XAC activity by phosphorylation is important for optimal outgrowth. Enhancement of membrane ruffling in fibroblasts and epithelial cells is not accompanied by a net change in ADF phosphorylation, but rather by a change in the rate of phosphate turnover (Meberg et al., 1998). Thus, the phosphocycling of ADF may be important for its function in driving actin turnover (Chen et al., 2000).
An unexpected result was the preferential phosphorylation of XAC over endogenous ADF. Perhaps related to this, basal phosphorylation of cofilin is higher than ADF in multiple cell types (Minamide et al., 1997; Meberg et al., 1998). This occurs despite ADF and cofilin having a similar cellular localization, highly conserved sequences around the phosphorylation site, and qualitatively similar signal-induced changes in phosphorylation. In terms of basal phosphorylation, XAC may therefore act more like mammalian cofilin than ADF. Alternatively, if ADF/cofilin phosphorylation preferentially occurs at the cell body, then high adenoviral-mediated production of XAC near the cell body would result in it being preferentially phosphorylated over endogenous ADF already residing and highly enriched in growth cones. Both pADF and ADF travel together with actin in axonal transport (Mills et al., 1996). Because the affinity of pADF for actin is minimal, it seems probable that the complex stays together only through a rapid turnover of phosphate on ADF. This strongly suggests that LIM kinase is distributed throughout the neuron and phosphorylation–dephosphorylation of XAC occurs continuously during transport.
The integral role played by ADF in actin-dependent processes is becoming more and more apparent. Regulated ADF activity has been found essential for cytokinesis in multiple organisms (Iida et al., 1993;Gunsalus et al., 1995; Abe et al., 1996). ADF also plays a major role in actin-based motility, being essential for the actin-based motility of Listeria (Carlier et al., 1997; Rosenblatt et al., 1997). Overexpression of ADF/cofilin also increasesDictyostelium cell motility (Aizawa et al., 1996) and the outgrowth of neurites (present results). ADF is likely to also be important for other actin-based processes in neurons, such as morphological plasticity of spines and synapses (Fischer et al., 1998) and regulation of ion channels by actin filaments (Johnson and Byerly, 1993; Rosenmund and Westbrook, 1993). The potential importance of ADF in neuronal development is also underscored by findings that visuospatial cognitive defects found in Williams syndrome may be attributable to hemizygosity of the LIM kinase 1 gene (Frangiskakis et al., 1996). ADF is a substrate for LIM kinase, and no other in vivo substrates have yet been found (Arber et al., 1998; Yang et al., 1998).
Increased ADF/cofilin expression likely increases neurite outgrowth by influencing actin dynamics and promoting lamellipodia extension in growth cones. Consistent with this, XAC(wt) expression slightly increased growth cone area and the number of growth cone filopodia. Because ADF/cofilin is known to increase actin treadmilling in non-neuronal cells (Carlier et al., 1997), the greater expression of ADF/cofilin likely increases actin filament turnover and treadmilling, which in turn increases rates of lamellipodia protrusion, growth cone advance, and neurite extension. ADF/cofilin may also regulate treadmilling of branched actin arrays at the leading edge during lamellipodia protrusion in non-neuronal cells (Svitkina and Borisy, 1999). The lack of a significant change in F-actin levels after ADF/cofilin overexpression is therefore not surprising, because it is the increase in actin turnover, not F-actin levels per se, that may regulate lamellipodia protrusion and growth cone motility.
To determine whether F-actin turnover was increased, cultures were treated with inhibitors of actin polymerization, and the rate of F-actin loss was assayed. When XAC(wt) was expressed, F-actin in growth cones disappeared more rapidly after treatment with either cytochalasin D or LatA. These results suggest that increased ADF/cofilin activity does increase F-actin turnover in growth cones, but they are certainly not conclusive, because measures of F-actin in growth cones rely on phalloidin binding, and ADF/cofilin binding to F-actin blocks phalloidin binding (McGough et al., 1997). Therefore, we cannot rule out the possibility that the observed decreases simply reflect greater ADF/cofilin binding. However, because differences in phalloidin labeling of growth cones were much greater after drug treatment than before, it seems likely that the lower phalloidin labeling in XAC(wt)-expressing neurons was contributed to by increased turnover of F-actin. The decrease in filopodia found on neurite shafts is also suggestive of increased F-actin turnover. Hippocampal neurons from gelsolin-deficient mice exhibit an increase in filopodia number on the neurite shaft and a decrease in the rate of filopodia retraction (Lu et al., 1997), indicating that the activity of actin-depolymerizing proteins influences filopodia retraction behind the growth cone.
F-actin concentrations in growth cones decrease after contact with certain factors that induce growth cone collapse (Fan et al., 1993;Kuhn et al., 1999), and F-actin preferentially accumulates in growth cone regions that contact positive guidance cues (Lin and Forscher, 1993; O'Connor and Bentley, 1993). However, it is unlikely that neurite outgrowth is directly correlated with total F-actin levels in growth cones. Growth cone turning is likely driven by graded differences in actin polymerization across the growth cone, not total actin polymerization, because toxins that specifically promote the formation of either too much or too little F-actin inhibit growth cone advance (Forscher and Smith, 1988). In addition, introduction of either constitutively active or dominant negative Rac1 mutants inhibit growth cone advance, but one mutant increases F-actin levels in growth cones, whereas the other decreases F-actin levels (Luo et al., 1994;Kuhn et al., 1999). The small GTPase Rac1 is an activator of LIM kinase 1 (Arber et al., 1998; Yang et al., 1998), and Rac1 may exert its effects on neurite outgrowth in part by modulating ADF activity.
An intriguing alternative hypothesis for how ADF/cofilin overexpression increases neurite outgrowth is that ADF/cofilin disrupts the actin filament network in growth cones, thereby enhancing microtubule-based extension. Much evidence suggests that actin filaments inhibits and/or directs microtubule extension (for review, see Tanaka and Sabry, 1995;Challacombe et al., 1996). For example, cytochalasin disruption of F-actin can increase neurite extension in an undirected, microtubule-based manner (Marsh and Letourneau, 1984; Bentley and Toroian-Raymond, 1986). Because tubulin levels in growth cones were higher in neurons overexpressing ADF/cofilin, increased microtubule-based extension may contribute to enhanced neurite outgrowth.
In summary, increased expression–activity of ADF/cofilin increases neurite extension a significant amount but has minimal effects on growth cone morphology or total F-actin levels. Increased neurite extension was likely contributed to by the following: (1) increased lamellipodia and filopodia extension, as reflected in slightly larger growth cones with more filopodia; (2) an increased rate of actin treadmilling, as indicated by increased F-actin turnover in the presence of cytochalasin D and LatA; and (3) an increase in microtubule invasion into growth cones, as indicated by increased tubulin immunoreactivity in growth cones.
Footnotes
This work was supported in part by National Institutes of Health Grants GM35126 and GM54004 to J.R.B. and NS09583 to P.J.M., and an American Paralysis Association Research Award BB2–9601 to J.R.B. and P.J.M. We thank Dr. Christine Wilcox and Don Traul for generously supplying the EGFP adenovirus and valuable discussions on adenovirus construction, Dr. Hiroshi Abe for generously supplying the XAC cDNAs, Todd Verrastro for producing the XAC antibody, Barb Hietala for performing morphological analyses, Laurie Minamide for recurring general assistance, and Dr. Tom Kuhn for valuable discussions.
Correspondence should be addressed to Peter Meberg, Department of Biology, Box 9019, University of North Dakota, Grand Forks, ND 58202. E-mail: pmeberg@prairie.nodak.edu.
REFERENCES
- 1.Abe H, Ohshima S, Obinata T. A cofilin-like protein is involved in the regulation of actin assembly in developing skeletal muscle. J Biochem. 1989;106:696–702. doi: 10.1093/oxfordjournals.jbchem.a122919. [DOI] [PubMed] [Google Scholar]
- 2.Abe H, Verrastro TA, Brown MD, Minamide LS, Caddoo WS, Agnew BJ, Obinata T, Bamburg JR. Xenopus development is dependent upon regulation of ADF/cofilin by phosphorylation. Mol Biol Cell [Suppl] 1995;6:22a. [Google Scholar]
- 3.Abe H, Obinata T, Minamide LS, Bamburg JR. Xenopus laevis actin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J Cell Biol. 1996;132:871–885. doi: 10.1083/jcb.132.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agnew BJ. PhD thesis. Colorado State University, Fort Collins, CO; 1995. Identification of the regulatory phosphorylation site on actin depolymerizing factor. [Google Scholar]
- 5.Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 6.Aizawa H, Sutoh K, Yahara I. Overexpression of cofilin stimulates bundling of actin filaments, membrane ruffling, and cell movement in Dictyostelium. J Cell Biol. 1996;132:335–344. doi: 10.1083/jcb.132.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 8.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 9.Bamburg JR, Bray D. Distribution and cellular localization of actin depolymerizing factor. J Cell Biol. 1987;105:2817–2825. doi: 10.1083/jcb.105.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamburg JR, Minamide LS, Morgan TE, Hayden SM, Giuliano KA, Koffer A. Purification and characterization of low-molecular weight actin-depolymerizing proteins from brain and cultured cells. Methods Enzymol. 1991;196:125–140. doi: 10.1016/0076-6879(91)96014-i. [DOI] [PubMed] [Google Scholar]
- 11.Bentley D, Toroian-Raymond A. Disoriented pathfinding by pio-neer neurone growth cones deprived of filopodia by cytochalasin treatment. Nature. 1986;323:712–715. doi: 10.1038/323712a0. [DOI] [PubMed] [Google Scholar]
- 12.Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia G-X, Hong Y, Chua N-H, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Challacombe JF, Snow DM, Letourneau PC. Role of the cytoskeleton in growth cone motility and axonal elongation. Semin Neurosci. 1996;8:67–80. [Google Scholar]
- 14.Chen H, Bernstein BW, Bamburg JR. Regulating actin dynamics in vivo. Trends Biochem Sci. 2000;25:19–23. doi: 10.1016/s0968-0004(99)01511-x. [DOI] [PubMed] [Google Scholar]
- 15.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 17.Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Pröschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 1996;86:59–69. doi: 10.1016/s0092-8674(00)80077-x. [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Foix AM, Coats WS, Baqué S, Alam T, Gerard RD, Newgard CB. Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J Biol Chem. 1992;267:25129–25134. [PubMed] [Google Scholar]
- 20.Gunsalus K, Bonaccorsi S, Williams E, Verni F, Gatti M, Goldberg ML. Mutations in twinstar, a Drosophila gene encoding a cofilin/ADF homolog, results in defects in centrosome migration and cytokinesis. J Cell Biol. 1995;131:1243–1259. doi: 10.1083/jcb.131.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iida K, Moriyama K, Matsumoto S, Kawasaki H, Nishida E, Yahara I. Isolation of a yeast essential gene, COF1, that encodes a homologue of mammalian cofilin, a low-M(r) actin-binding and depolymerizing protein. Gene. 1993;124:115–120. doi: 10.1016/0378-1119(93)90770-4. [DOI] [PubMed] [Google Scholar]
- 22.Jensen JR, DeWit M, Bamburg JR. Reversible DMSO-induced translocation of actin and actin polymerizing factor from growth cones to nuclei of cultured hippocampal neurons. Soc Neurosci Abstr. 1993;19:64. [Google Scholar]
- 23.Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+. Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn TB, Brown MD, Wilcox CL, Raper JA, Bamburg JR. Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: inhibition of collapse by opposing mutants of rac1. J Neurosci. 1999;19:1965–1975. doi: 10.1523/JNEUROSCI.19-06-01965.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin CH, Forscher P. Cytoskeletal remodeling during growth cone-target interactions. J Cell Biol. 1993;121:1369–1383. doi: 10.1083/jcb.121.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin CH, Forscher P. Growth cone advance is inversely proportional to retrograde F-actin flow. Neuron. 1995;14:763–771. doi: 10.1016/0896-6273(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 27.Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Witke W, Kwiatkowski DJ, Kosik KS. Delayed retraction of filopodia in gelsolin mutant mice. J Cell Biol. 1997;138:1279–1287. doi: 10.1083/jcb.138.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsh L, Letourneau PC. Growth of neurites without filopodial or lamellipodial activity in the presence of cytochalasin B. J Cell Biol. 1984;99:2041–2047. doi: 10.1083/jcb.99.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGough A, Pope B, Chiu W, Weeds A. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 32.Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Mills RG, Minamide LS, Yuan A, Bamburg JR, Bray JJ. Slow axonal transport of soluble actin with actin depolymerizing factor, cofilin, and profilin suggests actin moves in an unassembled form. J Neurochem. 1996;67:1225–1234. doi: 10.1046/j.1471-4159.1996.67031225.x. [DOI] [PubMed] [Google Scholar]
- 34.Minamide LS, Bamburg JR. A filter paper dye-binding assay forquantitative determination of protein without interference from reducing agents or detergents. Anal Biochem. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- 35.Minamide LS, Painter WB, Schevzov G, Gunning P, Bamburg JR. Differential regulation of actin depolymerizing factor and cofilin in response to alterations in the actin monomer pool. J Cell Biol. 1997;272:8303–8309. doi: 10.1074/jbc.272.13.8303. [DOI] [PubMed] [Google Scholar]
- 36.Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connor TP, Bentley D. Accumulation of actin in subsets of pioneer growth cone filopodia in response to neural and epithelial guidance cues in situ. J Cell Biol. 1993;123:935–948. doi: 10.1083/jcb.123.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison T. Xenopus actin depolymerizing protein/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- 40.Svitkina TM, Borisy GG. Arp2/3 complex and ADF/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 42.Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid filament turnover in the dynamic cytoskeleton. J Cell Biol. 1997;136:1165–1168. doi: 10.1083/jcb.136.6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wessel D, Flügge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 44.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]