

Abstract
Injury to the CNS results in the formation of the glial scar, a primarily astrocytic structure that represents an obstacle to regrowing axons. Chondroitin sulfate proteoglycans (CSPG) are greatly upregulated in the glial scar, and a large body of evidence suggests that these molecules are inhibitory to axon regeneration. We show that the CSPG neurocan, which is expressed in the CNS, exerts a repulsive effect on growing cerebellar axons. Expression of neurocan was examined in the normal and damaged CNS. Frozen sections labeled with anti-neurocan monoclonal antibodies 7 d after a unilateral knife lesion to the cerebral cortex revealed an upregulation of neurocan around the lesion. Western blot analysis of extracts prepared from injured and uninjured tissue also revealed substantially more neurocan in the injured CNS. Western blot analysis revealed neurocan and the processed forms neurocan-C and neurocan-130 to be present in the conditioned medium of highly purified rat astrocytes. The amount detected was increased by transforming growth factor β and to a greater extent by epidermal growth factor and was decreased by platelet-derived growth factor and, to a lesser extent, by interferon γ. O-2A lineage cells were also capable of synthesizing and processing neurocan. Immunocytochemistry revealed neurocan to be deposited on the substrate around and under astrocytes but not on the cells. Astrocytes therefore lack the means to retain neurocan at the cell surface. These findings raise the possibility that neurocan interferes with axonal regeneration after CNS injury.
Keywords: chondroitin sulfate, EGF, extracellular matrix, glial scar, proteoglycan, regeneration, TGFβ
The inability of neurons to regenerate within the adult mammalian CNS is a result of the innately poor regenerative ability of many CNS neurons and the inhibitory nature of the adult mammalian CNS. This inhibition is attributable to molecules associated with CNS myelin and those associated with the glial scar, a structure that in its mature form contains mostly astrocytes and can constitute an impediment to regrowing axons (for review, see Fawcett and Asher, 1999).
Various in vitro experiments have shown that astrocytes can inhibit axon growth. Astrocytes cultured as a three-dimensional tissue, astrocytes from injured adult rat optic nerve and injured cerebral cortex, and astrocytes removed from the injured adult CNS attached to nitrocellulose filter material were all nonpermissive for the growth of various axonal types (Smith et al., 1986; Fawcett et al., 1989,Bähr et al., 1995, Le Roux and Reh, 1996). Various lines of evidence suggest that inhibition by astrocytes is attributable at least in part to chondroitin sulfate proteoglycans (CSPGs). Comparison between permissive and inhibitory astrocyte cell lines showed that inhibitory cells produced inhibitory CSPGs, the activity of which could be reduced by chondroitinase, xylosides, and chlorate, all of which affect the glycosaminoglycan (GAG) component of proteoglycans. Three-dimensional astrocyte cultures were also rendered more permissive by chlorate (Smith-Thomas et al., 1994, 1995), and axon growth on reactive astrocytes removed from the adult CNS on filter material was increased by treatment with chondroitinase (McKeon et al., 1991,1995).
CSPGs are implicated in the inhibition of axon regeneration in the injured CNS in vivo. There is substantial upregulation of CSPG production in the glial scar after CNS injury, as revealed by antibodies that bind to the chondroitin sulfate GAG chains (McKeon et al., 1991; Laywell et al., 1992; Frisen et al., 1995; McKeon et al., 1995; Barker et al., 1996; Gates et al., 1996). Experiments in which sensory neurons were implanted into adult white matter tract showed substantial regeneration, but growth stopped at CSPG-rich sites of injury (Davies et al., 1997, 1999). Treatment of an axotomy injury with chondroitinase allowed regeneration of CNS axons (Moon et al., 1999). Axon growth on cryosections of normal and injured spinal cord was improved by pretreatment of the sections with chondroitinase (Zuo et al., 1998). A proteoglycan preparation from injured brain also had outgrowth-inhibitory effects, which were relieved with chondroitinase (Bovolenta et al., 1993).
One effect of the inhibitory CSPGs is interference with the neuronal growth-promoting effects of laminin (McKeon et al., 1991, 1995). Pretreatment of explanted glial scar astrocytes with chondroitinase led to an increase in neurite outgrowth, which was inhibited by laminin function-blocking antibodies (McKeon et al., 1991, 1995). The increased growth on chondroitinase-treated cryosections described above was largely inhibited by laminin-blocking antibodies (Zuo et al., 1998), and inhibitory CSPGs produced by inhibitory astrocyte cell lines and astrocytes were able to block the axon growth-promoting effects of laminin (Smith-Thomas et al., 1994, 1995; Fidler et al., 1999).
In the present study we have examined the expression of neurocan, a CSPG with well documented axon growth-inhibitory properties in CNS injuries, in various glial cell types, and have determined the effects of injury-related cytokines on its production.
Some of these data have been published previously in abstract form (Asher et al., 1998).
MATERIALS AND METHODS
Surgical procedures
Adult female Sprague Dawley rats (Charles River, Margate, UK; approximate body weight, 200 gm) were anesthetized under halothane, and the head was held in a stereotaxic apparatus with the incisor bar 2.5 mm below the interaural line. A fine scalpel blade was inserted stereotaxically and vertically into the brain, through a parasagittal dorsal craniotomy, to a depth of 3 mm below the dura and 2–3 mm lateral to the midline. The knife was then moved in the horizontal plane to produce a lesion 5–6 mm in length, midway between lambda and bregma. The operation was performed unilaterally (on the right-hand side) with the other hemisphere serving as a control. After 7–8 d, the animals were terminally anesthetized with an intraperitoneal injection of sodium pentabarbitone and decapitated. The brain was rapidly removed and immediately frozen on dry ice and stored at −70°C or prepared for frozen sectioning. All procedures were conducted in compliance with the UK Animals (Scientific Procedures) Act 1986.
Immunolabeling of frozen sections
Frozen, coronal sections (10 μm) were cut from unfixed tissue 7 d after lesion. Labeling was performed without fixation. Nonspecific binding was blocked with PBS containing 3% BSA and 20 mml-lysine (PBS/BSA/Lys). The sections were incubated with the 1G2 anti-neurocan monoclonal antibody (mAb) (supernatant diluted 1:2) (Oohira et al., 1994), the 1F6 anti-neurocan mAb [supernatant diluted 1:2; Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA] (Meyer-Puttlitz et al., 1995), or mouse IgG1 (10 μg/ml in PBS/BSA/Lys) for 1 hr. Bound antibodies were visualized with biotinylated anti-mouse immunoglobulins (1:100; Amersham, Little Chalfont, Bucks, UK) and Cy3-streptavidin (1.0 μg/ml; Jackson, West Grove, PA) in the manner described previously (Asher et al., 1995).
Sample preparation for SDS-PAGE
Brain tissue. Dissection was performed as rapidly as possible while the brain was unfreezing. A piece of tissue ∼2 × 4 × 6 mm containing the lesion was excised. A similar-sized piece was dissected from the same location on the contralateral cortex. The tissue was immediately placed in 1.0 ml ice-cold extraction buffer (0.05 m Tris-HCl, 0.15 m NaCl, pH 7.0) containing protease inhibitor cocktail (25× in 0.1 mphosphate buffer, pH 7.0; Roche, Lewes, East Sussex, UK) and homogenized in a Teflon-glass Dounce homogenizer. The homogenate was centrifuged at 13,000 × g for 10 min at 4°C. Two additional extracts were made from the resultant pellet in the same way. The pellet was then rehomogenized in extraction buffer containing, additionally, 1% Triton X-100 (Fluka, Gillingham, Dorset, UK) and incubated in an end-over-end shaker at 4°C for 1 hr. Another detergent extract was then made from the resultant pellet in the same manner. Thus, the tissue was extracted three times with saline and then twice with detergent. The protein content was determined according to the method of Bradford (1976) using the Coomassie Plus Protein Assay Reagent (Pierce, Chester, Cheshire, UK) and BSA to generate the standard curve. One-half of each sample was treated with 0.02 U chondroitinase ABC (protease-free, Roche) for 3 hr at 37°C.
Cultured cells. Conditioned medium was removed from the cells (astrocytes, O-2A progenitor cells, macrophages, or meningeal cells) and added to a tube containing protease inhibitor cocktail. The conditioned medium was then centrifuged at 1000 × gfor 10 min at 4°C and concentrated in a Centricon 100 (Millipore, Watford, Herts, UK) to one-tenth of its initial volume. The protein content of the extracts was determined in the manner described above. Chondroitinase ABC digestion was performed for 3 hr at 37°C using 0.01 U of enzyme per milliliter of unconcentrated conditioned medium.
Electrophoresis and Western blotting
SDS-PAGE was performed in 4 or 5% polyacrylamide gels with a 3% stacking gel under nonreducing conditions. Proteins were transferred to nitrocellulose (Hybond-C pure, Amersham) at 4°C for 15–18 hr, at a constant current of 150 mA.
All subsequent procedures were performed at room temperature. The blots were rinsed twice in Tris-buffered saline (TBS, 0.9% NaCl, 10 mm Tris-HCl, pH 7.5) containing 0.05% Tween 20 (TBS/Tween) and incubated for an additional 40 min in TBS/Tween. All washes and antibody incubations were performed in TBS/Tween. The blots were then incubated with one of the following antibodies: the 1G2 anti-neurocan mAb (supernatant diluted 1:20–1:40), the 1F6 anti-neurocan mAb (supernatant diluted 1:20–1:40), the rabbit anti-neurocan-N-terminal fragment pAb 291 (1:1000) (Matsui et al., 1994), the anti-versican mAb 12C5 (supernatant diluted 1:20) (Asher et al., 1991), a mouse myeloma-derived IgG1 (1.1 μg/ml; Sigma, Poole, Dorset, UK), or rabbit immunoglobulins (1.0 μg/ml; Dako, High Wycombe, Bucks, UK) for 2 hr. For labeling with rabbit antibodies, blocking and dilution of the primary antibody was performed in TBS/Tween containing 5% dried milk powder and 20 mml-lysine. Reactive species were visualized with either peroxidase-conjugated anti-mouse or anti-rabbit IgG (Vector, Peterborough, Cambs, UK) and a chemiluminescent substrate (National Diagnostics, Hull, Humberside, UK). The amount of protein in a given band was estimated by densitometry using a Quantimet 500 instrument.
Glial cell culture
All cell culture reagents were purchased from Life Technologies (Paisley, UK), unless stated otherwise. Glial cell cultures were prepared from the brains of newborn rats <3 d old. The cerebral cortices of six to eight rats were freed of meninges and collected in HBSS. The tissue was then transferred to DMEM (with 4.5 g/ld-glucose, Glutamax I, and sodium pyruvate) containing 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, and 2.5 μg/ml amphotericin B (Fungizone), and dissociated by passage through needles of decreasing size (19, 21, 23, and 25 ga). The suspension was filtered (without force) through 70 μm nylon mesh (BioDesign, Carmel, NY) and dispensed into four or five 75 cm2 poly-d-lysine-coated tissue culture flasks. Ninety percent of the medium was replaced the next day with fresh medium lacking the antibiotics and anti-mycotic, and the cells were subsequently fed every third day.
Between the 8th and 12th days, the cultures were shaken to remove the macrophages and progenitor cells that sit on top of the (astroglial) monolayer. The adherent cells were passaged (with the use of 0.25% trypsin, 1 mm EDTA) into 25 cm2 poly-d-lysine-coated flasks at a split ratio of 1:1 and maintained in serum-containing medium. Two days after a confluent monolayer had been reestablished, the serum-containing medium was replaced with serum-free DMEM containing 6.25 μg/ml insulin, 6.25 μg/ml transferrin, 6.25 ng/ml selenious acid, 1.25 mg/ml bovine serum albumin, 5.35 μg/ml linoleic acid (as ITS+; Collaborative Biomedical Products, Bedford, MA), 6.3 ng/ml progesterone (Sigma), 16 μg/ml putrescine (Sigma), 50 μg/mll-ascorbic acid (Sigma), and 16 μm cytosine arabinoside (Ara-C, Fluka). The cells were maintained for two periods of 4 d in this medium, at the end of which they were invariably free of any contaminating progenitor cells. Occasionally, microglial-like cells were visible, and these were eliminated by treatment with 10 mml-leucine methyl ester (Sigma) for 30 min (Giulian and Baker, 1986). The cells were grown for an additional 2–3 d in the same medium lacking Ara-C.
For the preparation of astrocyte-conditioned medium, each flask was washed once with DMEM, and 2.5 ml of serum-free DMEM containing the cytokine or growth factor under investigation was added for 2–8 d. Information pertaining to the source and concentration of the cytokines and growth factors is presented in Table 1. To investigate neurocan processing, astrocytes were maintained for 4 d in serum-free DMEM containing one of the following protease inhibitors: anti-thrombin III (0.1 U/ml), aprotinin (1.0 μg/ml), tissue inhibitor of metalloproteinase-2 (TIMP-2, 0.2 μg/ml), leupeptin (1.0 μg/ml), E-64 (1.0 μg/ml; Calbiochem, Nottingham, Notts, UK), cathepsin B inhibitor II (1.0 μg/ml; Calbiochem), or α2-macroglobulin (10 μg/ml). Unless stated otherwise, inhibitors were purchased from Roche and added fresh each day.
Table 1.
Sources and concentrations of cytokines and growth factors
| Cytokine/growth factor, species | Source | Stock solution concentration | Final concentration |
|---|---|---|---|
| CNTF, rat | R & D Systems | 10 μg/ml | 10 ng/ml |
| EGF, human | R & D Systems | 200 μg/ml | 0.1–10 ng/ml |
| FGF2, human | Boehringer Mannheim (Indianapolis, IN) | 10 μg/ml | 10 ng/ml |
| IFN-γ, rat | R & D Systems | 100 μg/ml | 10 ng/ml |
| IL-1α, mouse | Genzyme (Boston, MA) | 5 μg/ml | 5 ng/ml |
| IL-1β, rat | R & D Systems | 10 μg/ml | 10 ng/ml |
| IL-6, mouse | Boehringer | 2 μg/ml | 1 ng/ml |
| LIF, human | Boehringer | 5 μg/ml | 10 ng/ml |
| PDGF-AB, human | R & D Systems | 10 μg/ml | 10 ng/ml |
| TGFβ-1, human | Genzyme | 1 μg/ml | 0.1–10 ng/ml |
| TNF-α, rat | R & D Systems | 10 μg/ml | 10 ng/ml |
Cultures enriched for O-2A progenitor cells were derived from the cells dislodged during the shaking of the primary culture. The cell suspension was filtered through 35 and 15 μm nylon mesh and then preplated on uncoated tissue culture plastic to deplete it of macrophages and astrocytes. The nonattached cells from one 75 cm2 flask were dispensed into one poly-d-lysine-coated 25 cm2flask in DMEM containing 10% FCS. The cells were allowed to adhere for 2–3 hr, and the medium was changed to serum-free DMEM containing ITS+, 10 ng/ml fibroblast growth factor 2 (FGF2; Roche), and 10 ng/ml platelet-derived growth factor (PDGF-AB; R & D, Abingdon, Oxon, UK). This was collected after 2 d and replaced with the same medium, or with the same medium lacking the growth factors.
Cultures of meningeal cells were established from the meninges of four to six newborn rat brains by enzymatic dissociation with 0.2% collagenase and 0.1% trypsin. These cells were grown to confluence in DMEM/10% FCS. For the preparation of conditioned medium, the cells were grown for 4 d in DMEM containing 1% FCS and ITS+.
Stripe assays
Neurocan, phosphacan, NG2, and L1 were purified as described previously (Rauch et al., 1991; Dou and Levine 1994). The stripe assay was carried out as described by Baier and Klostermann (1994), with minor modifications. Acid-washed glass coverslips were coated with poly-l-lysine (100 μg/ml in water) overnight, washed, and then coated again with immunoaffinity-purified L1 (2 μg/ml, 2–3 hr, 37°C). The coverslips were washed in PBS and air-dried, and narrow stripes of the test proteins (10 μg/ml) were applied using a silicon matrix. After 1 hr at 37°C, the coverslips were removed from the matrices, washed in complete medium (DMEM, 10% FCS), and stored in complete medium until ready for use.
Explants of postnatal day (P) 4–5 rat cerebellum or P0 cortex were placed on top of the coverslips and grown for 48 hr in complete medium. For the cerebellar explants, the medium was supplemented with 20 mm KCl. Patterns of neurite outgrowth were visualized with either phase-contrast or fluorescence optics after staining the living cultures with carboxy-methyl fluorescein (Molecular Probes, Eugene, OR). In some cases, trace amounts of fluorescein-conjugated dextran (Molecular Probes) were added to the stripe-making solution to aid in the visualization of the stripes.
Immunocytochemistry
Astrocytes and O-2A progenitor cells were grown on poly-d-lysine-coated glass coverslips. Neurocan labeling was performed at room temperature on living cells in Liebovitz's L-15 medium (Life Technologies) containing 2% FCS (L-15/FCS). The cells were washed once in L-15/FCS and incubated with the 1G2 anti-neurocan mAb (supernatant diluted 1:2), the 1F6 anti-neurocan mAb (supernatant diluted 1:2), the 1D1 anti-neurocan mAb (ascites diluted 1:100; DSHB), or the rabbit anti-neurocan–N-terminal fragment polyclonal antibody (pAb) 291 (1:1000) for 20 min. The cells were washed three times with L-15/FCS and then incubated with biotinylated anti-mouse immunoglobulins or biotinylated anti-rabbit immunoglobulins (1:100; Amersham) for 20 min. The cells were washed as before and incubated with Cy3-streptavidin (1 μg/ml) for 20 min. Finally, the cells were washed three times in L-15/FCS and once in PBS and fixed in cold (−20°C) methanol for 2 min. Glial fibrillary acidic protein (GFAP) labeling was performed on methanol-fixed cells with rabbit anti-GFAP (20 μg/ml, Dako) and FITC anti-rabbit IgG (7.5 μg/ml, Jackson). Labeling with the A2B5 (an O-2A cell marker; American Type Culture Collection, Manassas, VA) and 5A5 (anti-polysialylated N-CAM, DSHB) mAbs was also performed on living cells, in the manner described above. Nuclei were labeled with Hoechst No. 33342 (Sigma) for 30 min. Sterile testicular hyaluronidase (type IV, Sigma) was added directly to astrocytes growing on glass coverslips in serum-free medium to a final concentration of 20 or 100 μg/ml, and incubation continued for 3 hr at 37°C in a CO2-containing atmosphere.
RESULTS
Neurocan inhibits neurite outgrowth
To determine whether neurocan is inhibitory to axon outgrowth, we used a stripe assay as described in Materials and Methods. In this assay, as axons grow out of small explants of developing neural tissue, they can choose to grow on different regions of the substrate. Molecules that are inhibitory or nonpermissive for axonal extension are avoided in this assay (Baier and Klostermann, 1994). As shown Figure 1,C and D, the axons that extend out of either cerebellar or cortical explants avoid the neurocan stripes and elongate preferentially on L1. Similar results were obtained when the stripes were made with either phosphacan or NG2, two other CSPGs that are inhibitory to neurite outgrowth (data not shown). The pattern of neurite outgrowth on surfaces coated with stripes of BSA on top of L1 was indistinguishable from that observed on coverslips coated uniformly with L1. Thus, when given a choice, growing axons avoid neurocan.
Fig. 1.

Neurocan is avoided by growing axons.A, A cerebellar explant growing on an L1-coated coverslip. Note the uniform halo of growing neurites. B, A cerebellar explant growing on an L1-coated coverslip with stripes of BSA (25 μg/ml). As in A, neurite outgrowth is uniform.C, A cerebellar explant growing on an L1-coated coverslip with stripes of neurocan (10 μg/ml). Axons extend in thin bundles with wider gaps between the bundles. As shown inE, the wide gaps contain the added molecules (in this case neurocan), and the narrow bands are the L1-coated substrate.D, A cortical explant growing on an L1-coated coverslip with neurocan (10 μg/ml) stripes. As in C, wide gaps separate narrow bands of outgrowth. E, Fluorescein–dextran-containing stripes (+) form wide gaps that alternate with narrow bands of uncoated substrate (−). A, B,C, and E are epifluorescence illumination; D is phase-contrast optics.
Neurocan expression is upregulated in response to CNS injury
Neurocan expression was examined in frozen sections cut 7 d after a unilateral knife lesion to the cerebral cortex. This revealed a substantial increase in neurocan expression around the lesion, in comparison to the uninjured side (Fig.2). The 1G2 (anti-neurocan-C) and 1F6 (anti-neurocan–N-terminal fragment) mAbs labeled in an identical manner. Neurocan expression was increased for a distance of ∼100 μm from the wound edge in the cortical gray matter, but the increased expression was spread over a greater distance in the underlying white matter, extending for up to 0.5 mm on either side of the injury. In the surrounding normal brain, there was generally much higher expression in white matter than in gray, a notable exception being the labeling of the gray matter adjacent to the midline and medial to the lesion known as the retrosplenial agranular cortex.
Fig. 2.
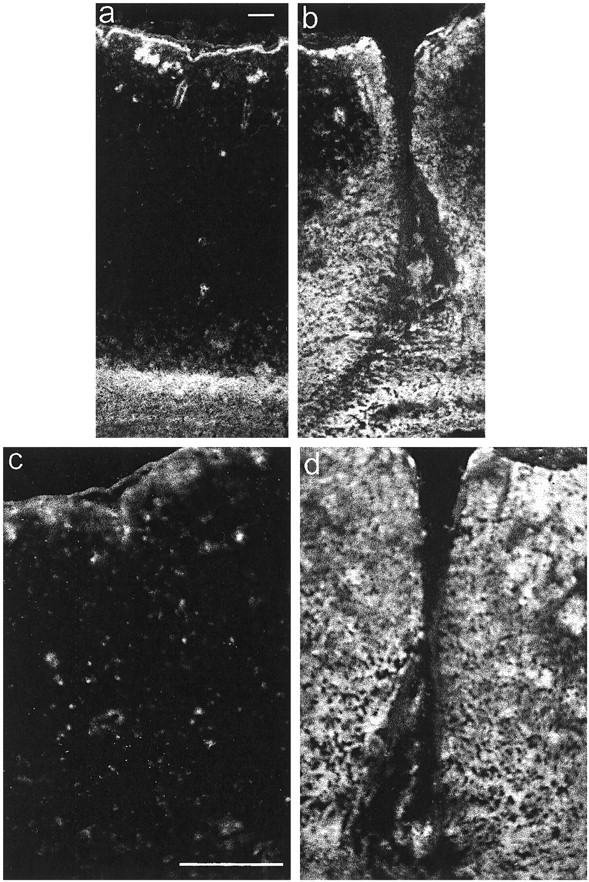
Immunolocalization of neurocan in a CNS lesion. Coronal frozen sections were labeled with the anti-neurocan mAb 1G2 7 d after a knife lesion to the cerebral cortex. The images ina and b were taken with a 10× objective, and those in c and d were taken with a 20× objective. The dorsal surface of the brain isuppermost. Labeling is apparent around the lesion (b, d), which is clearly lacking on the uninjured side (a, c). Scale bar, 100 μm.
Western blot analysis of neurocan expression in the brain
Neurocan has been described in at least four forms: intact neurocan and three smaller forms resulting from proteolytic cleavage of the intact molecule. The C-terminal fraction of the molecule is known as neurocan-C, whereas there are two species from the N-terminal end of 130 and 90 kDa. The 130 kDa fragment contains the 1F6 antibody binding site and was therefore detected in our experiments, whereas the 90 kDa fragment does not (Meyer-Puttlitz et al., 1995). We refer to these N-terminal fragments as neurocan-130 and neurocan-90. All of the species carry chondroitin sulfate. Neurocan-C and neurocan-130 are thought to arise as a result of a single proteolytic cleavage between amino acids 638 and 639 (Rauch et al., 1992; Matsui et al., 1994). This event is referred to as processing, and the smaller neurocan fragments are referred to as the processed forms. The domain structure of neurocan is shown in Figure 3. On Western blots with the antibodies used in our experiments, neurocan is seen as broad indistinct smears, which on chondroitinase treatment resolve into bands of 270 kDa (intact neurocan), 150–163 kDa (neurocan-C), and 122–130 kDa (neurocan-130). To detect these bands we have used three antibodies. Monoclonal antibody 1G2 recognizes intact neurocan and neurocan-C, and mAb 1F6 and pAb 291 recognize intact neurocan and neurocan-130.
Fig. 3.

Domain structure of neurocan. The two processed fragments detected in the present experiments, neurocan-130 and neurocan-C, are thought to arise as a result of a single proteolytic cleavage on the C-terminal side of met638. Both fragments carry chondroitin sulfate. The 1G2 and 1D1 mAbs recognize epitopes in the C-terminal half of neurocan and react with neurocan-C. The 1F6 mAb and pAb291 recognize structures in the N-terminal half of neurocan and react with neurocan-130. Ig, Immunoglobulin; PTR, proteoglycan tandem repeat;CS, chondroitin sulfate; EGF, epidermal growth factor; CRP, complement regulatory protein.
Neurocan expression in the injured CNS was examined by Western blotting using extracts made from tissue dissected from around the lesion and from control unlesioned cortex. Three serial Tris-buffered saline extracts and two Triton X-100 extracts were prepared from injured brain and its uninjured equivalent in the manner described in Materials and Methods. In extracts from normal and damaged CNS, the vast majority of all three forms of neurocan was present in the first saline extract, although it was also detectable in the two subsequent saline extracts (results not shown). Very little neurocan was found in the detergent extracts.
Comparison of the first saline extract from injured brain with that from uninjured brain revealed a clear increase in intact neurocan expression in response to the lesion (Fig.4). The two extracts were equalized for total protein. In the absence of a reducing agent (Fig. 4a), detection of the two smaller processed forms was hampered by nonspecific binding; this problem was greatly alleviated by the use of a reducing agent. In blots made under reducing conditions, it was possible to see that intact neurocan was greatly upregulated in the injured CNS, with a slight increase in neurocan-C (Fig. 4b). Under reducing conditions, CNS-derived intact neurocan migrated at ∼275 kDa, neurocan-C at 163 kDa, and neurocan-130 at 122 kDa.
Fig. 4.

Western blot analysis of neurocan expression in injured brain. Tris-buffered saline extracts were prepared from injured and uninjured cerebral cortex, and either left untreated (−) or treated with (+) chondroitinase ABC (chABC). The extracts were equalized for protein (15 μg in a; 8 μg in b,c), run in a 5% gel either with (b,c) or without (a) a reducing agent (0.2 m DTT), and transferred to nitrocellulose. The blots were labeled for neurocan with either the 1G2 or 1F6 mAb. Both mAbs react with intact (275 kDa) neurocan. The 1G2 mAb recognizes an epitope in the C-terminal half of neurocan and so reacts with neurocan-C, whereas 1F6 recognizes an epitope in the N-terminal half of neurocan and so reacts with neurocan-130. An upregulation of intact neurocan was clearly evident in the injured brain extracts. This difference was apparent whether or not the chondroitin sulfate was removed with chondroitinase ABC (a). The level of neurocan-C was also slightly increased (b). The band just below the 170 kDa marker (arrowhead) is nonspecific. Size determinations were made in a 5% gel by comparison of their relative mobilities with those of laminin (400 kDa), nonreduced α2-macroglobulin (340 kDa), myosin (204 kDa), α2-macroglobulin (170 kDa), and β-galactosidase (116 kDa).
Densitometric analysis of the intact neurocan core protein revealed a ∼4.8-fold increase in response to the injury (from Fig.4a, without a reducing agent). On shorter exposures, the chondroitinase-treated intact neurocan band was seen to consist of three discrete bands. These are likely to result from differences in glycosylation. Neurocan is produced by various cell types (see below), and these are likely to attach different types and/or amounts of carbohydrate.
Production of purified glial cultures
The glial scar that develops after CNS injury contains astrocytes, oligodendrocyte precursors, microglia, and meningeal cells. To ascertain which of these cell types is responsible for the increased amounts of neurocan, we examined its expression in cultures of purified glial cell types. Astrocyte cultures had to be as pure as possible and free of O-2A progenitor cells, which are also capable of secreting neurocan (see below). Although shaking the culture was found to be an effective means of removing O-2A cells and macrophages, some small, round, phase-bright cells remained attached to the monolayer. These cells were identified as pre-O-2A cells (Ben-Hur et al., 1998), rather than O-2A cells, on the basis of their reactivity with the 5A5 mAb against polysialylated N-CAM (Dodd et al., 1988). The use of an anti-mitotic was found to be a highly effective means of ridding a (confluent) astrocyte monolayer of these highly proliferative cells. The combination of shaking and treatment with leucine methyl ester removed all of the visible, top-dwelling macrophages. More than 95% of the monolayer cells were astrocytes, insofar as they were GFAP positive. Cultures of O-2A lineage cells were generated by shaking the top-dwelling cells from mixed glial cell cultures, removing microglia and astrocytes by preplating on plastic, and then driving O-2A cell division with FGF2 and PDGF. These cultures contained small numbers of astrocytes, generally <5%. Meningeal cell cultures were produced by dissociation of meninges stripped from the P0 rat brain. Microglial cultures were made from cells dislodged from mixed glial cultures by light shaking and purified by selecting cells that adhered to plastic that was not tissue culture-treated.
Neurocan is present in astrocyte-conditioned medium
Western blot analysis of astrocyte-conditioned medium with the anti-neurocan mAb 1G2 revealed two broad regions of reactivity, one migrating between ∼290 and 425 kDa and the other between 170 and 220 kDa. After treatment with chondroitinase ABC, these smears were resolved into two discrete bands of ∼270 and 150 kDa (Fig.5). The 270 kDa band represents the intact neurocan core protein. The 150 kDa species corresponds to the C-terminal half of neurocan (Rauch et al., 1992; Matsui et al., 1994) and is therefore referred to as neurocan-C. A rabbit antiserum raised against a 16 amino acid peptide (amino acids 483–498) contained within the N-terminal half of rat neurocan (Matsui et al., 1994) was also reactive with the 270 kDa species and with an additional species of (approximately) 130 kDa (Fig. 5). The 1F6 mAb, which recognizes an epitope in the N-terminal half of neurocan (Meyer-Puttlitz et al., 1995), behaved in a similar manner, i.e., it was reactive with the 270 and 130 kDa species (Fig. 5). This 130 kDa fragment corresponds to a fragment from the N-terminal half of neurocan and is referred to as neurocan-130. Astrocyte-conditioned medium therefore contains three species that react with neurocan antibodies: intact neurocan (270 kDa) and two fragments generated by proteolytic cleavage (processing) of the intact molecule, neurocan C (150 kDa) and neurocan-130 (130 kDa).
Fig. 5.

Three distinct neurocan species are present in astrocyte-conditioned medium. Western blot analysis of astrocyte (astro) and O-2A lineage cells (O-2A) conditioned medium with anti-neurocan-C (1G2), anti-neurocan-130 (1F6), and anti-versican (12C5) mAbs, and a rabbit antiserum raised against neurocan-130 (pAb291). In chondroitinase-treated (+) conditioned medium, the 1G2 and 1F6 mAbs and pAb291 all recognized the same 270 kDa species (neurocan core protein). The 1G2 mAb was also reactive with a 150 kDa species (neurocan-C), whereas the 1F6 mAb and pAb291 were also reactive with a 130 kDa species (neurocan-130). Without previous chondroitinase treatment, neurocan migrated as a highMr, polydisperse species (neurocan + GAG). Neurocan core protein and neurocan-C were also detected in O-2A cell-conditioned medium. To demonstrate the specificity of these antibodies, the same samples were probed with an anti-versican mAb. No versican reactivity was detected in astrocyte-conditioned medium. A single, highMr species was seen in chondroitinase-treated O-2A cell-conditioned medium. This does not correspond to any of those recognized by the neurocan antibodies. No reactivity was seen when a mouse monoclonal IgG1 was used as the primary antibody (data not shown). Size determinations were made in a 4% gel by comparison of relative mobilities with those of the following proteins: laminin (400 kDa), nonreduced α2-macroglobulin (340 kDa), myosin (212 kDa), α2-macroglobulin (170 kDa), and β-galactosidase (116 kDa).
An antibody (12C5) against the closely-related CSPG versican and a myeloma-derived IgG1(MOPC21) were used as negative controls. No 12C5-reactive species were detected in astrocyte-conditioned medium, although a single, high Mr species was seen in O-2A-conditioned medium (Fig. 5 and see below). No reactivity whatsoever was seen when the MOPC21IgG1 was used as the primary antibody.
Astrocytes process neurocan
To be certain that the 130 and 150 kDa species arose during the culture period and were not an artifact of the storage, concentration, and/or chondroitinase treatment, astrocyte-conditioned medium was added, immediately after collection, to a tube containing protease inhibitor cocktail and centrifuged to remove any detached cells and debris. Half of the conditioned medium was then added to hot, two times sample buffer, and boiled immediately for 5 min. The other half was treated with protease-free chondroitinase ABC for 3 hr at 37°C. Both samples were then subjected to Western blot analysis without being concentrated or frozen (Fig.6a).
Fig. 6.

a, The presence of the three forms of neurocan is not an artifact of storage, concentration, or chondroitinase treatment. The same three bands are seen whether conditioned medium is analyzed before or after storing, concentrating, or chondroitinase treatment. b, Protease inhibitors do not affect neurocan processing. Astrocytes were grown in the presence of aprotinin (1.0 μg/ml), anti-thrombin III (0.1 inhibitor U/ml), or TIMP-2 (0.2 μg/ml) for 4 d. Each inhibitor was added fresh each day. Conditioned medium was concentrated and treated with chondroitinase ABC, and an equal amount of total protein (150 μg) was applied to each lane. The blot was labeled with the 1G2 mAb (left) and then relabeled with the 1F6 mAb (right). Neither serine protease (aprotinin and anti-thrombin III) nor metalloproteinase (TIMP-2) inhibitors prevented the processing of neurocan.
The results of this experiment were identical to those described above. Importantly, in the absence of chondroitinase treatment, the 1G2 mAb was reactive with two polydisperse species, the smaller of which (170–220 kDa) corresponds to neurocan-C with attached GAG (Fig.6a). As before, treatment with chondroitinase ABC resolved these polydisperse species into two discrete bands of ∼270 and 150 kDa. These findings indicate that (GAG-bearing) neurocan-C exists in astrocyte-conditioned medium before chondroitinase ABC digestion and that astrocytes are therefore capable of generating the smaller neurocan species.
The processing of neurocan is thought to involve a single proteolytic cleavage between amino acids 638 and 639. To find out whether inhibition of extracellular proteases might reduce or prevent neurocan processing by astrocytes, we investigated the effects of adding serine protease inhibitors (aprotinin and anti-thrombin III) and a metalloproteinase inhibitor (TIMP-2) to astrocyte cultures. None of these inhibitors had any effect on neurocan processing, in that the levels of both processed fragments appeared unchanged (Fig.6b). We also investigated the effects of cysteine protease inhibitors (leupeptin, E-64, and cathepsin B inhibitor II) and α2-macroglobulin, an endoproteinase inhibitor. None of these inhibitors prevented neurocan processing, in that levels of neurocan-C appeared unaltered (data not shown).
Neurocan is found on the substrate around astrocytes but not on the cell surface
To obtain further evidence for the astrocytic origin of neurocan, astrocyte cultures were double-labeled for neurocan and GFAP. Labeling of living cells revealed deposits of substrate-bound neurocan in the immediate vicinity of most GFAP-positive cells (Fig.7). The cells themselves were not labeled. The application of the same procedure to methanol-fixed cells, which gives the antibody access to the inside of the cell and its ventral (lower) surface, revealed that neurocan was located under and/or inside astrocytes. With time, neurocan came to cover the entire substrate, although it remained visibly concentrated around astrocytes. The 1G2, 1F6, and 1D1 mAbs and the polyclonal anti-neurocan–N-terminal fragment labeled in an identical manner. Treatment with transforming growth factor (TGF) β, epidermal growth factor (EGF), or interferon γ (IFN-γ) had no effect on this staining pattern. Neurocan labeling with either the 1G2 or 1F6 mAb was not affected by pretreatment of the cells with testicular hyaluronidase, indicating that the attachment of neurocan to the substrate is not dependent on hyaluronate.
Fig. 7.

Astrocytes incorporate neurocan into a pericellular, substrate-bound ECM in vitro. Living astrocytes were labeled with the anti-neurocan mAb 1G2 (b), fixed in methanol, and labeled with rabbit antibodies against GFAP (a). Labeling for neurocan was seen on the substrate around GFAP-positive cells. Scale bar, 25 μm.
Neurocan is present in the conditioned medium of O-2A cells
We examined conditioned medium from O-2A cell cultures with the 1G2 antibody. Intact neurocan and neurocan-C were detectable in the conditioned medium of O-2A lineage cells (Fig. 5). The amount of neurocan in O-2A-conditioned medium was comparable or greater than that found in astrocyte-conditioned medium, so the few contaminating astrocytes could not have been responsible for more than a fraction of the neurocan present in O-2A-conditioned medium. Versican was also detected in the conditioned medium of O-2A lineage cells. The anti-versican mAb 12C5 recognized a single species of highMr that required chondroitinase ABC treatment to enter the gel (Fig. 5).
As with astrocytes, O-2A lineage cells did not label for neurocan. Unlike astrocytes, however, neurocan was not seen on the substrate around these cells; neither was it seen under the cells in permeabilized cultures. This implies that O-2A lineage cells produce and process neurocan but do not produce the other extracellular matrix component that mediates the attachment of neurocan to the substrate.
Other cell types
Neurocan was not detectable in the conditioned medium of macrophages or meningeal cells derived from newborn rat brain (data not shown).
Cytokines influence neurocan expression in astrocytes
Cytokines and growth factors implicated in CNS injury and astrogliosis were screened for their effect on neurocan expression in astrocytes. The amount of neurocan in astrocyte-conditioned medium was assessed by immunoblotting with the 1G2 mAb. Screening experiments indicated that TGFβ and EGF led to an increase in the amount of neurocan present in astrocyte-conditioned medium and that IFN-γ, interleukin-1 β (IL-1β), and PDGF-AB led to a decrease (Fig.8). Tumor necrosis factor α, ciliary neurotrophic factor, leukemia inhibitory factor, interleukin-6, and FGF2 were without obvious effect. Astrocytes were exposed to cytokines for either 4 or 6 d at the concentrations given in Table1.
Fig. 8.

Cytokines influence neurocan expression in cultured astrocytes. The cells were grown for 6 d in the presence of 10 ng/ml of the cytokine, and the conditioned medium was concentrated and digested with chondroitinase ABC and an equal volume (65 μl) was applied to each lane. The blot was labeled with the anti-neurocan mAb 1G2. An increase in the amount of neurocan was seen in response to TGFβ and to a greater extent with EGF. PDGF, IFN-γ, and IL-1β brought about a decline in the amount of neurocan that was detected.
The effect of TGFβ was most apparent after 2 and 4 d but rather less so after 6 and 8 d (Fig. 9). The more robust response to EGF was evident at all time points, however. Dose–response experiments indicated that 1.0 ng/ml TGFβ was optimal, insofar as 0.1 ng/ml had no discernible effect, and 10 ng/ml did not have a significantly greater effect than 1.0 ng/ml (Fig.10). EGF was ineffective at 0.1 and 1.0 ng/ml and only brought about an increase in neurocan expression at 10 ng/ml (Fig. 10). When EGF and TGFβ were used in combination, the effect was no greater than that seen with EGF alone. IFN-γ and IL-1β, and to a lesser extent PDGF, partially negated the effects of EGF (Fig. 10).
Fig. 9.

Time course of effects of TGFβ and EGF on neurocan expression in astrocytes. Cultured astrocytes were treated with TGFβ (10 ng/ml) or EGF (10 ng/ml) for 2, 4, 6, or 8 d. The conditioned medium was concentrated and treated with chondroitinase ABC, and an equal amount of protein (200 μg) was applied to each lane. The blot was labeled with the anti-neurocan mAb 1F6. TGFβ and EGF led to an increase in the amount of neurocan detected at all time points. The EGF-induced increase was at all time points greater than that of TGFβ.
Fig. 10.

Effects of the concentration of TGFβ and EGF on neurocan expression in astrocytes. Cultured astrocytes were treated with TGFβ or EGF alone (0.1, 1.0, or 10 ng/ml), or with EGF (10 ng/ml) and IFN-γ (10 ng/ml), IL-1β (10 ng/ml), or PDGF (10 ng/ml) for 4 d. The conditioned medium was concentrated and treated with chondroitinase ABC, and an equal volume (30 μl) was applied to each lane. The blot was labeled first with the anti-neurocan-C mAb 1G2 (above) and then with the anti-neurocan-130 mAb 1F6 (below). TGFβ was effective at 1.0 and 10 ng/ml but not 0.1 ng/ml, whereas EGF was only effective in bringing about an increase in neurocan expression at 10 ng/ml. IFN-γ and IL-1β appeared to override the effects of EGF. The EGF/IL-1β combination led to an increase in the amounts of neurocan-C and neurocan-130, relative to that of unprocessed neurocan.
The effects of TGFβ, EGF, IFN-γ, and PDGF were investigated in more detail (Fig. 11). Three separate astrocyte cultures (in 25 cm2 flasks) were treated with each cytokine (10 ng/ml) for 3 d. The effects seen previously were clearly reproduced. Each sample was equalized for total protein, and the results are therefore indicative of either an increase in neurocan synthesis or a decrease in its degradation or both. The scale of these effects was quantified by densitometry. TGFβ brought about a ninefold increase and EGF a 23-fold increase in the amount of neurocan detected. PDGF reduced neurocan levels to ∼20% of control, and IFN-γ brought about a 50% reduction. TGFα appeared as effective as EGF in stimulating neurocan synthesis (data not shown). None of the treatments consistently altered the ratios of intact to processed neurocan. To determine whether these effects were caused simply by an effect on astrocyte proliferation, nuclei were counted in one of the cultures that were used for the neurocan analysis. TGFβ, EGF, IFN-γ, and PDGF had no effect on cell number, in comparison with untreated astrocytes, under these conditions. These experiments were conducted on well established, confluent monolayers of astrocytes. These cells proliferate rapidly to form a monolayer, but cease to do so once it has been established (i.e., they exhibit contact inhibition). None of the factors tested had any effect on cell number, presumably because they were unable to overcome the effects of contact inhibition.
Fig. 11.

Quantification of the effects of TGFβ, EGF, IFNγ, and PDGF on neurocan expression in astrocytes. Three flasks of astrocytes were grown in the presence of each cytokine (10 ng/ml) for 3 d. The conditioned medium was treated with chondroitinase ABC, and an equal amount of protein (50 μg in a; 200 μg in b) was applied to each lane. The blot was labeled with the anti-neurocan mAb 1G2. The amount of neurocan core protein in each lane was quantified by densitometry. TGFβ brought about a ninefold increase in the amount of neurocan detected, and EGF caused a 23-fold increase (c). PDGF reduced neurocan levels to ∼20% of control, and IFNγ brought about a 50% reduction (d). The error bars represent SEM. By Student'st test, the effects of TGFβ, EGF, and PDGF were significant with p < 0.01. The effects of IFNγ were less significant (p < 0.05).
DISCUSSION
Neurocan is an inhibitory CSPG
We demonstrate in this paper that neurocan arranged as stripes on an L1 substrate repels axons, causing them to be guided along the neurocan-free stripes. Previous in vitro experiments have also shown that neurocan has inhibitory properties (Margolis and Margolis, 1997). Neurocan inhibits outgrowth from embryonic chick CNS neurons and from PC12D cells, probably through direct neuronal effects (Freidlander et al., 1994; Katoh-Semba et al., 1998). Neurocan binds with high affinity to the cell adhesion molecules N-CAM, L1/Ng-CAM, and TAG-1/axonin-1 (Friedlander et al., 1994; Milev et al., 1996), inhibits the homophilic binding of N-CAM and L1/Ng-CAM (Grumet et al., 1993), inhibits neuronal attachment to N-CAM and L1/Ng-CAM (Friedlander et al., 1994), and inhibits L1/Ng-CAM-dependent neurite outgrowth from embryonic chick neurons (Friedlander et al., 1994). Neurocan also binds with high affinity to tenascin (Grumet et al., 1994; Aspberg et al., 1997; Rauch et al., 1997; Milev et al., 1998a). In many cases, the inhibitory properties of CSPGs have been attributed to the chondroitin sulfate (Fichard et al., 1991; Brittis et al., 1992; Brittis and Silver, 1994; Smith-Thomas et al., 1994, 1995; Emerling and Lander, 1996). The binding of neurocan to L1/Ng-CAM was largely dependent on chondroitin sulfate (Friedlander et al., 1994), implying that the ability of neurocan to interfere with neurite outgrowth on L1/Ng-CAM may also depend on chondroitin sulfate. The combination of adhesion molecule and matrix binding may create large complexes with widespread effects on cell surface and matrix interactions and therefore multiple effects on axon growth.
In vivo neurocan is present in many of the structures that guide axons by repulsion during development. It has been suggested that the presence of neurocan in the roof plate prevents the incoming dorsal root ganglion axons from crossing the midline (Katoh-Semba et al., 1998), that neurocan repels retinal axons from the hypothalamus and epithalamus (Tuttle et al., 1998), and that neurocan is involved in the formation of the barrel fields in the somatosensory cortex (Watanabe et al., 1995). The presence of neurocan in glial barriers in the developing nervous system and its inhibitory effects in vitro make it entirely plausible that it would exert similar effects on regenerating axons in the glial scar.
Neurocan is upregulated in the injured CNS
Many previous studies have shown an upregulation of CSPG in CNS injuries. We have demonstrated that one of these CSPGs is neurocan. Immunolabeling showed that neurocan expression was greatly increasedin vivo around a knife lesion to the cerebral cortex, and Western blot analysis of neurocan in saline extracts prepared from injured and noninjured tissue revealed considerably more neurocan in the injured brain. It has been reported that neurocan is upregulated in response to spinal cord injury and entorhinal cortex lesions (Plant et al., 1998; Haas et al., 1999).
Source of neurocan
At various times the glial scar contains microglia, oligodendrocyte precursors, meningeal cells, astrocytes, and other cell types. Of these, we show that astrocytes and oligodendrocyte precursors are able to produce neurocan.
Oohira et al. (1994) showed that astrocyte-conditioned medium contains neurocan, and we have confirmed this. We made strenuous efforts to ensure that these cultures were devoid of pre-O-2A and O-2A progenitor cells and macrophages, and the few remaining contaminating cells are likely to be meningeal cells, which we have shown do not produce neurocan. In low-density astrocyte cultures, the only neurocan detectable by immunocytochemistry was found on the substrate around (and under) GFAP-positive cells. Such a distribution makes it highly likely that astrocytes are the source of this neurocan. The pericellular, substrate-bound distribution of neurocan in cultured astrocytes is somewhat reminiscent of that of hyaluronic acid (Asher and Bignami, 1991), and given the ability of neurocan to bind hyaluronic acid (Rauch et al., 1991), one might have expected that the attachment of neurocan to the substrate is mediated by hyaluronic acid. This is evidently not the case, however, because hyaluronidase had no effect on the neurocan labeling around astrocytes. Neurocan was also detected in the conditioned medium of O-2A lineage cells, and they too did not label for neurocan. Neither did we observe labeling of the substrate around these cells. The binding of neurocan to the substrate must therefore be mediated by something that astrocytes, but not O-2A cells, produce.
Although neurocan is produced by astrocytes, it is not retained on the astrocyte cell surface. This may explain why conventional astroglial monolayers support robust neurite outgrowth (Noble et al., 1984), whereas three-dimensional cultures do not (Fawcett et al., 1989). We have shown previously that proteoglycans impede the growth of axons in three-dimensional cultures, because sodium chlorate, which prevents GAG sulfation, renders them more permissive (Smith-Thomas et al., 1995). In conventional monolayer cultures, neurocan is not retained at the cell surface and will be greatly diluted by the medium, whereas in three-dimensional cultures it is trapped between the cells, where it would be in a position to interfere with access to neurite outgrowth-promoting molecules on astrocytes.
In situ hybridization has shown that neurocan mRNA is present in neurons in the postnatal cerebellum (Engel et al., 1996). Neurocan mRNA is also widely expressed in the late fetal forebrain, especially in the major neuronal cell layers (Engel et al., 1996). Curiously, neurocan immunoreactivity is much less widespread than the mRNA in late fetal forebrain, possibly because of translational block (Meyer-Puttlitz et al., 1996). Immediately after birth, neurocan immunoreactivity spreads throughout the cerebral cortex, and the cellular origin(s) of this neurocan has not been identified (Oohira et al., 1994; Tuttle et al., 1995). The ability of newborn rat brain astrocytes to produce neurocan in vitro suggests that some of this neurocan may be produced by astrocytes (Milev et al., 1998b). In addition, oligodendrocyte precursors appear in large numbers in the CNS at this time and may be another source, because these cells also produce neurocan in vitro. The strong neurocan immunoreactivity that we have seen in white matter tracts argues in favor of a glial origin.
In the injured CNS, the appearance of the neurocan labeling is consistent with a glial origin and could be from astrocytes or from oligodendrocyte precursor cells, which are recruited to CNS injuries in large numbers (Levine, 1994). A recent study has demonstrated that in the injured CNS astrocytes can produce neurocan (Haas et al., 1999).
Neurocan processing
Neurocan exists as the full-length, intact form and as two smaller chondroitin sulfate-bearing species (Fig. 3). The two smaller species are thought to arise as a result of a single proteolytic cleavage between amino acids 638 and 639 (Rauch et al., 1992; Matsui et al., 1994). Northern blot analysis has shown a single (7.5 kb) neurocan transcript in P4 and adult rat brain (Rauch et al., 1992). This and the structure of the neurocan gene (Rauch et al., 1995) argue against the generation of the three forms of neurocan by alternative splicing. The most likely scenario is that the two smaller forms arise as a result of a single proteolytic cleavage. In vitro, astrocytes (and oligodendrocyte precursors) were themselves capable of processing neurocan. The generation of the smaller fragments was not affected, however, by the addition of (non-membrane permeant) serine or cysteine protease or metalloproteinase inhibitors to the culture medium. These findings suggest that processing occurs intracellularly. In the normal adult CNS the processed forms of the molecule predominate, but after injury it was primarily the intact form of neurocan that was upregulated.
Control of neurocan expression
What might cause the upregulation of neurocan in CNS lesions? TGFβ has been implicated in the increased deposition of ECM in the glial scar. TGFβ-neutralizing antibodies reduced the deposition of laminin, fibronectin (Logan and Berry, 1993; Lagord et al., 1999), and CSPG (Griffith and McKeon, 1999) in the glial scar. TGFβ also increased the expression of tenascin (Smith and Hale, 1997), laminin, and fibronectin (Baghdassarian et al., 1993) in cultured astrocytes. We therefore investigated the control of neurocan expression in astrocytes by cytokines known to be released in response to CNS injury. EGF/TGFα and TGFβ brought about an increase in the amount of neurocan relative to the total protein content in the conditioned medium of these cells, whereas PDGF and IFN-γ brought about a decrease.
EGF has been shown to increase TGFβ expression in astrocytes (Lindholm et al., 1992), and it is therefore possible that the effects of EGF on astrocytes are mediated by TGFβ. Alternatively, it is conceivable that EGF mimics the effects of TGFα on astrocytes, the overexpression of which is sufficient to induce astrogliosis in vivo (Rabchevsky et al., 1998).
Inhibition in the glial scar
Neurocan is not the only CSPG that is upregulated in response to CNS injury. Other studies have shown increased expression of NG2 (Levine, 1994; Grill et al., 1998), versican (Asher et al., 1999), decorin (Stichel et al., 1995), biglycan (Stichel et al., 1995), and phosphacan (McKeon et al., 1995; Barker et al., 1996). We hypothesize that TGFβ or TGFα/EGF or both induce an increase in the expression of neurocan in (reactive) astrocytes and that this and recruitment of neurocan-producing oligodendrocyte precursors lead to the accumulation of this inhibitory CSPG at the site of injury. The many complex effects of neurocan and other CSPGs then impede axonal regeneration.
Footnotes
This work was supported by the Wellcome Trust, the International Spinal Research Trust, the Medical Research Council, and Action Research. We thank Dr. S. Jenner (Department of Physiology, Cambridge) for help with densitometry and Dr. R. W. Farndale (Department of Biochemistry, Cambridge) for use of the densitometer. The 5A5 monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank, which was developed under the auspices of the National Institute of Child Health and Human Development and is maintained by the Department of Biological Sciences at the University of Iowa, Iowa City, IA.
Correspondence should be addressed to James Fawcett, Physiological Laboratory, University of Cambridge, Downing Street, Cambridge CB2 3EG, UK. E-mail: jwf108@cam.ac.uk.
REFERENCES
- 1.Asher R, Bignami A. Localization of hyaluronate in primary glial cell cultures derived from newborn rat brain. Exp Cell Res. 1991;195:401–411. doi: 10.1016/0014-4827(91)90390-g. [DOI] [PubMed] [Google Scholar]
- 2.Asher R, Perides G, Vanderhaeghen J-J, Bignami A. Extracellular matrix of central nervous system white matter: demonstration of an hyaluronate-protein complex. J Neurosci Res. 1991;28:410–421. doi: 10.1002/jnr.490280314. [DOI] [PubMed] [Google Scholar]
- 3.Asher RA, Scheibe RJ, Keiser HD, Bignami A. On the existence of a cartilage-like proteoglycan and link proteins in the central nervous system. Glia. 1995;13:294–308. doi: 10.1002/glia.440130406. [DOI] [PubMed] [Google Scholar]
- 4.Asher RA, Fidler PS, Rogers JH, Fawcett JW. TGFbeta stimulates neurocan synthesis in cultured rat astrocytes. Soc Neurosci Abstr. 1998;24:56. [Google Scholar]
- 5.Asher RA, Morgenstern DA, Adcock KH, Rogers JH, Fawcett JW. Versican is up-regulated in CNS injury and is a product of O-2A lineage cells. Soc Neurosci Abstr. 1999;25:750. [Google Scholar]
- 6.Aspberg A, Miura R, Bourdoulous S, Shimonaka M, Heinegård D, Schachner M, Ruoslahti E, Yamaguchi Y. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc Natl Acad Sci USA. 1997;94:10116–10121. doi: 10.1073/pnas.94.19.10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baghdassarian D, Toru-Delbauffe D, Gavaret JM, Pierre M. Effects of transforming growth factor-β1 on the extracellular matrix and cytoskeleton of cultured astrocytes. Glia. 1993;7:193–202. doi: 10.1002/glia.440070302. [DOI] [PubMed] [Google Scholar]
- 8.Bähr M, Przyrembel C, Bastmeyer M. Astrocytes from adult rat optic nerves are nonpermissive for regenerating retinal ganglion cell axons. Exp Neurol. 1995;131:211–220. doi: 10.1016/0014-4886(95)90043-8. [DOI] [PubMed] [Google Scholar]
- 9.Baier H, Klostermann S. Axon guidance and growth cone collapse in vitro. Neuroprotocols. 1994;4:96–105. [Google Scholar]
- 10.Barker RA, Dunnett SB, Faissner A, Fawcett JW. The time course of loss of dopaminergic neurons and the gliotic reaction surrounding grafts of embryonic mesencephalon to the striatum. Exp Neurol. 1996;141:79–93. doi: 10.1006/exnr.1996.0141. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Hur T, Rogister B, Murray K, Rougon G, Dubois-Dalcq M. Growth and fate of PSA-NCAM+ precursors of the postnatal brain. J Neurosci. 1998;18:5777–5788. doi: 10.1523/JNEUROSCI.18-15-05777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bovolenta P, Wandosell F, Nieto-Sampedro M. Characterization of a neurite outgrowth inhibitor expressed after CNS injury. Eur J Neurosci. 1993;5:454–465. doi: 10.1111/j.1460-9568.1993.tb00512.x. [DOI] [PubMed] [Google Scholar]
- 13.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Brittis PA, Silver J. Exogenous glycosaminoglycans induce complete inversion of retinal ganglion cell bodies and their axons within the retinal neuroepithelium. Proc Natl Acad Sci USA. 1994;91:7539–7542. doi: 10.1073/pnas.91.16.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brittis PA, Canning DR, Silver J. Chondroitin sulfate as a regulator of neuronal patterning in the retina. Science. 1992;255:733–736. doi: 10.1126/science.1738848. [DOI] [PubMed] [Google Scholar]
- 16.Davies SJA, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- 17.Davies SJA, Goucher DR, Doller C, Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;19:5810–5822. doi: 10.1523/JNEUROSCI.19-14-05810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodd J, Morton SB, Karagogeos D, Yamamoto M, Jessell TM. Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron. 1988;1:105–116. doi: 10.1016/0896-6273(88)90194-8. [DOI] [PubMed] [Google Scholar]
- 19.Dou C-L, Levine JM. Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J Neurosci. 1994;14:7616–7628. doi: 10.1523/JNEUROSCI.14-12-07616.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emerling DE, Lander AD. Inhibitors and promoters of thalamic neuron adhesion and outgrowth in embryonic neocortex: functional association with chondroitin sulfate. Neuron. 1996;17:1089–1100. doi: 10.1016/s0896-6273(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 21.Engel M, Maurel P, Margolis RU, Margolis RK. Chondroitin sulfate proteoglycans in the developing central nervous system. I. Cellular sites of synthesis of neurocan and phosphacan. J Comp Neurol. 1996;366:34–43. doi: 10.1002/(SICI)1096-9861(19960226)366:1<34::AID-CNE3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 22.Fawcett JW, Asher RA. The glial scar and CNS repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- 23.Fawcett JW, Housden E, Smith-Thomas LC, Meyer RL. The growth of axons in three-dimensional astrocyte cultures. Dev Biol. 1989;135:449–458. doi: 10.1016/0012-1606(89)90193-0. [DOI] [PubMed] [Google Scholar]
- 24.Fichard A, Verna JM, Olivares J, Saxod R. Involvement of a chondroitin sulfate proteoglycan in the avoidance of chick epidermis by dorsal root ganglia fibers: a study using β-d-xyloside. Dev Biol. 1991;148:1–9. doi: 10.1016/0012-1606(91)90312-q. [DOI] [PubMed] [Google Scholar]
- 25.Fidler PS, Schuette K, Asher RA, Dobbertin A, Thornton SR, Calle-Patino Y, Muir E, Levine JM, Geller HM, Rogers JH, Faissner A, Fawcett JW. Comparing astrocytic cells lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci. 1999;19:8778–8788. doi: 10.1523/JNEUROSCI.19-20-08778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frisen J, Haegerstrand A, Risling M, Fried K, Johansson CB, Hammarberg H, Elde R, Hokfelt T, Cullheim S. Spinal axons in central nervous system scar tissue are closely related to laminin-immunoreactive astrocytes. Neuroscience. 1995;65:293–304. doi: 10.1016/0306-4522(94)00467-j. [DOI] [PubMed] [Google Scholar]
- 28.Gates MA, Fillmore H, Steindler DA. Chondroitin sulfate proteoglycan and tenascin in the wounded adult mouse neostriatum in vitro: dopamine neuron attachment and process outgrowth. J Neurosci. 1996;16:8005–8018. doi: 10.1523/JNEUROSCI.16-24-08005.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffith RW, McKeon RJ. Immunoneutralization of TGFbeta attenuates CS-PG synthesis and enhances process outgrowth in vitro and in vivo. Soc Neurosci Abstr. 1999;25:749. [Google Scholar]
- 31.Grill RJ, Stallcup WB, Tuszynski MH. Temporal upregulation and spatial distribution of putative inhibitory and growth permissive substrate molecules in the injured adult rat spinal cord. Soc Neurosci Abstr. 1998;24:1054. [Google Scholar]
- 32.Grumet M, Flaccus A, Margolis RU. Functional characterization of chondroitin sulfate proteoglycans of brain: interactions with neurons and neural cell adhesion molecules. J Cell Biol. 1993;120:815–824. doi: 10.1083/jcb.120.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grumet M, Milev P, Sakurai T, Karthikeyan L, Bourdon M, Margolis RK, Margolis RU. Interactions with tenascin and differential effects on cell adhesion of neurocan and phosphacan, two major chondroitin sulfate proteoglycans of nervous tissue. J Biol Chem. 1994;269:12142–12146. [PubMed] [Google Scholar]
- 34.Haas CA, Rauch U, Thon N, Merten T, Deller T. Entorhinal cortex lesion in adult rats induces the expression the neuronal chondroitin sulfate proteoglycan neurocan in reactive astrocytes. J Neurosci. 1999;19:9953–9963. doi: 10.1523/JNEUROSCI.19-22-09953.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh-Semba R, Matsuda M, Watanabe E, Maeda N, Oohira A. Two types of brain chondroitin sulfate proteoglycan: their distribution and possible functions in the rat embryo. Neurosci Res. 1998;31:273–282. doi: 10.1016/s0168-0102(98)00047-9. [DOI] [PubMed] [Google Scholar]
- 36.Lagord C, Gonzalez AM, Clarke WE, Jackson RH, Baird A, Logan A. Antagonizing TGFbeta activity in CNS wounds inhibits glial scarring. Soc Neurosci Abstr. 1999;25:749. [Google Scholar]
- 37.Laywell ED, Dorries U, Bartsch U, Faissner A, Schachner M, Steindler DA. Enhanced expression of the developmentally regulated extracellular matrix molecule tenascin following adult brain injury. Proc Natl Acad Sci USA. 1992;89:2634–2638. doi: 10.1073/pnas.89.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Roux PD, Reh TA. Reactive astroglia support primary dendritic but not axonal outgrowth from mouse cortical neurons in vitro. Exp Neurol. 1996;137:49–65. doi: 10.1006/exnr.1996.0006. [DOI] [PubMed] [Google Scholar]
- 39.Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci. 1994;14:4716–4730. doi: 10.1523/JNEUROSCI.14-08-04716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-β1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Logan A, Berry M. Transforming growth factor-β1 and basic fibroblast growth factor in the injured CNS. Trends Neurosci. 1993;14:337–343. doi: 10.1016/0165-6147(93)90007-7. [DOI] [PubMed] [Google Scholar]
- 42.Margolis RU, Margolis RK. Chondroitin sulfate proteoglycans as mediators of axon growth and pathfinding. Cell Tissue Res. 1997;290:343–348. doi: 10.1007/s004410050939. [DOI] [PubMed] [Google Scholar]
- 43.Matsui F, Watanabe E, Oohira A. Immunological identification of two proteoglycan fragments derived from neurocan, a brain-specific chondroitin sulfate proteoglycan. Neurochem Int. 1994;25:425–431. doi: 10.1016/0197-0186(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 44.McKeon RJ, Schreibe RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKeon RJ, Höke A, Silver J. Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol. 1995;136:32–43. doi: 10.1006/exnr.1995.1081. [DOI] [PubMed] [Google Scholar]
- 46.Meyer-Puttlitz B, Milev P, Junker E, Zimmer I, Margolis RU, Margolis RK. Chondroitin sulfate and chondroitin/keratan sulfate proteoglycans of nervous tissue: developmental changes of neurocan and phosphacan. J Neurochem. 1995;65:2327–2337. doi: 10.1046/j.1471-4159.1995.65052327.x. [DOI] [PubMed] [Google Scholar]
- 47.Meyer-Puttlitz B, Junker E, Margolis RU, Margolis RK. Chondroitin sulfate proteoglycans in the developing central nervous system. II. Immunocytochemical localization of neurocan and phosphacan. J Comp Neurol. 1996;366:44–54. doi: 10.1002/(SICI)1096-9861(19960226)366:1<44::AID-CNE4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 48.Milev P, Maurel P, Häring M, Margolis RK, Margolis RU. TAG-1/axonin-1 is a high-affinity ligand of neurocan, phosphacan/protein-tyrosine phosphatase-ζ/β, and N-CAM. J Biol Chem. 1996;271:15716–15723. doi: 10.1074/jbc.271.26.15716. [DOI] [PubMed] [Google Scholar]
- 49.Milev P, Chiba A, Häring M, Rauvala H, Schachner M, Ranscht B, Margolis RK, Margolis RU. High affinity binding and overlapping localization of neurocan and phosphacan/protein-tyrosine phosphatase-ζ/β with tenascin-R, amphoterin, and the heparin-binding growth-associated molecule. J Biol Chem. 1998a;273:6998–7005. doi: 10.1074/jbc.273.12.6998. [DOI] [PubMed] [Google Scholar]
- 50.Milev P, Maurel P, Chiba A, Mevissen M, Popp S, Yamaguchi Y, Margolis RK, Margolis RU. Differential regulation of expression of hyaluronan-binding proteoglycans in brain: aggrecan, versican, neurocan, and brevican. Biochem Biophys Res Commun. 1998b;247:207–212. doi: 10.1006/bbrc.1998.8759. [DOI] [PubMed] [Google Scholar]
- 51.Moon LDF, Rhodes K, Fawcett JW. Extensive axon regeneration following in vivo treatment of rat brain the chondroitinase ABC. Soc Neurosci Abstr. 1999;25:751. [Google Scholar]
- 52.Noble M, Fok-Seang J, Cohen J. Glia are a unique substrate for the in vitro growth of central nervous system neurons. J Neurosci. 1984;4:1892–1903. doi: 10.1523/JNEUROSCI.04-07-01892.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oohira A, Matsui F, Watanabe E, Kushima Y, Maeda N. Developmentally regulated expression of a brain specific species of chondroitin sulfate proteoglycan, neurocan, identified with a monoclonal antibody 1G2 in the rat cerebrum. Neuroscience. 1994;60:145–157. doi: 10.1016/0306-4522(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 54.Pindzola RR, Doller C, Silver J. Putative inhibitory extracellular matrix molecules at the dorsal root entry zone of the spinal cord during development and after root and sciatic nerve lesions. Dev Biol. 1993;156:34–48. doi: 10.1006/dbio.1993.1057. [DOI] [PubMed] [Google Scholar]
- 55.Plant GW, Dimitropolou A, Bates ML, Bunge MB. The expression of inhibitory proteoglycans following transplantation of Schwann cell grafts into completely transected rat spinal cord. Soc Neurosci Abstr. 1998;24:69. [Google Scholar]
- 56.Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier M-P. A role for transforming growth factor α as an inducer of astrogliosis. J Neurosci. 1998;18:10541–10552. doi: 10.1523/JNEUROSCI.18-24-10541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rauch U, Gao P, Janetzko A, Flaccus A, Hilgenberg L, Tekotte H, Margolis RK, Margolis RU. Isolation and characterization of developmentally regulated chondroitin sulfate and chondroitin/keratan sulfate proteoglycans of brain identified with monoclonal antibodies. J Biol Chem. 1991;266:14785–14801. [PubMed] [Google Scholar]
- 58.Rauch U, Karthikeyan L, Maurel P, Margolis RU, Margolis RK. Cloning and primary structure of neurocan, a developmentally regulated, aggregating chondroitin sulfate proteoglycan of brain. J Biol Chem. 1992;267:19536–19547. [PubMed] [Google Scholar]
- 59.Rauch U, Grimpe B, Kulbe G, Arnold-Ammer I, Beier DR, Fassler R. Structure and chromosomal localization of the mouse neurocan gene. Genomics. 1995;28:405–410. doi: 10.1006/geno.1995.1168. [DOI] [PubMed] [Google Scholar]
- 60.Rauch U, Clement A, Retzler C, Frohlich L, Fassler R, Gohring W, Faissner A. Mapping of a defined neurocan binding site to distinct domains of tenascin-C. J Biol Chem. 1997;272:26905–26912. doi: 10.1074/jbc.272.43.26905. [DOI] [PubMed] [Google Scholar]
- 61.Smith GM, Hale JH. Macrophage/microglia regulation of astrocytic tenascin: synergistic action of transforming growth factor-β and basic fibroblast growth factor. J Neurosci. 1997;17:9624–9633. doi: 10.1523/JNEUROSCI.17-24-09624.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith GM, Miller RH, Silver J. Changing role of forebrain astrocytes during development, regenerative failure, and induced regeneration upon transplantation. J Comp Neurol. 1986;251:23–43. doi: 10.1002/cne.902510103. [DOI] [PubMed] [Google Scholar]
- 63.Smith-Thomas LC, Fok-Seang J, Stevens J, Du J-S, Muir E, Faissner A, Geller HM, Rogers JH, Fawcett JW. An inhibitor of neurite outgrowth produced by astrocytes. J Cell Sci. 1994;107:1687–1695. doi: 10.1242/jcs.107.6.1687. [DOI] [PubMed] [Google Scholar]
- 64.Smith-Thomas LC, Stevens J, Fok-Seang J, Faissner A, Rogers JH, Fawcett JW. Increased axon regeneration in astrocytes grown in the presence of proteoglycan synthesis inhibitors. J Cell Sci. 1995;108:1307–1315. doi: 10.1242/jcs.108.3.1307. [DOI] [PubMed] [Google Scholar]
- 65.Stichel CC, Kappler J, Junghans U, Koops A, Kresse H, Müller HW. Differential expression of the small chondroitin/dermatan sulfate proteoglycans decorin and biglycan after injury of the adult rat brain. Brain Res. 1995;704:263–274. doi: 10.1016/0006-8993(95)01131-5. [DOI] [PubMed] [Google Scholar]
- 66.Tuttle R, Schlaggar BL, Braisted JE, O'Leary DDM. Maturation-dependent upregulation of growth-promoting molecules in developing cortical plate controls thalamic and cortical neurite growth. J Neurosci. 1995;15:3039–3052. doi: 10.1523/JNEUROSCI.15-04-03039.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tuttle R, Braisted JE, Richards LJ, O'Leary DDM. Retinal axon guidance by region-specific cues in diencephalon. Development. 1998;125:791–801. doi: 10.1242/dev.125.5.791. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe E, Aono S, Matsui F, Yamada Y, Naruse I, Oohira A. Distribution of a brain-specific proteoglycan, neurocan, and the corresponding mRNA during the formation of barrels in the rat somatosensory cortex. Eur J Neurosci. 1995;7:547–554. doi: 10.1111/j.1460-9568.1995.tb00659.x. [DOI] [PubMed] [Google Scholar]
- 69.Zuo J, Neubauer D, Dyess K, Ferguson TA, Muir D. Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp Neurol. 1998;154:654–662. doi: 10.1006/exnr.1998.6951. [DOI] [PubMed] [Google Scholar]