

Abstract
NMDA receptors in mice were mutated by gene targeting to substitute asparagine (N) in position 598 of the NR1 subunit to glutamine (Q) or arginine (R). Animals expressing exclusively the mutatedNR1 alleles, NR1Q/Q andNR1−/R mice, developed a perinatally lethal phenotype mainly characterized by respiratory failure. The dysfunctions were partially rescued in heterozygous mice by the presence of pure wild-type receptors. Thus, NR1+/Q mice exhibited reduced life expectancy, with females being impaired in nurturing; NR1+/R mice displayed signs of underdevelopment such as growth retardation and impaired righting reflex, and died before weaning. We analyzed the key properties of NMDA receptors, high Ca2+ permeability, and voltage-dependent Mg2+ block, in the mutant mice. Comparison of the complex physiological and phenotypical changes observed in the different mutants indicates that properties controlled by NR1 subunit residue N598 are important for autonomic brain functions at birth and during postnatal development. We conclude that disturbed NMDA receptor signaling mediates a variety of neurological phenotypes.
Keywords: NMDA receptor, gene targeting, Cre-loxP, Mg2+ block, Ca2+ influx, coincidence detection, respiration, nurturing, barrel cortex, LTP
NMDA receptors are glutamate-gated ion channels expressed by the majority of central neurons at all developmental stages. They are best characterized by a slow response tol-glutamate, the major excitatory central neurotransmitter, high permeability to Ca2+, and voltage-dependent gating (Mayer and Westbrook, 1987; Ascher and Nowak, 1988). During development, NMDA receptors are important for neuronal survival, differentiation, and migration (Balázs et al., 1989;Brewer and Cotman, 1989; Komuro and Rakic, 1993) and for formation and stabilization of synapses and circuits (Constantine-Paton et al., 1990;Fox and Daw, 1993). In the postnatal and adult brain, NMDA receptors are coincidence detectors of presynaptic and postsynaptic activity, because channel gating requires presynaptic glutamate release and simultaneous depolarization of the postsynaptic membrane. Coincidence detection by the NMDA receptor rests on a voltage-dependent channel block by extracellular Mg2+. The voltage-controlled Ca2+ influx by the NMDA receptor is thought to be essential for activity-dependent modulations in synaptic strength (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993).
Functional NMDA receptors are heteromeric assemblies (Hollmann and Heinemann, 1994; Dingledine et al., 1999) of the principal NR1 subunit (Moriyoshi et al., 1991) with the modulatory NR2 subunits (NR2A to 2D) (Kutsuwada et al., 1992; Meguro et al., 1992; Monyer et al., 1992;Ishii et al., 1993). Studies on recombinant NMDA receptors identified a single amino acid residue in the NR1 subunit, asparagine 598 (N598), as a critical determinant for the key properties of the NMDA receptor, high Ca2+ permeability, and voltage-dependent Mg2+ block (Burnashev et al., 1992). It was subsequently found that N598, which contributes to the narrow constriction of the channel pore (Kuner et al., 1996;Wollmuth et al., 1996), also controls gating properties, potentiation and block by polyamines, inhibition by protons and Zn2+, and affinity to glutamate and glycine (Kashiwagi et al., 1997; Schneggenburger and Ascher, 1997;Traynelis et al., 1998; Zheng et al., 1999).
Mice deficient in NMDA receptors demonstrated the importance of this receptor for neuronal development and plasticity. NMDA receptor “knock-out” mice (NR1−/−), which lack the NR1 subunit, do not feed, fail to develop whisker-related patterns (barrelettes) in the brainstem trigeminal complex (BSTC), and die 10 hr after birth from respiratory failure (Forrest et al., 1994;Li et al., 1994). NR2B-deficient mice, which lack most embryonic NMDA receptors, do not suckle and starve to death within a day after birth. When handfed to live for several days, the mutant mice fail to form the barrelette structure in the BSTC (Kutsuwada et al., 1996). Mice expressing low levels of NMDA receptors are impaired in barrel structure formation in the somatosensory cortex (Iwasato et al., 1997), and mice that lack the NMDA receptor specifically in hippocampal CA1 pyramidal cells fail to establish long-lasting changes in synaptic strength of these neurons (Tsien et al., 1996).
In this study, we generated mice that express mutant NMDA receptors as a consequence of NR1 gene targeting-assisted codon substitutions N598Q and N598R for the critical channel site. We analyzed key physiological parameters of the NMDA receptor in mice that express exclusively mutant receptors and determined in heterozygotes the dominance of the mutated NR1 subunits. Based on the phenotypic appearance of the mutant mice, the activity-dependent Ca2+ influx through the NMDA receptor is likely to play an essential role in autonomic brain functions. Moreover, disturbed NMDA receptor-mediated signaling in combination with reduced numbers of pure wild-type receptors leads to dysfunctions of the nervous system, the severity of which depends on the dominance of the mutation.
MATERIALS AND METHODS
Animal experiments. Animal care was in compliance with the institutional guidelines at the animal facility of the Center for Molecular Biology, INF 282, D-69120 Heidelberg, Germany. Transgenic manipulations were performed according to a license (37–9185.81/35/97) of the Regierungspräsidium (Karlsruhe, Germany).
Generation and analysis of mutant alleles and mice. The NR1 gene-targeting vector was constructed from genomic 129/Sv mouse strain DNA. Codon exchanges and restriction sites for cloning and diagnostic purposes were introduced by PCR mutagenesis with primers: N1in10Ndo (5′-CGGAATTCGCGGCCGCTTGGGATTTACTGCAGCAC-3′) for the uniqueNotI site in intron 10 used for linearization of the targeting vector; N1LQSdo (5′-GGCGTCCTGCTGCAGTCTGGCATTGG-3′) and N1LQSup (5′-CCAATGCCAGACTGCAGCAGGACGCC-3′) for the N598Q codon exchange and the diagnostic PstI site in exon 15; N1LRSdo (5′-GGCGTCCTGCTCAGATCTGGCATTGG-3′) and N1LRSup (5′-CCAATGCCAGATCTGAGCAGGACGCC-3′) for the N598R codon exchange and the diagnostic BglII site in exon 15; N1in18Xdo (5′-CACCAAACTACTCGAGCCCTGGCCTGGC-3′) and N1in18Xup(5′-GCCAGGCCAGGGCTCGAGTAGTTTGGTG-3′) for the uniqueXhoI site in intron 18 used for in-sense insertion of aloxP-flanked neomycin phosphotransferase gene (neo) as a selection marker. The final targeting vector comprised ∼2.2 kb of 5′ and ∼8 kb of 3′ sequences relative to theneo gene (Fig.1B). R1 mouse embryonic stem (ES) cells (Nagy et al., 1993) were electroporated [107 cells; Bio-Rad (Hercules, CA) gene pulser set at 240 V and 500 μF] with 40 μg ofNotI-linearized construct. G418-resistant (250 μg/ml) colonies were screened for homologous recombination by nested PCR with first primer pair, primers 1 (N1in10do1, 5′-GGATCTGTCCCCAAGGGTAGC-3′) and 2 (pgkprom1, 5′-GAATGTGTGCGAGGCCAGAGG-3′), and second primer pair, primers 3 (N1in10do2, 5′-CTAGCCATGTCAGAAGGATGTG-3′) and 4 (pgkprom2, 5′-CAGACTGCCTTGGGAAAAGCG-3′). Integration of the point mutations was assessed by restriction analysis of the resultant 2.5 kb PCR product with PstI and BglII for the NR1(N598Q) and NR1(N598R) mutations, respectively, and was confirmed by DNA sequence analysis. For neo gene elimination, the recombinant ES cells were electroporated with 30 μg of Cre-encoding plasmid pMC-Cre (Gu et al., 1993). Cre recombination events were detected by PCR with primers 5 (N1ex18do1, 5′-CTGGGACTCAGCTGTGCTGG-3′) and 6 (N1in18up1, 5′-AGGGGAGGCAACACTGTGGAC-3′). PCR products were 455 and 532 bp DNA fragments for the wild-type and mutant alleles, respectively (numbering and location of primers as in Fig. 1B). Genotypes of the PCR-positive clones were confirmed by Southern blot analysis probed with a 830 bp AvrII–EcoRV rat NR1cDNA fragment (Fig. 1A,C). Targeting-positive ES cells were injected into C57Bl/6 mouse blastocysts, and chimeric animals were backcrossed to C57Bl/6 mice. In progeny analysis at postnatal day zero (P0) all mutant NR1 alleles were distributed at Mendelian frequency. For maintenance of mouse lines, tail DNA was genotyped by PCR. The NR1Qand NR1R alleles were detected with primers 5 and 6, as used for the ES cell analysis. TheNR1Qneo andNR1Rneo alleles were identified by amplification of neo gene sequences with primers rspneo4 (5′-GGCTATTCGGCTATGACTGGGC-3′) and rspneo5 (5′-GGGTAGCCAACGCTATGTCCTG-3′), resulting in a 624 bp DNA fragment. The presence of the cre gene allele was determined with primers rspcre1 (5′-ACCAGGTTCGTTCACTCATGG-3′) and rspcre2 (5′-AGGCTAAGTGCCTTCTCTACAC-3′) for a 216 bp cre gene amplicon. The NR1− allele was identified by multiplex PCR using primers NR1–301 (5′-CCAACGCCATACAGATGGCCCTGT-3′), neo2300R (5′-GTGCCAGCGGGGCTGCTAAAG-3′), and NR1–445R (5′-CCAGCCTGCACACTTTAGGTCACATTG-3′), generating PCR products of 1138 bp for the wild-type and 477 bp for the mutant allele.
Fig. 1.

Targeted manipulation of the NR1gene. A, Schematic representation of the NR1 subunit. The signal peptide (S), putative membrane segments (M1–M4), and the N598 position (N) in M2 are indicated. The horizontal bar delineates the cDNA probe used for Southern analysis inC. B, Exon–intron organization of gene segments corresponding to NR1+ (wild-type allele), NR1Q(R)neo (targeted alleles), andNR1Q(R) (targeted alleles after Cre recombination). Boxes represent exons 11–22. Coding regions are gray, M1–M4 encoding exons areblack. Correspondence to NR1 subunit domains is indicated by dashed lines to A. The codon 598 in exon 15 for the wild-type (N) and mutated (Q or R) amino acids in M2 is marked by an arrowhead above the respective alleles. Relevant restriction sites are indicated and marked by asteriskswhen introduced for cloning or diagnostic purposes: E,EcoRI; X*, XhoI;P*, PstI; B*,BglII. loxP elements are shown asfilled triangles, the neo gene as anopen box. Filled circles in theNR1Q(R)neo allele delineate the 5′ and 3′ termini of the targeting construct. PCR primers for screening G418-resistant ES cell clones (primers 1–4) and for Cre recombination events (primers 5 and 6) are indicated by horizontal arrows above the NR1Q(R)neo allele. The EcoRI restriction fragments in kilobases (kb) from the different alleles are represented by horizontal lines below the alleles. C, Southern blot ofEcoRI-digested genomic ES cell DNA from the clones injected into blastocytes. The cDNA probe indicated in Adetects the wild-type allele (16.8 kb), the 5′ (6.2 kb), and 3′ (12.7 kb and 10.7 kb after neo gene elimination) homologous recombination events as well as random integrations.
In vivo deletion of the neo gene in mice.The dominant lethal effect of the NR1Rallele forced us to create the NR1−/Rgenotype by first crossing NR1+/− mice with a cre-transgenic “deleter” strain, in which the recombinase is expressed during early embryogenesis (Schwenk et al., 1995). We then crossedNR1+/−cre+/−animals with NR1+/Rneo mutants to obtainNR1−/Rcre+/−mice, in which the allele-silencing neo gene is removed from the NR1Rneo allele. For simplicity, this genotype is designated as NR1−/R.NR1+/R mice were obtained by crossing NR1+/Rneo mutants with the deleter strain. The genetic background ofNR1+/− mice was 129/Sv × C57Bl/6, and that of the deleter strain was C57Bl/6 × DBA2, backcrossed to CD1 or C57Bl/6 mice.
Immunoblot analysis. Crude membrane protein preparation from P0 total brain tissue and immunoblotting were performed as described (Sprengel et al., 1998). Proteins (20 μg/lane) were separated by SDS/PAGE (10%). NR1 protein was visualized by use of anti-rat NMDA receptor-1 mouse monoclonal antibody mAb 54.1 at 0.5 μg/ml (Siegel et al., 1994) and ECL detection (Amersham-Pharmacia, Braunschweig, Germany).
mRNA quantification. From total brain RNA, NR1cDNA fragments were amplified by RT-PCR with primers N1PCR7 (5′-TGTGGAATTCA-ATGAGGATGGGGA-3′) and N1ex20up1 (5′-CCAGCTGCATCTGC-TTCCTAC-3′). The amplified DNA fragment of 1542 bp was digested by EcoRI and BsrfI, and the 1093 bp restriction fragment encoding the M2 segment was inserted intoEcoRI- and XmaI-digested M13mp19 replicative form DNA. Ligated products were subcloned in Escherichia coliJM101 cells, and the plaques containing NR1 wild-type or mutant-derived cDNA inserts were detected by differential oligonucleotide hybridization (Higuchi et al., 1993), with oligonucleotides N1LQSup and N1LRSup for the mutant alleles and N1M2hyb (5′-CCAATGCCAGAGTTGAGCAGGACGCC-3′) for the wild-type allele.
Electrophysiology. Coronal or transverse slices (250 μm) were prepared from the brains of P0 or P14 mice, respectively. Currents evoked by fast application of NMDA (100 μm) in the presence of glycine (10 μm) were measured in nucleated soma patches pulled from identified hippocampal CA1 pyramidal cells as described (Brusa et al., 1995). Duration of the NMDA pulse was 50–100 msec. The standard extracellular solution was (in mm): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES–NaOH, pH 7.2. The intracellular solution contained (in mm): 140 CsCl, 10 EGTA, 2 MgCl2, 2 adenosine triphosphate (disodium salt), and 10 HEPES–CsOH, pH 7.3. In some experiments Ca2+-free and/or Mg2+-free extracellular solutions were used. High Ca2+ extracellular solution contained (in mm): 105N-methyl-d-glucamine, 30 CaCl2, and 5 HEPES–HCl, pH 7.2. Relative Ca2+ to Na+permeabilities were determined as described (Brusa et al., 1995). Fractional Ca2+ currents through recombinant NR1(N598Q)/NR2A receptors expressed in HEK293 cells were measured as described (Burnashev et al., 1995). All recordings were at 22–24°C. Values are given as mean ± SEM. Statistics were determined by two-tailed t test.
Long-term potentiation experiments. Transverse hippocampal slices from adult (3 month) male mouse brains were prepared as described (Feldmeyer et al., 1999). Orthodromic synaptic stimulation (50 μsec, <100 μA, 0.2 Hz) was delivered alternately through tungsten electrodes to two independent pathways in the CA1 region, one activating synapses in the apical (stratum radiatum), the other in the basal (stratum oriens) dendrites. Extracellular responses were recorded by two glass electrodes placed in the corresponding layers. After a stable recording period of at least 15 min, one of the pathways was tetanized (100 Hz, 1 sec) with a strength just above the threshold for generation of a population spike in response to a single test stimulus. The synaptic strength was assessed by measuring the slope of the field EPSP in the middle third of its rising phase. Six consecutive measurements (1 min) were averaged and normalized to the values obtained 4–7 min before tetanic stimulation. Mice were semirandomly selected from different groups, and at the end of the experimental series their identity was revealed, and data were pooled across animals of the same genotype. Data are given as mean ± SEM. Statistics were determined by two-tailed t test.
Histology of barrel cortex. Mouse brains were fixed for 1 hr in 4% paraformaldehyde and cryoprotected for 24 hr in 30% sucrose in 0.1% PBS containing (in mm): 137 NaCl, 6.5 Na2HPO4, 2.7 KCl, and 1.5 KH2PO4, pH 7.4. Cytochrome oxidase activity was visualized by incubation of mounted tangential cryosections (50 μm) in 4% sucrose, 0.05% cytochrome C, and 0.05% diaminobenzidine (Cases et al., 1996).
Chest plethysmography. Pups were removed from a 37°C, humidified environment, and after 20 min placed with their chest between a platform electrode and a flexible electrode at diaphragm level. Electrodes were covered with contact gel and submitted to alternating current (300 mA, 10 kHz). Breathing rhythm was monitored by recording the change in resistance resulting from changes in distance between the electrodes during breathing movements of the chest. Conductivity changes were documented with an oscillographic recorder.
RESULTS
Generation of NR1 mutant genotypes
We replaced by gene targeting in mouse ES cells codon 598 for asparagine (N, AAC) by codons for glutamine (Q, CAG) and arginine (R, AGA) in exon 15 of the NR1 gene (Hollmann et al., 1993) (Fig. 1B). Homologous replacement was confirmed by Southern blot analysis of ES cell DNA (Fig. 1B,C) and sequence analysis of PCR-amplified gene segments. For each mutation, two independent mouse lines were established in which the mutated NR1 allele still carried (NR1Qneo,NR1Rneo), or had lost by Cre-mediated deletion (NR1Q,NR1R), the loxP-flankedneo gene (Fig. 1B).
In addition to heterozygous and homozygous mutants, we generated hemizygous NR1−/Q andNR1−/R mice by employing theNR1− knock-out allele ofNR1+/− mice (Forrest et al., 1994). As the NR1R allele was dominant lethal but was silenced by the neo gene insertion (see below), mice carrying the NR1R allele were produced from NR1+/Rneo mice by crossing with the cre-transgenic deleter strain (Schwenk et al., 1995).
Expression of the mutant NR1 alleles is silenced by theneo gene insertion in intron 11
The expression of the sequence-manipulated NR1 alleles was monitored by immunoblot analysis (Fig.2). We found that the neogene, but not the single loxP element, silenced the manipulated NR1 alleles, possibly by interfering with transcript processing (Nagy et al., 1998; Feldmeyer et al., 1999). NR1 protein levels were strongly reduced in brains ofNR1Qneo/Qneo andNR1Rneo/Rneo mice, but were unchanged in mice expressing the NR1Q orNR1R alleles (Fig.2A,B).
Fig. 2.

Insertion of the neo gene silences expression of the targeted NR1 alleles. Comparison by immunoblot analysis of NR1 subunit levels in brains of wild-type versus mutant mice with and without the neo gene in the targeted NR1 alleles in A, andB, respectively. Size markers indicated on theleft are in kilodaltons. Arrows indicate NR1-specific signals.
For quantification, NR1-specific RT-PCRs were performed on total brain RNA from heterozygotes. RT-PCR products were subcloned, and the number of clones derived from the wild-type and mutant alleles was determined by allele-specific differential oligonucleotide hybridization (Higuchi et al., 1993). Of the entire NR1 mRNA population (100%), NR1Q mRNA constituted 48.2 ± 6% (mean ± SD, n = 7),NR1R mRNA 50.9 ± 1.8% (n = 5), NR1Qneo mRNA 0.6 ± 0.6% (n = 3), andNR1Rneo mRNA 1.5 ± 1.4% (n = 3) in the respective heterozygotes. Thus, theneo gene-containing NR1 alleles can be viewed as null alleles. The wild-type NR1 allele was not upregulated in the presence of the mutant alleles.
NMDA receptors of NR1Q/Q mice are fourfold reduced in Ca2+ permeability and altered in Mg2+ block, whereas inNR1−/R mice NMDA receptor currents are undetectable
NMDA receptor physiology was analyzed in nucleated soma patches (Sather et al., 1992) of hippocampal CA1 pyramidal cells in acute brain slices from homozygous and hemizygous mutants.
In NR1Q/Q mice, the NMDA receptor-mediated mean current amplitude at +60 mV was 243 ± 107 pA (n = 6), not significantly different from that obtained in wild-type littermates (267 ± 76 pA; n = 5;p = 0.85) at P0 (Table1). This demonstrated functional receptor expression in NR1Q/Q mice, consistent with the comparable single-channel conductance of recombinantly expressed NR1/NR2B and NR1(N598Q)/NR2B receptors (Premkumar and Auerbach, 1997). In hippocampal CA1 pyramidal cells ofNR1−/R mice, NMDA receptor-mediated currents could not be recorded, probably because of the approximately 30-fold reduced single-channel conductance of recombinant NR1(N598R)/NR2A receptors (Béhé et al., 1995).
Table 1.
Properties of NMDA receptors in NR1gene-targeted mice
| NR1genotypes | INMDA in pA | Ca2+ reversal potential in mV | PCa/PNa | |
|---|---|---|---|---|
| P0 | P14 | |||
| +/+ | 267 ± 76 (n = 5) | 413 ± 90 (n = 8) | 18.2 ± 0.3 (n = 10) | 6.3 ± 0.2 (n = 10) |
| Q/Q | 243 ± 107 (n = 6) | — | −8.5 ± 1.3 (n = 4) | 1.4 ± 0.1 (n = 4) |
| +/Q | 171 ± 78 (n = 4) | 418 ± 56 (n = 7) | 15.8 ± 0.2 (n = 4) | 5.4 ± 0.04 (n = 4) |
| +/R | ND | 71 ± 33 (n = 4) | −3.8 ± 1.6 (n = 6) | 1.8 ± 0.2 (n = 6) |
Averaged NMDA receptor-mediated peak currents were recorded at +60 mV holding potential from hippocampal CA1 pyramidal cells of animals at P0 and P14. The Ca2+ to Na+permeability ratios (PCa/PNa) were derived from the shift in Ca2+ reversal potential when switching from high extracellular Na+ to high extracellular Ca2+ solution. Values are given as mean ± SEM. ND, Not determined.
As predicted from in vitro studies (Burnashev et al., 1992), in NR1Q/Q mice, the Ca2+ permeability of NMDA receptors was decreased approximately fourfold, when estimated from the shift in the Ca2+ reversal potential from 18.2 ± 0.3 mV (n = 10) in wild-type to -8.5 ± 1.3 mV (n = 4) in NR1Q/Q mice (Table 1). In addition, the mutation altered the voltage dependence of NMDA receptor-mediated currents. The Mg2+block appeared to be enhanced at depolarizing potentials (−20 to −50 mV) and incomplete at resting potential (−70 mV) (Fig.3A). These changes resulted from two characteristics of NR1(N598Q)/NR2 receptors, which could be dissected by measurements in Ca2+-free or Mg2+-free conditions. The incomplete block by Mg2+ at resting potential was also observed in Ca2+-free conditions (Fig.3B), whereas Mg2+-free conditions revealed a strong and voltage-dependent block by Ca2+ (Fig. 3B,C) which, under physiological conditions (1.8 mmCa2+, 1 mmMg2+), dominated the Mg2+ block at depolarizing potentials (Fig. 3B). In addition, we found that recombinantly expressed NR1(N598Q)/NR2A receptors showed reduced and voltage-dependent Ca2+ permeability with fractional Ca2+ currents (Pf) in 1.8 mmCa2+ of 4.7, 5.3, and 6.5% (mean of 2 each) at the respective membrane potentials of −20, −40, and −60 mV. This differs for recombinant wild-type (NR1/NR2A) receptors (Burnashev et al., 1995), which display an almost constant value of ∼11% at potentials more negative than −20 mV. Thus, when compared to wild type, NMDA receptors in neurons of NR1Q/Qmice show altered responses with properties seen in recombinant receptors. After glutamate stimulation at depolarizing potentials, the NMDA receptor-mediated influx of Ca2+ and Na+ is reduced in this mutant, and at resting potential Ca2+ and Na+ pass the channel that is blocked by Mg2+ in wild type.
Fig. 3.

Altered NMDA receptor properties inNR1 mutant mice. Panels present current–voltage (I–V) relationships measured in nucleated soma patches of hippocampal CA1 pyramidal cells for the genotypes indicated. Currents are normalized to peak response at +60 mV holding potential. Concentrations of Ca2+ and Mg2+ions in the extracellular solution are given in millimolar concentration for each panel (concentration of monovalent cations was 140 mm in all measurements). A, Differences in voltage dependence of NMDA receptors in NR1Q/Q, NR1+/Q, andNR1+/R mice under physiological ionic conditions, in comparison with wild type (thin red line). B,I–V relationships of NMDA receptor-mediated currents in NR1Q/Qmice in either Ca2+-free, Mg2+-free, or physiological conditions.C, Comparison of different mutant mice to wild type with respect to voltage-dependent block by Ca2+ in Mg2+-free conditions.
NR1Q/Q andNR1−/R mice develop a perinatally lethal phenotype
The altered NMDA receptor properties induced a perinatally lethal phenotype in both NR1Q/Q andNR1−/R mice. In contrast to knock-out mice that died within 10 hr after birth (Forrest et al., 1994; Li et al., 1994) (Table 2)NR1Q/Q pups died within the first hour after birth (Table 2) from strong respiratory distress. They were cyanotic, gasped for air (Fig.4A), and exhibited irregular breathing patterns (Fig. 4B). At 37°C in a humidified environment, breathing became more frequent and regular, and NR1Q/Q pups lived up to 10 hr. Moreover, NR1Q/Q pups did not feed (Table2), as indicated by the lack of milk in their stomachs (Fig.4A). We observed that they failed to attach to the mother's nipples. The same phenotype was observed forNR1−/Q, and, more severely, for NR1−/R mice (Fig.4A, Table 2). These results indicated that the altered channel behavior affected essential NMDA receptor functions for autonomic brain stem circuits.
Table 2.
Phenotypes of NR1 gene-targeted mice
| NR1genotypes | Phenotypes |
|---|---|
| −/− | |
| Qneo/Qneo | Death within 10 hr after birth; respiratory deficits; |
| Rneo/Rneo | no feeding |
| Q/Q | |
| −/Q | Death within 1 hr after birth; respiratory distress; |
| −/R | no feeding |
| +/R | Death within 4 weeks after birth; growth retarda tion; poor righting reflex; poor feeding |
| +/Q | Increased mortality; females with nurturing deficit |
| +/− | |
| +/Qneo | No obvious difference to wild type |
| +/Rneo |
Fig. 4.

Homozygous and hemizygous mutants suffer from respiratory failure. A, Respiratory distressed and cyanotic NR1Q/Q andNR1−/R mice compared to wild type shortly after birth. B, Chest plethysmography shows the different breathing pattern of wild-type andNR1Q/Q mice at P0.
Perinatal lethality is rescued by the presence of wild-type NMDA receptors in heterozygotes
To evaluate channel parameters that lead to dysfunction of NMDA receptors early in development, we included heterozygous mice in our analysis. In all heterozygotes, the perinatally lethal phenotype was rescued by the presence of wild-type NMDA receptors.
Channel analysis in heterozygotes needs to consider that NMDA receptors coassemble two NR1 subunits (Béhé et al., 1995). Hence, neurons of NR1+/Q andNR1+/R mice harbor a heterogeneous NMDA receptor population, composed of pure wild-type, pure mutant, and “mixed” receptors. These mixed receptors contain one wild-type and one mutant NR1 subunit. They constitute at least half of the entire NMDA receptor population and display properties that depend on the functional contribution of the NR1(N598Q) or NR1(N598R) mutation in the channel pore. The electrophysiological profile of the mixed receptors was evaluated from changes in current amplitude, Ca2+ reversal potential, andI–V relationship, which were determined by nucleated soma patch recordings of hippocampal CA1 pyramidal cells from heterozygous mice.
The NR1(N598Q) subunit is not dominant in the mixed NMDA receptors of NR1+/Q mice
In NR1+/Q mice, the NMDA receptor-mediated responses were comparable to those observed in wild type. The NMDA receptor-mediated mean current amplitude in hippocampal CA1 pyramidal cells at +60 mV was not significantly different at P0 (171 ± 78 pA; n = 4) and P14 (418 ± 56 pA; n = 7) from wild-type mice at P0 (267 ± 76 pA; n = 5; p = 0.36) and P14 (413 ± 90 pA; n = 8; p = 0.96). This was consistent with similar values inNR1Q/Q mice (Table 1). The small shift in the Ca2+ reversal potential from 18.2 ± 0.3 mV (n = 10) in wild-type mice to 15.8 ± 0.2 mV (n = 4) inNR1+/Q mice (Table 1) can be explained by the presence of the pure mutant receptors within the NMDA receptor population of heterozygotes. Furthermore, the small shift demonstrated that the mixed receptors are Ca2+-permeable like wild-type receptors. The voltage dependence of the entire NMDA receptor-mediated current was also not affected, and the block by Mg2+at resting potential (Fig. 3A) appeared to be normal. However, in Mg2+-free conditions we observed a Ca2+ block intermediate to that in wild-type and NR1Q/Q mice (Fig.3C), but this block was not strong enough to dominate the Mg2+ block under physiological conditions (Fig. 3A).
In summary, in NR1+/Q mice, the response of the mixed receptors seems comparable to that of the pure wild-type receptors. Therefore, phenotypic abnormalities most likely result from the altered electrophysiological responses of the pure mutant receptors as described above for theNR1Q/Q mice.
NR1+Q mice exhibit increased mortality and impaired maternal behavior
The phenotype of NR1+/Qmice was characterized by increased mortality and impaired maternal behavior (Fig.5A,B, Table 2). Pregnant NR1+/Qfemales were often hyperactive before delivery. After delivery, these females performed poorly on maternal tasks such as nest building, disconnecting and eating the placenta, cleaning the newborn, and retrieving and crouching over the pups. The mothers turned aggressive toward the newborn, which lay scattered (Fig. 5B), displayed bruises and bites, and were sometimes cannibalized. Typically, litters were underfed and died, or were killed, within 2 d. Maternal performance did not improve after repeated breeding. Litters were occasionally raised with these mothers by help with nest building, collecting the pups, and placing the mothers repeatedly over their offspring. Thus, appropriate NMDA receptor-regulated signaling appears to be required for adaptive neuronal responses, which might underlie the induction of instinctive behavior, such as nurturing. Similar nurturing deficiencies were described for fosB,Dbh, and Peg3 knock-out mice (Brown et al., 1996;Thomas and Palmiter, 1997; Li et al., 1999), but it remains unclear to what extent the similar mutant phenotypes result from defects in shared signaling pathways.
Fig. 5.
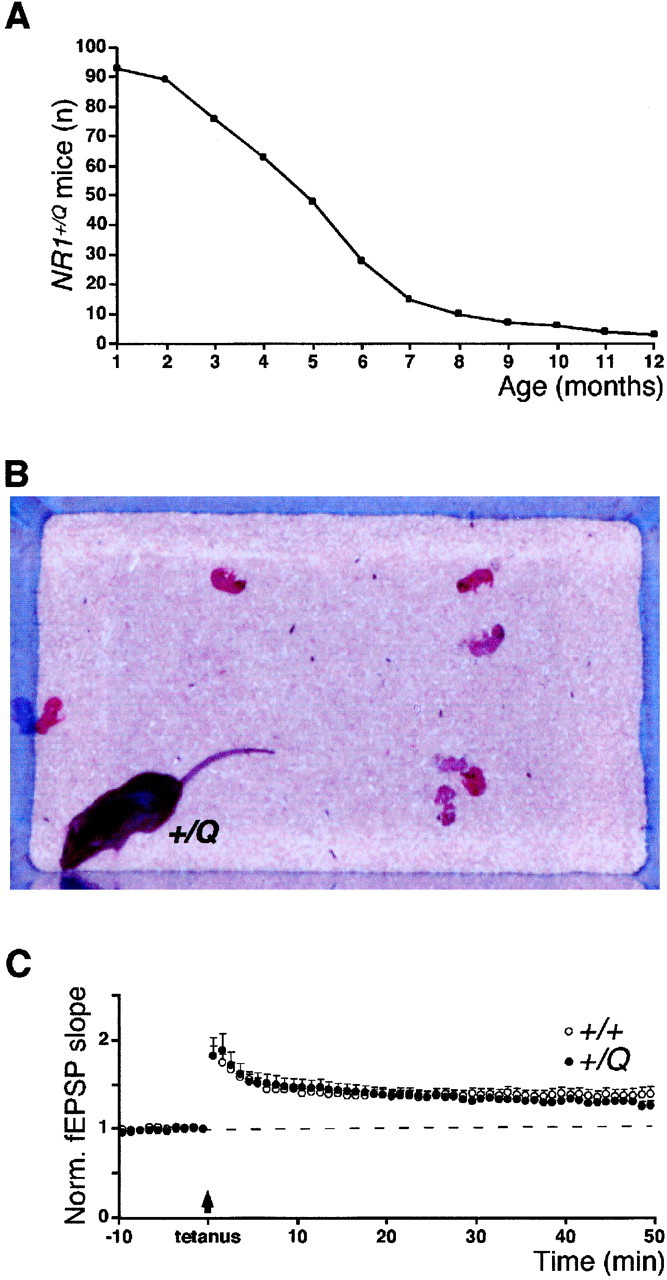
Increased mortality, nurturing deficits, and normal CA3/CA1 synapse LTP in NR1+/Q mice.A, Survival curve of NR1+/Qmice (n = 93). Numbers on ordinate indicate NR1+/Q animals alive at the respective age. B, NR1+/Qmother with pups after delivery. C, LTP experiments at hippocampal CA3/CA1 synapses. Normalized and pooled field EPSP slope measurements after tetanic stimulation (arrow) are shown for wild-type and NR1+/Q mice.Vertical bars indicate SEM. The dashed line symbolizes the control pathway.
Long-term potentiation at CA3/CA1 synapses was not affected in NR1+/Q mice
In contrast to wild type, the pure mutant receptors inNR1+/Q mice allow Ca2+ influx at resting potential after glutamate stimulation. To evaluate the impact of this Ca2+ influx on synaptic plasticity in the hippocampal CA1 region, long-term potentiation (LTP) studies were performed on brain slices from adult mice. In both wild-type andNR1+/Q mice, tetanic activation of the Schaffercollateral pathway produced a persistent homosynaptic potentiation of the synaptic responses, characteristic for LTP (Fig. 5C). The magnitude of LTP 40–45 min after tetanization was 137 ± 7% (control pathway, 102 ± 6%;n = 23) in wild-type mice, and similar in the mutants (131 ± 6%; control pathway, 98 ± 3%; n = 13; p = 0.55). In both groups, LTP was blocked by AP-5 (100 μm), a selective NMDA receptor antagonist (data not shown). Thus, the Ca2+ influx in the mutant at resting potential has no effect on LTP at hippocampal CA3/CA1 synapses.
The NR1(N598R) subunit is dominant in the mixed NMDA receptors ofNR1+/Q mice
In NR1+/R mice, the overall NMDA receptor-mediated responses were affected by the dominance of the NR1(N598R) subunit in the mixed receptors whose single-channel conductance approximates one-fourth of the pure wild-type receptors, as determined for recombinant NR1(N598R)/NR2A receptors (Béhéet al., 1995). The pure mutant receptors show highly reduced single-channel conductance (Béhé et al., 1995), and therefore contribute little to the whole soma current. Indeed, in hippocampal CA1 pyramidal cells ofNR1+/R mice, the NMDA receptor-mediated mean current amplitude at +60 mV was significantly lower (71 ± 33 pA; n = 4) than in wild type (413 ± 90 pA; n = 8; p = 0.02) at P14 (Table 1). In addition, the strong shift in the Ca2+ reversal potential from 18.2 ± 0.3 mV (n = 10) in wild-type to −3.8 ± 1.6 mV (n = 6) inNR1+/R mice (Table 1) showed that the mixed receptors are Ca2+-impermeable like the pure mutant receptors (Burnashev et al., 1992), and that the remaining Ca2+ influx (∼25% of wild type, see Table 1) is exclusively mediated by the pure wild-type receptors. Similarly, the strong reduction of the Mg2+ block (Fig. 3A) reflected the absence of the block in the mixed receptors.
In summary, in NR1+/R mice, the pure wild-type receptors signal normally, whereas the mixed receptors after glutamate stimulation flux Na+ but not Ca2+, and lack Mg2+ block.
NR1+/R mice survive P0 but die prematurely
The phenotype of NR1+/Rmice reflected the dominance of the mutant NR1 subunit in the mixed receptors. NR1+/R mice breathe and feed, and survive P0. However, approximately two-thirds of allNR1+/R mice died within 2 d after birth (Fig. 6A) from inefficient feeding (Table 2). Survival increased with reduced litter size. Longer livingNR1+/R mice were delayed in development (Table 2), as judged from growth retardation (Fig.6B), poor righting reflex, and decreased activity. None of the NR1+/R mice survived 4 weeks (Fig. 6A, Table 2).
Fig. 6.
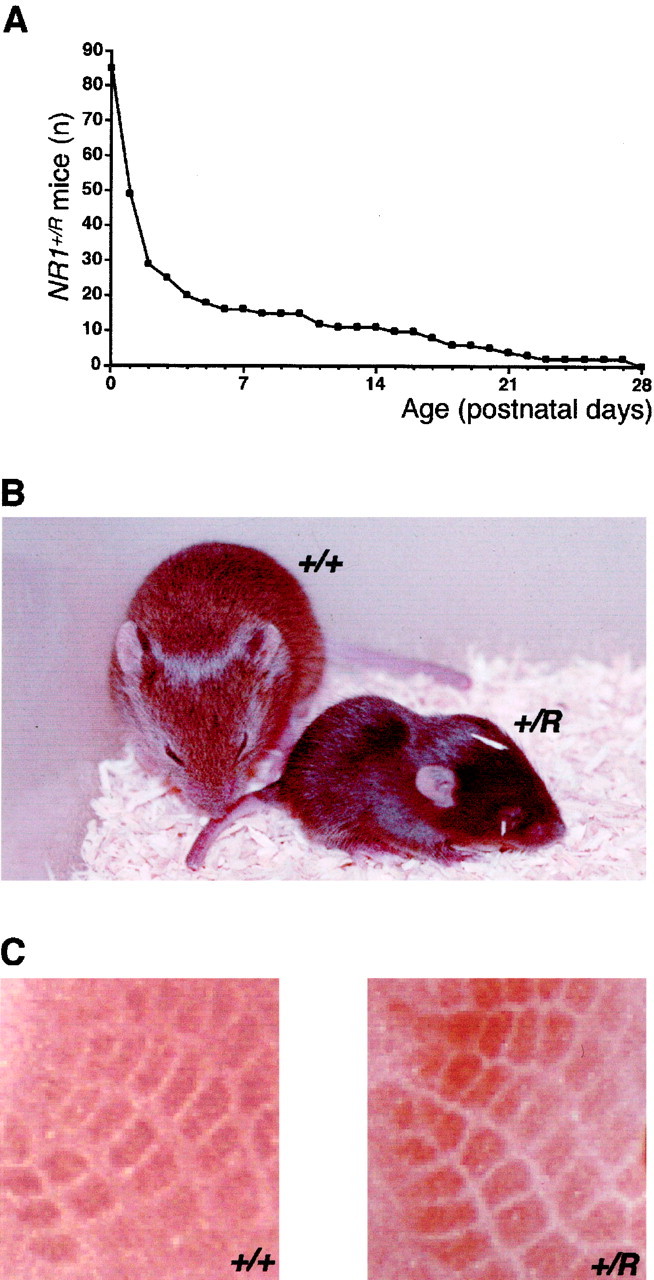
Premature death, poor growth, and normal whisker barrel formation in NR1+/R mice.A, Survival curve ofNR1+/R mice (n = 85). Numbers on ordinate indicateNR1+/R animals alive at the respective postnatal day. B, Size difference ofNR1+/R and wild-type mice at P14.C, Comparable whisker barrel architecture in the primary somatosensory neocortex in NR1+/R and wild-type mice at P8, revealed by cytochrome oxidase staining.
Barrel cortex is formed with 25% of pure wild-type NMDA receptors in NR1+/R mice
The formation of periphery-related somatosensory patterns in the neocortex, dependent on NMDA receptor-mediated neural activity (Mitrovic et al., 1996), is lacking in mice with highly reduced NMDA receptor expression (Iwasato et al., 1997). InNR1+/R mice, whisker barrel formation was evident in the primary somatosensory cortex (Fig.6C). This indicated that 25% pure wild-type receptors in the entire NMDA receptor population was sufficed for the anatomical development of this cortical structure. Furthermore, it supported our hypothesis that in NR1+/Rmice, the activity-controlled Ca2+ influx mediated by the residual pure wild-type receptors can rescue the malfunctional mixed and pure mutant receptors.
DISCUSSION
The analysis of our NMDA receptor mutant mice indicated that residue N598 in the NR1 subunit is essential for NMDA receptor function. In mice with NR1(N598Q) and NR1(N598R) mutations, the altered electrophysiological properties of NMDA receptors correlate with phenotypic severity of the mutation.
Homozygous NR1Q/Q and hemizygousNR1−/R mice
In newborn NR1Q/Q mice, we observed typical symptoms of NMDA receptor dysfunction with death at P0. The phenotype of NR1Q/Q mice is even more severe than that of NR1−/−knock-out mice, which live several hours longer. It can be converted to the knock-out phenotype when the expression of the mutant allele is downregulated, as in NR1Qneo/Qneo mice (Table 2). This indicates that in the absence of NMDA receptors, compensatory mechanisms achieve a partial rescue. Therefore, the improper signaling of the mutant NMDA receptors seems to be more detrimental than no signaling, as similarly observed for other ion channel mutants (Brusa et al., 1995; Surmeier et al., 1996; Zuo et al., 1997) when compared to the respective knock-outs (Kashiwabuchi et al., 1995; Jia et al., 1996; Signorini et al., 1997). This observation also holds true for NR1−/R mice, in which we found no indications for compensatory mechanisms, even though NMDA receptor-mediated currents escaped our method of detection. The phenotype of NR1−/R mice was similar to that of NR1Q/Q mice and converted to the knock-out phenotype when the NR1(N598R) mutation was silenced inNR1Rneo/Rneo mice (Table 2).
Heterozygous NR1+/Q andNR1+/R mice
Heterozygotes coexpressing wild-type and mutant NR1 subunits at comparable levels exhibit phenotypes, the severity of which correlates with the dominance of the mutant subunit in the mixed receptors. These mixed receptors constitute at least half of the NMDA receptor population, with the pure wild-type and the pure mutant receptors representing the other half.
The phenotype of NR1+/Q mice, characterized mainly by increased mortality and impaired maternal instincts, combined with the altered electrophysiological properties of NMDA receptors in hippocampal CA1 pyramidal cells, indicates that the NR1(N598Q) mutation is not dominant in the mixed receptors. Therefore, the distinctive phenotype seems to be generated by the pure mutant receptors, which have reduced Ca2+ permeability and flux Ca2+ at resting potential. This is supported by a similar phenotype with increased mortality of mouse mutants that show Ca2+ influx at resting potential by unedited AMPA receptors in hippocampal CA1 pyramidal cells (Feldmeyer et al., 1999).
By contrast, the premature death ofNR1+/R mice (∼85% do not survive 2 weeks) reflects dominance of the NR1(N598R) subunit. Mutant animals derived from the chimeric founder had the same phenotype as those derived via the deleter strain, indicating that differences in genetic background contribute little to theNR1+/R phenotype. In these mutants, most NMDA receptors are Ca2+-impermeable, lack Mg2+ block, and have small single-channel conductance, generating an approximately fourfold reduction in NMDA receptor-mediated Ca2+ influx and macroscopic current. However, the presence of ∼25% of pure wild-type receptors can overcome the dysfunction of the mutated receptors and can rescue the functioning of perinatal autonomic circuits and the formation of topographically patterned whisker barrels in the primary somatosensory neocortex.
Conclusion
A comparison of the NR1Q/Q andNR1+/R mutants is instructive regarding the link between altered NMDA receptor properties and severity of phenotype. In both mutants, the altered channel site in the NMDA receptors leads to an approximately fourfold reduced Ca2+ influx. Both mutants exhibit lethal phenotypes. However, whereas NR1Q/Q mice die perinatally, NR1+/R mice can survive 3 weeks. The phenotypic difference can be explained by NMDA receptors, which constitute in NR1Q/Q mice a pure mutant, but in NR1+/Rmice a heterogeneous receptor population.
In NR1+/R mice, up to 25% of the NMDA receptors are pure wild-type receptors with regular signaling and properly controlled Ca2+ influx. The majority (75%) of the NMDA receptors inNR1+/R mice are Ca2+-impermeable and do not interfere with the proper Ca2+ signaling by the pure wild-type receptors. The mutant receptors can flux only monovalent ions in a voltage-independent manner, similar to AMPA receptors, which are mostly colocalized with NMDA receptors at synapses. Apparently, the fourfold reduced number of wild-type NMDA receptors is sufficient to sustain autonomic brain functions required from birth onwards. This was corroborated recently by a hypomorphic NR1 allele in gene-manipulated mice, in which strongly reduced NMDA receptor expression has minor effects on the phenotype (Mohn et al., 1999). During subsequent postnatal life, the suboptimal Ca2+ influx or voltage-independent NMDA receptor-mediated Na+ influx accompanied by developmental deficiencies, might underlie the premature mortality of NR1+/R mice.
The variability of phenotypes in heterozygotes, in particular the wide window of mortality, might mirror the stochastic incorporation into synapses of NMDA receptors bearing wild-type or mutant NR1 subunits. Thus, neurons with few synapses and low number of NMDA receptors per synapse could become functionally compromised during transient synaptic underrepresentation of the pure wild-type receptor.
NR1Q/Q mice show NMDA receptor-mediated Ca2+ influx comparable toNR1+/R mice, but fail to develop autonomic functions, such as breathing and feeding. We therefore assume that the deficient coincidence detection of the mutated channels, caused by the incomplete Mg2+ block at resting potential, is responsible for the perinatally lethal phenotype. Thus, tightly voltage-controlled Ca2+ influx as a determinant of coincidence detection of presynaptic and postsynaptic activity is likely to be an essential property of the NMDA receptor function in developing neurons of the mouse brain.
Although the present study focused on Ca2+permeability and voltage-dependent Mg2+block as the key properties of NMDA receptors and suggested a link between changes in these properties and severity of phenotype, we cannot exclude that other functional changes in the mutant NMDA receptors (Kashiwagi et al., 1997; Schneggenburger and Ascher, 1997;Traynelis et al., 1998; Zheng et al., 1999) may have contributed to the phenotypes. However, because these other changes affect macroscopic current and because it is known that the number of NMDA receptors can be strongly reduced with minor effects on the phenotype (Mohn et al., 1999), we propose that the voltage-independent, but not the reduced, Ca2+ influx is the most detrimental property change in our mutant mice.
The early death of our mutant mice precluded studies on the effect of NR1 subunit residue N598 substitutions on NMDA receptor functions in the mature brain. An evaluation of these effects should become feasible with the use of the floxed NR1Qneo andNR1Rneo genes as null alleles along with a growing number of inducible or subregion-specificcre-transgenic mouse lines.
Footnotes
This work was supported in part by the Volkswagenstiftung to R.S. and P.H.S., the Bristol-Myers Squibb company to P.H.S., the National Institutes of Health Cancer Center Support Grant P30 CA21765 and the American Lebanese Syrian Associated Charities to T.C., and a Sinsheimer Scholarship to D.F. D.F.H. was a recipient of an Alexander von Humboldt research award. We thank Andràs Nagy for the R1 ES cell line, Hua Gu for the plasmid pMC-Cre, William G. Janssen for the monoclonal antibody 54.1 to NR1, Rita Pfeffer for animal care, Johannes Vogel for advice and help with chest plethysmography, and Lonnie P. Wollmuth for critical comments on this manuscript. Cre-expressing mice were used under a noncommercial research license agreement between DuPont Pharmaceutical Company and the Max-Planck Society.
Correspondence should be addressed to Dr. Rolf Sprengel, Max-Planck-Institute for Medical Research, Jahnstraβe 29, D-69120 Heidelberg, Germany. E-mail: sprengel@mpimf-heidelberg.mpg.de.
REFERENCES
- 1.Ascher P, Nowak L. The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol (Lond) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balázs R, Hack N, Jørgensen OS, Cotman CW. N-methyl-d-aspartate promotes the survival of cerebellar granule cells: pharmacological characterization. Neurosci Lett. 1989;101:241–246. doi: 10.1016/0304-3940(89)90539-9. [DOI] [PubMed] [Google Scholar]
- 3.Béhé P, Stern P, Wyllie DJA, Nassar M, Schoepfer R, Colquhoun D. Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc R Soc Lond B Biol Sci. 1995;262:205–213. doi: 10.1098/rspb.1995.0197. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Brewer GJ, Cotman CW. NMDA receptor regulation of neuronal morphology in cultured hippocampal neurons. Neurosci Lett. 1989;99:268–273. doi: 10.1016/0304-3940(89)90458-8. [DOI] [PubMed] [Google Scholar]
- 6.Brown JR, Ye H, Bronson RT, Dikkes P, Greenberg ME. A defect in nurturing in mice lacking the immediate early gene fosB. Cell. 1996;86:297–309. doi: 10.1016/s0092-8674(00)80101-4. [DOI] [PubMed] [Google Scholar]
- 7.Brusa R, Zimmermann F, Koh D-S, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 8.Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Günther W, Seeburg PH, Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- 9.Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol (Lond) 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 11.Constantine-Paton M, Cline HT, Debski E. Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu Rev Neurosci. 1990;13:129–154. doi: 10.1146/annurev.ne.13.030190.001021. [DOI] [PubMed] [Google Scholar]
- 12.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 13.Feldmeyer D, Kask K, Brusa R, Kornau H-C, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby Ø, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 14.Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- 15.Fox K, Daw NW. Do NMDA receptors have a critical function in visual cortical plasticity? Trends Neurosci. 1993;16:116–122. doi: 10.1016/0166-2236(93)90136-a. [DOI] [PubMed] [Google Scholar]
- 16.Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 18.Hollmann M, Boulter J, Maron C, Beasley L, Sullivan J, Pecht G, Heinemann S. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron. 1993;10:943–954. doi: 10.1016/0896-6273(93)90209-a. [DOI] [PubMed] [Google Scholar]
- 19.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M, Nakanishi S. Molecular characterization of the family of the N-methyl-d-aspartate receptor subunits. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- 21.Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S. NMDA receptor-dependent refinement of somatotopic maps. Neuron. 1997;19:1201–1210. doi: 10.1016/s0896-6273(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 22.Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 23.Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- 24.Kashiwagi K, Pahk AJ, Masuko T, Igarashi K, Williams K. Block and modulation of N-methyl-d-aspartate receptors by polyamines and protons: role of amino acid residues in the transmembrane and pore-forming regions of NR1 and NR2 subunits. Mol Pharmacol. 1997;52:701–713. doi: 10.1124/mol.52.4.701. [DOI] [PubMed] [Google Scholar]
- 25.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–97. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 26.Kuner T, Wollmuth LP, Karlin A, Seeburg PH, Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 27.Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 28.Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 29.Li L-L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S. Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell. 1994;76:427–437. doi: 10.1016/0092-8674(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 31.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 32.Mayer ML, Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- 33.Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M. Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature. 1992;357:70–74. doi: 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- 34.Mitrovic N, Mohajeri H, Schachner M. Effects of NMDA receptor blockade in the developing rat somatosensory cortex on the expression of the glia-derived extracellular matrix glycoprotein tenascin-C. Eur J Neurosci. 1996;8:1793–1802. doi: 10.1111/j.1460-9568.1996.tb01323.x. [DOI] [PubMed] [Google Scholar]
- 35.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 36.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 37.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 38.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagy A, Moens C, Ivanyi E, Pawling J, Gertsenstein M, Hadjantonakis A-K, Pirity M, Rossant J. Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr Biol. 1998;8:661–664. doi: 10.1016/s0960-9822(98)70254-4. [DOI] [PubMed] [Google Scholar]
- 40.Premkumar LS, Auerbach A. Stoichiometry of recombinant N-methyl-d-aspartate receptor channels inferred from single-channel current patterns. J Gen Physiol. 1997;110:485–502. doi: 10.1085/jgp.110.5.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sather W, Dieudonné S, MacDonald JF, Ascher P. Activation and desensitization of N-methyl-d-aspartate receptors in nucleated outside-out patches from mouse neurones. J Physiol (Lond) 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneggenburger R, Ascher P. Coupling of permeation and gating in an NMDA-channel pore mutant. Neuron. 1997;18:167–177. doi: 10.1016/s0896-6273(01)80055-6. [DOI] [PubMed] [Google Scholar]
- 43.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siegel SJ, Brose N, Janssen WG, Gasic GP, Jahn R, Heinemann SF, Morrison JH. Regional, cellular, and ultrastructural distribution of N-methyl-d-aspartate receptor subunit 1 in monkey hippocampus. Proc Natl Acad Sci USA. 1994;91:564–568. doi: 10.1073/pnas.91.2.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci USA. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby Ø, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SGN, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- 47.Surmeier DJ, Mermelstein PG, Goldowitz D. The weaver mutation of GIRK2 results in a loss of inwardly rectifying K+ current in cerebellar granule cells. Proc Natl Acad Sci USA. 1996;93:11191–11195. doi: 10.1073/pnas.93.20.11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas SA, Palmiter RD. Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell. 1997;91:583–592. doi: 10.1016/s0092-8674(00)80446-8. [DOI] [PubMed] [Google Scholar]
- 49.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 51.Wollmuth LP, Kuner T, Seeburg PH, Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. J Physiol (Lond) 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng X, Zhang L, Wang AP, Araneda RC, Lin Y, Zukin RS, Bennett MVL. Mutation of structural determinants lining the N-methyl-d-aspartate receptor channel differentially affects phencyclidine block and spermine potentiation and block. Neuroscience. 1999;93:125–134. doi: 10.1016/s0306-4522(99)00154-2. [DOI] [PubMed] [Google Scholar]
- 53.Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]