

Abstract
The addition or loss of synapses in response to changes in activity, disease, or aging is a major aspect of nervous system plasticity in the adult. The mechanisms that affect the turnover and maintenance of synapses in the adult are poorly understood and are difficult to investigate in the brain. Here, we exploited a unique anatomical arrangement in the neuromuscular system to determine whether subtypes of synapses can differ in anatomical plasticity and vulnerability. In three genetic mouse models of motoneuron disease of diverse origin and severity, we observed a gradual and selective loss of synaptic connections that begun long before the onset of clinical deficits and correlated with the timing of disease progression. A subgroup of fast-type (fast-fatiguable) neuromuscular synapses was highly vulnerable and was lost very early on. In contrast, slow-type synapses resisted up to the terminal phase of the disease. Muscle-specific differences were also evident. Similar selective losses were detected in aged mice. These selective vulnerability properties of synapses coincided with hitherto unrecognized major differences in stimulus-induced anatomical plasticity that could also be revealed in healthy mice. Using paralysis and/or growth-associated protein 43 overexpression to induce synaptic sprouting, we found that slow-type, disease-resistant synapses were particularly plastic. In contrast, fast-type synapses with the highest vulnerability failed to exhibit any stimulus-induced change. The results reveal pronounced subtype specificity in the anatomical plasticity and susceptibility to loss of neuromuscular synapses and suggest that degenerative motoneuron diseases involve a common early pathway of selective and progressive synaptic weakening also associated with aging.
Keywords: selective vulnerability, motoneuron disease, synapse subtypes, neuromuscular junction, GAP43, anatomical plasticity, Botulinum toxin A
The functioning and plasticity of neural systems depends critically on the specific properties of their neuronal components. Anatomical and molecular studies have revealed a great degree of diversity among neuronal types in the adult (Stevens, 1998), but the plasticity properties of their synapses are difficult to investigate in brain tissue. Because of the unique experimental advantages associated with this system, intrinsically distinct subtypes of neurons and their specific functions have been best characterized in motor pools, the groups of motoneurons that innervate one defined skeletal muscle (Henneman and Mendell, 1991;Burke, 1994). Each muscle unit (one muscle and its motor pool) consists of a defined set of functionally distinct motor units (one motoneuron and the muscle fibers it innervates). Mammalian motor units are subdivided into the three main functional types: slow (S), fast fatigue-resistant (FR), and fast fatigable (FF). Whereas slow motor units develop relatively little force but are extremely resistant to fatigue, FF units are powerful but rapidly exhausted. In addition, there are functionally intermediate forms within these categories, particularly among FF and FR motor units. During muscle activity, motor units are recruited in a stereotyped and task-related manner, which is achieved by distinct degrees of input-related excitability of the motoneuron subtypes (S > FR > FF) (Henneman and Mendell, 1991; Burke, 1994). For any major functional subtype of motor unit, muscle fibers express characteristic type-specific muscle protein isoforms. Thus, motor units represent functionally distinct subunits of the peripheral motor system whose synapses on muscle can be studied with unique spatial resolution.
The specific motor unit complement of any particular muscle is determined genetically and has species-specific features that reflect behavioral requirements (Burke 1994). Although motor unit identities are considerably stable, their projections and synapses (Waerhaug and Lomo, 1994) within a muscle can exhibit functional and anatomical plasticity in the adult and expand in response to local signals through synaptic (preterminal and ultraterminal) and nodal (from nodes of Ranvier) nerve sprouting (Brown, 1984; Caroni, 1997). This anatomical plasticity is potentiated by intrinsic growth-promoting proteins, such as growth-associated protein 43 (GAP43) and cortical cytoskeleton-associated protein 23 (Aigner et al., 1995; Caroni, 1997). In diseases that target motoneurons, extensive nerve sprouting and synaptic remodeling occurs as part of compensatory reinnervation processes and possibly also of motoneuron pathology. Evidence from animal models indicates that, in these diseases, neuronal dysfunction precedes the clinical phase of the disease, but the mechanisms underlying disease progression still need to be defined (Kong and Xu, 1998; Wong et al., 1998; Williamson and Cleveland, 1999). Such knowledge may lead to earlier detection and more effective treatments.
Here, we performed a detailed analysis of the anatomical plasticity and susceptibility to loss of neuromuscular synapse subtypes in mice. This was greatly facilitated by the observation that on triceps surae muscles, subtypes of synapses are arranged topographically. We demonstrate that neuromuscular synapses differ profoundly in their anatomical plasticity properties, that they are very early targets in motoneuron diseases, and that there is a striking correspondence between anatomical plasticity and resistance to disease.
MATERIALS AND METHODS
Mouse models and treatments. Superoxide dismutase 1(G93A) [SOD1(G93A)] (high-copy number; Chiu et al., 1995) and motoneuron degeneration (Mnd) mice were from The Jackson Laboratory (Bar Harbor, ME); progressive motoneuropathy (pmn) mice were kindly provided by A. Kato (University of Geneva, Geneva, Switzerland). Thy1-GAP43 transgenic mice were as described previously (Aigner et al., 1995). Purified Botulinum toxin A (BotA) (Botox, clinical grade; Allergan AG, Lachen, Switzerland) was applied at 0.01 U/gm mouse (1–3 months old). To assess muscle strength, mice were allowed to grip a small grid loaded with weights and were then lifted by the tail. The behavioral score Σ(tw × W) was compiled from the times tw during which the mouse was able to carry the weights W (40, 30, 20, or 10 gm). A maximum period of 30 sec was allowed for each weight.
Immunocytochemistry and histology. For most experiments involving p75 immunocytochemistry, mice were perfused with 4% paraformaldehyde, and cryostat sections were treated with antibodies as described previously (Aigner et al., 1995). An antiserum to p75 was a kind gift from U. Müller (Friedrich Miescher Institute, Basel, Switzerland). Subtypes of skeletal muscle fibers were analyzed by immunocytochemistry using the subtype-specific myosin heavy chain monoclonal antibodies BF-F3 (type IIb, fast fatiguable), SC-71 (type IIa, fast fatigue resistant), and BA-D5 (type I, slow), which were a kind gift from S. Schiaffino (Muscle Research Laboratory, University of Padova, Padova, Italy). Frozen cryostat sections were post-fixed for 2 min with 3.7% formaldehyde in PBS and subsequently treated with 0.1% trypsin for 3 min, before the application of the monoclonal antibodies. For some experiments, the sections were counterstained for neurofilament protein (NF-200; Sigma, St. Louis, MO), p75, and/or rhodamine isothiocyanate–α-bungarotoxin (Molecular Probes, Eugene, OR). For electron microscopy, mice were perfused with glutaraldehyde, and muscle was processed according to standard procedures.
Analysis of histology data. The triceps surae muscles [medial gastrocnemius (MGC), lateral gastrocnemius (LGC), and soleus] from one mouse were placed in PBS with 10 mm EDTA and mounted for cryosectioning with the side that faced the bone down and parallel to the plane of section. Fifty micrometer sections of the entire muscles were collected and processed for a combined silver-esterase reaction as described previously (Aigner et al., 1995). The section shown in the figures was defined as that including the largest proximal part of the main muscle branches of the MGC and LGC nerves. It was also the first or second section with soleus synapses. On average, in a postnatal day 24 (P24) mouse, this corresponded to section 19 of a total of 35 sections (from a total of 22 P24 mice analyzed, only three deviated from this average by more than one section).
To quantitate muscle fiber type, sprouting, or denervation with respect to position along the synaptic regions of the triceps surae, we subdivided the synaptic band according to muscles and LGC compartments as shown in Figure 1. Each such segment was then further subdivided into 8–20 subsegments of approximately equal length, and all synapses within such subsegments were scored to yield one point on the graph. The first point on the left of the graph represents the most peripheral (Fig. 1, top of the synaptic band) subsegment of the MGC, followed by the adjacent subsegment, up to the end of the synaptic band at the most lateral end of the LGC. The synaptic band of the soleus was analyzed accordingly and plotted on the right of the graph. At least four mice were analyzed for each graph, and one representative example is shown. The distributions of denervated and sprouting synapses were highly reproducible. In SOD1 mice, the absolute extent of the denervation varied somewhat among individuals, and the points on the graphs are averages from the two muscles of a representative mouse.
Fig. 1.

Topographic distribution of muscle fiber subtypes in mouse triceps surae. a, Left, Schematic of section orientation, with respect to hindlimb axis.Right, Arrangement of muscles (MGC and LGC, with medial, intermediate and lateral compartment), tendons, and synapses on a representative section (Achilles tendon is on theleft) (see Materials and Methods for details).b, Quantitative analysis of muscle fiber type distributions along the synaptic band of a section as shown ina. The vertical bars are muscle and/or compartment borders. The points represent segments along the synaptic band (first segment to the left corresponds to top of the synaptic band in a) (see Materials and Methods for details). The values are averages from both muscles of one mouse.
RESULTS
Topographic arrangement of muscle fiber subtypes in mouse triceps surae muscles
To investigate anatomical plasticity and the vulnerability properties of functionally defined subtypes of neuromuscular synapses, we first mapped the distribution of muscle fiber subtypes in the mouse triceps surae. This well characterized and accessible mixed-type hindlimb muscle consists of MGC, LGC, and soleus (Fig. 1a). The LGC is further subdivided into the three major subcompartments medial, intermediate, and lateral LGC (Fig. 1a). Sections (50 μm) through the entire muscle were cut parallel to the hindlimb bones, and one of the two to three sections that included the largest proximal part of the main muscle branches of the MGC and LGC nerves was analyzed in detail (Fig. 1a; see also Materials and Methods). As shown in Figure 1b, fast-fatiguable (type IIb), fast fatigue-resistant (type IIa), and slow (type I) muscle fibers were distributed in a characteristic pattern with respect to the arrangement of tendons and muscle compartments. Generally, type IIb fibers were located most laterally, and type I fibers most medially. The fractions of each muscle containing significant proportions of either IIb, IIa, or I fibers were remarkably well defined, with sharp boundaries (Fig.1b) and variations among individuals (C57Bl6 and CB6 mouse strains) not exceeding 10% along a muscle cross-section (data not shown). Variations among strains and individuals were mainly restricted to relative contents of muscle fiber types within transition zones. Therefore, muscle fiber, and thus neuromuscular synapse subtypes in mouse triceps surae, are distributed in a predictable and topographic manner.
Profound differences in the competence and control of synaptic sprouting at neuromuscular synapse subtypes
To determine whether neuromuscular synapse subtypes (Waerhaug and Lomo, 1994) differ in their competence for synaptic growth, we mapped BotA-induced ultraterminal nerve sprouting, a well characterized form of synaptic sprouting induced by blockade of transmitter release (Brown, 1984). As shown in Figure2a, sprouting synapses exhibited a striking topographic distribution reminiscent of slow-type muscle fibers, whereas MGC and LGC synapses located in predominantly type IIb regions failed to sprout. In addition, muscle-specific differences were detectable, with sprouting in soleus being consistently more pronounced than in slow-type regions of MGC and LGC (Fig. 2a). Comparable differences were detected at any time after toxin treatment, with no detectable ultraterminal sprouting in type IIb regions, even after prolonged paralysis (Fig. 2a). To investigate the responses of synapses in type IIb muscle regions to BotA in more detail, we stained muscle sections with an antibody against the low-affinity NGF receptor p75, a gene that is induced in synapse-associated Schwann cells (tSCs) when neuromuscular transmission is impaired (Hassan et al., 1994). As shown in Figure 2b, in contrast to the type-specific nerve sprouting response, the BotA treatment induced inactivation-sensitive p75 expression and process outgrowth in tSCs from all regions of MGC and soleus.
Fig. 2.
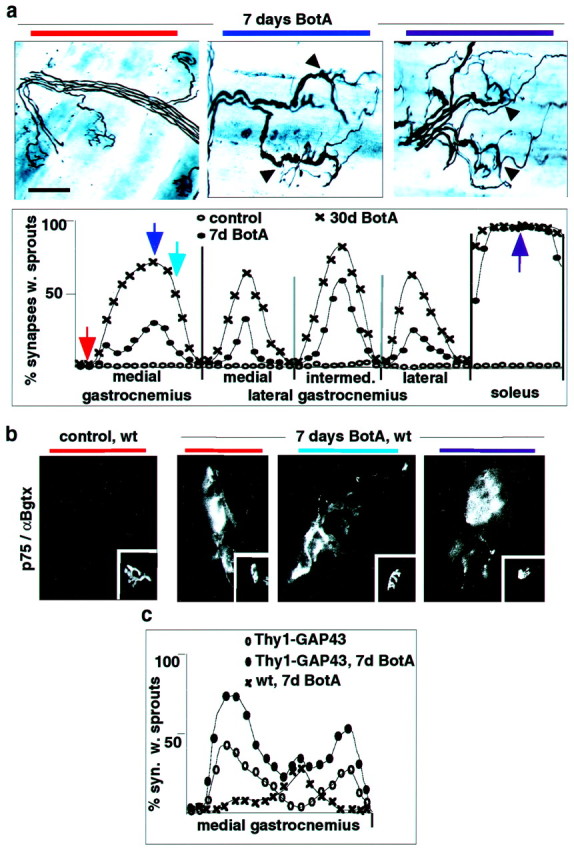
Profound differences in the growth response of neuromuscular synapse subtypes in response to paralysis and/or GAP43 overexpression. a, Distribution of synapses with sprouts in triceps surae of BotA-treated wild-type mice. Top row, Silver-esterase stains (arrowheads, sprouting synapses); bottom row, quantitative analysis as in Figure 1b (n = 4). Positional color code in the figure (horizontal bars) refers to the position of the arrows in the graph. b, Upregulation of tSC p75 immunoreactivity by BotA-induced paralysis at all types of triceps surae synapses (insets, α-bungarotoxin-positive synapses). c, Distribution of synapses with sprouts in MGC of Thy1-GAP43 mice, with and without BotA treatment. Scale bar: a, 55 μm; b, 25 μm.
To determine whether overexpression of a protein that promotes sprouting can overcome the incompetence of type IIb synapses to sprout, we analyzed spontaneous and BotA-induced synaptic sprouting in transgenic mice overexpressing the growth-promoting protein GAP43 specifically in adult neurons (Aigner et al., 1995). Detailed analysis of such Thy1-GAP43 transgenic mice revealed the presence of synaptic sprouting in MGC regions enriched in type IIa muscle fibers but not in its most lateral type IIb regions (Fig. 2c). Treating Thy1-GAP43 mice with BotA led to great potentiation of sprouting in those regions in which GAP43 was already effective without paralysis but to no potentiation of sprouting in the other sections of MGC (Fig.2c).
Early and selective loss of neuromuscular synapse subtypes in mice overexpressing SOD1(G93A)
We next determined whether neuromuscular synapse subtypes may differ in their vulnerability to disease. One genetic model to study motoneuron disease is transgenic mice overexpressing high levels of mutated human Cu,Zn SOD1(G93A) associated with familial amyotrophic lateral sclerosis (ALS) (Chiu et al., 1995). These mice develop clinical symptoms of muscle weakness at approximately P90, loss of spinal motoneurons after P100, and die on average at P136 (Chiu et al., 1995). Unexpectedly, and as shown in Figure3a, they already exhibited extensive local muscle denervation at P50. Systematic mapping of the innervation pattern in the triceps surae revealed a highly regionalized and topographic distribution of the denervation (Fig. 3b) highly reminiscent of that of type IIb muscle fibers. Topographic distributions of denervation resembling that of type IIb fibers were also detected in other muscles, such as gluteus and gracilis (data not shown). Analysis of SOD1(G93A) mice at P80 revealed extensive atrophy and degeneration of peripheral muscle fibers in the medial gastrocnemius, consistent with persistent denervation (data not shown). To provide molecular evidence for the selective loss of synapses, we analyzed corresponding muscle sections for p75 immunoreactivity. In agreement with the denervation pattern, at P50 labeling of tSC processes was prominent at peripheral synapses of MGC, less frequent in more medial sections of the MGC, and absent in the soleus (Fig.3c). In control experiments, nontransgenic mice exhibited no labeling of synapses in the MGC or soleus, whereas substantial labeling was induced by denervation in all muscles (data not shown). Double-labeling with myosin isoform antibodies revealed that, at P58, loss of nerve-associated neurofilament immunoreactivity (Fig.3d) and upregulation of p75 (data not shown) in mixed muscle regions was selective for synapses on type IIb and some type IIa muscle fibers.
Fig. 3.
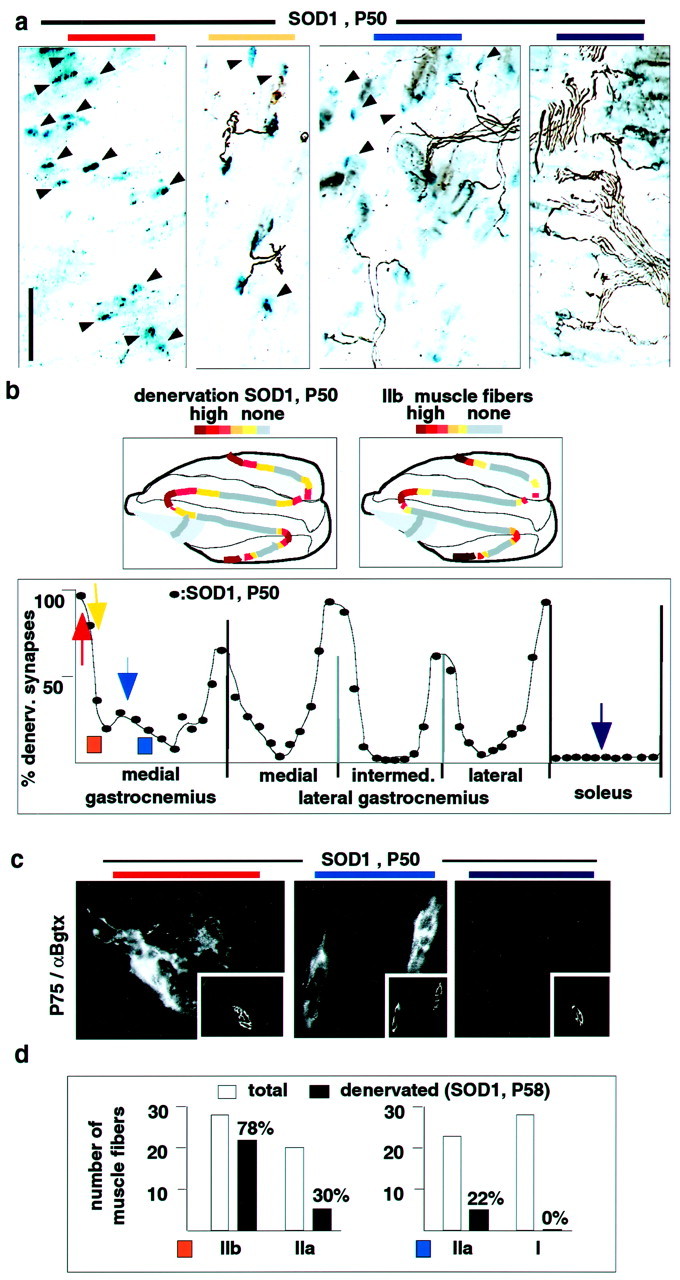
Early selective denervation of neuromuscular synapses in SOD1(G93A) mice. a, Innervation pattern in different triceps surae regions (one muscle; combined silver-esterase stain). Positional color code like in Figure 2.Arrowheads, Denervated synapses [blue-esterase reaction product, but no nerve (black)]. b,Top row, Schematic of distribution of denervated synapses [SOD1(G93A), P50] and type IIb muscle fibers. Bottom row, Distribution of denervated synapses in a representative SOD1 mouse at P50 (quantitative analysis; see Materials and Methods for details) (vertical bars, muscle or compartment borders).c, Upregulation of synaptic p75 immunoreactivity in muscle regions with high contents of denervated synapses; α-bungarotoxin (insets) visualizes synapses.d, Selective denervation of type IIb and type IIa fibers in SOD1(G93A) mice. Two mixed fiber regions of MGC (see colored rectangles in b for their positions) from two mice (total of 4 muscles) were analyzed for myosin heavy chain isoform expression and the presence or absence of neurofilament-positive nerve at α-bungarotoxin-positive synapses. In two control mice, all synapses were innervated by this criterion (data not shown). The region labeled in orange (left) contained no type I fibers; the region labeled in blue(right) contained no type IIb fibers. Thenumbers represent denervation percentages for the particular fiber type. Scale bar: a, 180 μm;c, 32 μm.
To determine whether early selective disconnection of certain neuromuscular synapses was attributable to early vulnerability of the corresponding motoneurons to the mutated SOD1, we analyzed the pathology of neuromuscular synapses in muscle regions that were prone or resistant to denervation. Ultrastructural analysis of nerve terminals in the MGC and the soleus at P50 revealed prominent vacuolation in nearly all terminals of both muscles (Fig.4a). Selective disconnection was also not attributable to differential expression of the transgene, because undistinguishably high levels of human SOD1 immunoreactivity were detected in all intramuscular MGC, LGC, and soleus nerves (data not shown). Therefore, mutated SOD1 induced comparable pathology in all motoneuron subtypes. Nevertheless, only some of these exhibited early peripheral denervation.
Fig. 4.
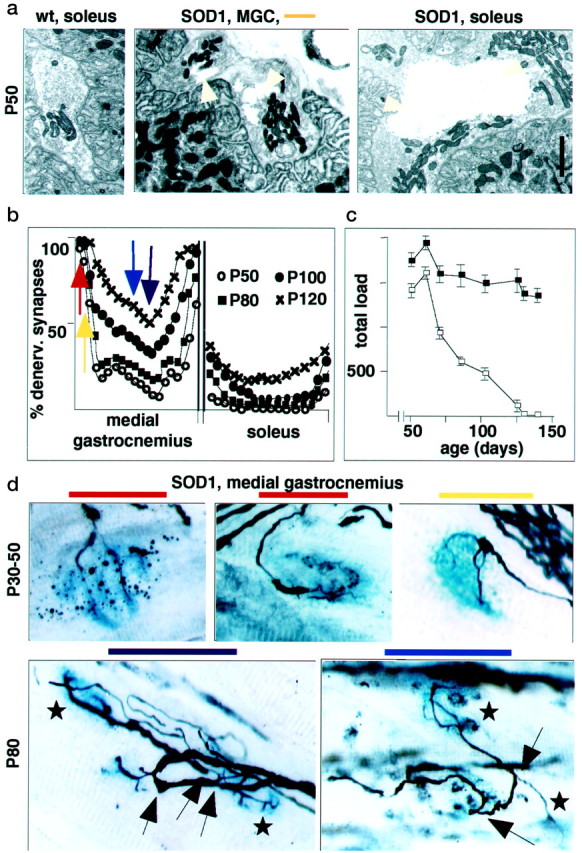
Selective vulnerability of fast-type MGC synapses in SOD1 model. a, Widespread vacuole formation in vulnerable and resistant synapses of SOD1 mice. Ultrastructure of neuromuscular junctions at P50. Note presence of large vacuoles (arrowheads) at nerve terminals of MGC and soleus.b, Distribution of denervated synapses in SOD1 mice from P50 (nonsymptomatic) to P120 (extensive paralysis and motoneuron death). Color code as in Figure 2. c, Time course of force decline in limbs of SOD1 mice (open squares) and wild-type littermates (filled squares). Loaded grid test; total load, Σ(tw ×W); n = 26.d, Dying-back features of denervation (top row) and sprouting with reinnervation (bottom row; arrows, sprouts; stars, synapses innervated by sprouts) in SOD1 mice. Scale bar:a, 1.1 μm; d, 16 μm.
To determine how selective denervation in the SOD1 mice evolved with time, we mapped MGC and soleus innervation patterns between P30, soon after motor units have acquired their adult properties, and P120, when these mice exhibit pronounced motoneuron loss (Fig.4b,d). Between P30 and P40, we detected progressive thinning of neurofilament-positive terminal branches, characteristic of an axonal dying-back pathology, predominantly in the peripheral section of the MGC (Fig. 4d), but denervation was not pronounced. Between P45 and P80, dramatic denervation developed in the predominantly fast regions of the MGC, whereas slow regions and the soleus were spared (Fig. 4b). As shown in Figure4c, this progressive denervation correlated with loss of muscle strength, as monitored in the loaded grid test. Beginning at approximately P60 and extending into the clinical phase of the disease, local reinnervation by collateral and ultraterminal sprouting was detected in medial muscle regions (Fig. 4d). This was reflected in a slight local increase in the proportion of muscle fibers immunoreactive for type I myosin heavy chain (data not shown). At P80, this increase was restricted to transition regions of muscle exhibiting mixed IIa/I muscle fibers (Fig. 1b), with no evidence for a significant expansion of slow motor units beyond their normal muscle territory. In the soleus, significant denervation was restricted to the very last phase of the disease (P120) (Fig. 4b). Therefore, throughout the disease process, triceps surae synapses on type IIb fibers are affected early on and usually before those on type IIa fibers, whereas synapses on type I fibers resist denervation and sprout to reinnervate nearby vacated synapses. In addition, in this SOD1(G93A) model, a progressive dying-back process involving selective synaptic weakening and denervation sets on long before the appearance of clinical symptoms and long before the loss of motoneurons.
A common pattern of early and selective synaptic weakening and loss in degenerative motoneuron diseases and aging
To determine whether the pattern of early, protracted, and selective synaptic weakening may be a general property of degenerative motoneuron diseases, we performed a similar analysis in progressive motoneuropathy (pmn) and motoneuron degeneration (Mnd) mice. Both strains carry spontaneous mutations leading to motoneuron degeneration in the adult, but the kinetics of the diseases is very different (Pioro and Mitsumoto, 1995). Pmn mice exhibit some muscle atrophy as soon as P15–P18, motoneuron loss from P20 on, and die at approximately P45, whereas Mnd mice exhibit first signs of muscle weakness at 6 months and die at ∼1 year.
As shown in Figure 5a, in pmn mice, denervation was already pronounced at P15, the earliest time point that we have studied. Although also detectable in the soleus and in slow sections of the MGC, denervation was much more extensive in MGC regions predominantly innervated by fast motoneurons. At later time points, further deterioration was detected, but slow muscle regions remained better preserved (Fig. 5a). In the slowly progressing Mnd model, atrophy of nerve terminal branches was the predominant early (3 months) anatomical deficit (data not shown). This was accompanied by the selective appearance of p75 immunoreactivity and extensive sprouting of tSC processes at affected fast synapses (Fig.5b). Reflecting the less aggressive progression of the disease, in these mice, denervation progressed much more gradually than in the pmn and SOD1 models. Again, selective dying-back-type deficits at the synapses of predominantly fast MGC regions were detected before the onset of clinical manifestations, whereas slow muscle regions and soleus synapses resisted and did not exhibit tSC reactions until the latest phases of the disease (Fig. 5a).
Fig. 5.
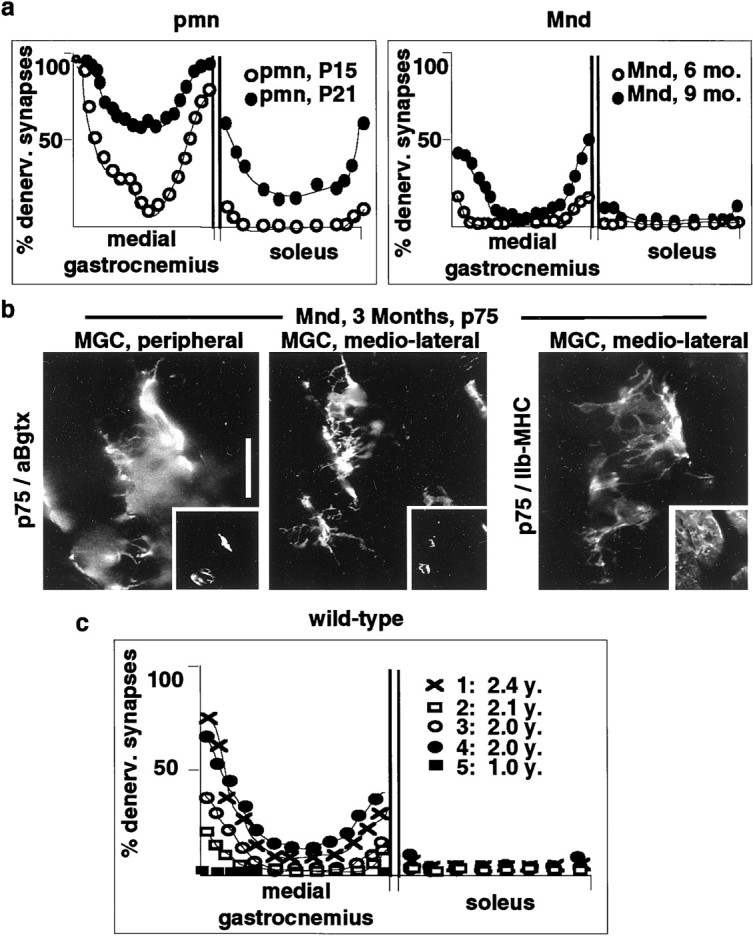
Selective and type-specific vulnerability of neuromuscular synapses in motoneuron diseases and aging.a, Distribution of denervated synapses in pmn and Mnd mice. b, Early upregulation of p75 at fast-type neuromuscular synapses of Mnd mice. Scale bar, 30 μm.c, Distribution of denervated synapses in aged wild-type mice. The four curves are averages of left and right muscles for four separate mice.
To determine whether synapse pathology and loss associated with age may affect preferentially the same types of synapses that are vulnerable in motoneuron disease, we analyzed corresponding patterns of innervation in aged wild-type mice. As shown in Figure 5c, although loss of synapses varied markedly among aged mice, there was a clear preference for synapse loss in lateral and predominantly type IIb regions of MGC and LGC, whereas neuromuscular junctions in the soleus were not denervated. In addition, and like in Mnd mice, we observed a pronounced thinning and atrophy of preterminal and terminal nerve branches in aged mice. This weakening was detected at nearly all synapses from predominantly type IIb regions and was not detectable in soleus or slow regions of gastrocnemius (data not shown). In affected muscle regions, we also observed obviously atrophic muscle fibers that had presumably been denervated for several months. Because on these fibers acetylcholine esterase reaction product could not be detected anymore, they were not included in the quantitation of denervated synapses.
Denervation resistant synapses in SOD1(G93A) and Mnd mice exhibit a progressive impairment in stimulus-induced synaptic sprouting
A comparison of SOD1-induced denervation and BotA-induced sprouting revealed the existence of a striking inverse relationship between susceptibility to synapse loss in motoneuron disease and competence for synaptic sprouting (Fig.6a). To determine whether disease progression affects anatomical plasticity at the synapse, we compared BotA-induced ultraterminal sprouting in healthy, SOD1(G93A), and Mnd mice. Soleus synapses in SOD1 (Fig. 6b) and Mnd (Fig. 6c) mice exhibited a progressive impairment in this form of stimulus-induced synaptic sprouting. In SOD1(G93A) mice, sprouting was already significantly impaired at P50, when mice were free of clinical disease symptoms. In the slowly progressing Mnd model, soleus synapses exhibited reduced sprouting at 3 months (presympthomatic) and absence of sprouting at 9 months (Fig.6c). In demonstrating that disease progression impairs anatomical plasticity, these findings support the notion that absence of anatomical plasticity and disease-induced synapse loss are mechanistically related.
Fig. 6.

Relationship between anatomical plasticity and vulnerability to disease at the neuromuscular junction.a, Inverse relationship between BotA-induced sprouting in wild-type mice and susceptibility to denervation in SOD1(G93A) motoneuron disease mice (bold line); the data were replotted from Figures 2a and 3b.b, Impairment of BotA-induced sprouting in the soleus of SOD1(G93A) mice. c, Impairment of BotA-induced sprouting in the soleus of Mnd mice.
DISCUSSION
The findings of this study provide novel insights into the mechanisms and factors that affect the progression of motoneuron diseases. The main results are as follows: (1) functionality and maintenance of neuromuscular synapses are affected very early in motoneuron diseases; (2) early synaptic deficits are detected in motoneuron diseases of diverse origin, and the exacerbation of these lesions correlates with the timing and speed of disease progression; (3) synapse types that fail to exhibit terminal sprouting are selectively vulnerable, whereas synapses undergoing robust paralysis-induced sprouting are selectively resistant; and (4) degenerative motoneuron diseases involve a gradual and protracted progression of functional losses that begin long before the onset of the clinical phase and long before the actual death of motoneurons. Based on the results of this study, we propose a selective axonal dying-back model, whereby several distinct disease form-specific pathological processes lead to a gradual reduction in motoneuron fitness, which in turn results in a common pattern of selective and progressive synaptic weakening and denervation (Fig.7). In this model, onset time and kinetics of disease progression are affected by upstream events specific to the particular pathological process, but the clinical features of motoneuron diseases are determined by the facts that the neuromuscular synapses in the periphery appear to be particularly sensitive to neuronal dysfunction, that the synapses on type IIb muscle fibers, which are least competent to sprout, are most vulnerable, and that those on type I fibers, which are most plastic, are particularly resistant. Similar weakening and selective loss of synapses with low sprouting competence appears to be associated with age. This model is consistent with reports of early neuromuscular junction transmission defects in a dog model of ALS (Pinter et al., 1995, 1997) and in post-polio syndrome patients (Cashman and Trojan, 1995; Larsson et al., 1995). Early transmission defects were also detected in ALS patients (Maselli et al., 1993), and in one study, weakening was particularly pronounced in high threshold, i.e., FF motor units (Dengler et al., 1990). The model is also consistent with the fact that, in pmn and SOD1 mice, treatments that prevent the loss of motoneurons have little effect on disease progression, whereas those that protect motoneuron axons do (Sagot et al., 1995, 1996, 1998; Couillard-Despres et al., 1998; Williamson et al., 1998; Williamson and Cleveland, 1999; W. Spooren, unpublished observations).
Fig. 7.

Proposed model of selective weakening, dying-back, and denervation progression in degenerative motoneuron diseases and aging (see Discussion).
Anatomical plasticity in the target region is a crucial adaptational and repair process in the adult, and there is a need to understand the factors that regulate this plasticity. This study demonstrates that, in the neuromuscular system, a major contributing factor is the existence of profound differences among neuromuscular synapse subtypes. Ultraterminal sprouting in soleus and slow synapses of gastrocnemius was induced by blockade of neuromuscular transmission. Different, presumably FR synapses in gastrocnemius exhibited GAP43-induced sprouting, which was potentiated by BotA. Finally, FF synapses in gastrocnemius failed to sprout under all these experimental conditions. These findings are consistent with and extend previous reports that, upon BotA treatment, synapses in slow muscles sprout more readily than those in fast ones (Duchen, 1970). Because mouse soleus contains both slow and fast fatigue-resistant motor units, synaptic sprouting in response to paralysis appears to be determined by both muscle and motor unit type. Because the electrophysiological properties of the motoneurons are so different, our findings suggest that episodes of major anatomical plasticity in the target region may alter significantly the physiological properties of muscles. Consistent with this prediction, shifts toward slow-types of muscle units have indeed been detected in association with pathology (Dengler et al., 1990;Larsson et al., 1995). Differential sprouting in the target region may ensure that, under conditions of partial dysfunction, essential muscle functions associated with maintenance of posture and motor coordination are conserved at the expense of muscle force.
Although the differences in the control of terminal sprouting by synapse subtypes could be attributable exclusively to intrinsic differences among motoneurons, it seems more likely that they also involve differential regulation of nerve sprouting by the local environment at the synapse. Thus, although motoneurons can determine muscle fiber properties through their specific activity patterns, local retrograde signaling does play a role in regulating nerve growth at the synapse (Nguyen et al., 1998). On the other hand, overexpression of GAP43 in adult motoneurons promoted nerve sprouting at certain, presumably FR-type synapses, but not in the soleus (P. Caroni, unpublished results). Because in these mice the transgene was expressed specifically in all adult motoneurons (Aigner et al., 1995), it must have selectively affected particular axon terminals, leading to nerve sprouting. Regulation of terminal growth may therefore result from an interplay between specific presynaptic and postsynaptic properties. Because we could already detect differences in BotA-induced sprouting at P10 (data not shown), specific regulation may also affect aspects of neuromuscular junction development.
With respect to clinical implications, these results show that peripheral synapses may serve as novel targets to treat motoneuron diseases, including ALS. Thus, partial denervation of muscle not only directly produces weakness but may also affect disease progression through overburdening and exhaustion of the remaining and expanded motor units. Candidate molecules for early treatment of peripheral synapses include neurotrophic factors (Haase et al., 1997; Sagot et al., 1998). Furthermore, the discovery of early functional deficits at accessible peripheral neuromuscular synapses should facilitate early detection and treatment of motoneuron diseases. Finally, a major goal will be to understand how properties of slow-type synapses (Burke, 1994; Waerhaug and Lomo, 1994) confer resistance to denervation in motoneuron diseases. Motoneuron diseases may involve aggregation-mediated pathogenic mechanisms (Brujin et al., 1998) also found in other late-onset and progressive neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases (Goedert et al., 1998). Accordingly, the model of early, selective, and progressive synaptic dysfunction emerging from this study may also apply to other neurodegenerative diseases.
Footnotes
We are grateful to S. Arber, J. Kapfhammer, W. Krek, U. Müller, M. Rüegg, M. E. Schwab, and G. Thomas for valuable comments on this manuscript. We thank T. Mennini (Istituto Mario Negri, Milano, Italy) for help with Mnd mice.
Correspondence should be addressed to P. Caroni, Friedrich Miescher Institut, Maulbeerstrasse 66, CH-4058 Basel, Switzerland. E-mail:caroni@fmi.ch.
REFERENCES
- 1.Aigner L, Arber S, Kapfhammer J, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 2.Brown MC. Sprouting of motor nerves in adult muscles: a recapitulation of ontogeny. Trends Neurosci. 1984;7:10–14. [Google Scholar]
- 3.Brujin LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 4.Burke RE. Physiology of motor units. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York: 1994. pp. 464–484. [Google Scholar]
- 5.Caroni P. Intrinsic neuronal determinants that promote axonal sprouting and elongation. BioEssays. 1997;19:767–775. doi: 10.1002/bies.950190906. [DOI] [PubMed] [Google Scholar]
- 6.Cashman NR, Trojan DA. Correlation of electrophysiology with pathology, pathogenesis, and anticholinesterase therapy in post-polio syndrome. Ann NY Acad Sci. 1995;753:138–150. doi: 10.1111/j.1749-6632.1995.tb27540.x. [DOI] [PubMed] [Google Scholar]
- 7.Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- 8.Couillard-Despres S, Zhu K, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dengler R, Konstanzer A, Kuther G, Hesse S, Wolf W, Struppler A. Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve. 1990;13:545–550. doi: 10.1002/mus.880130612. [DOI] [PubMed] [Google Scholar]
- 10.Duchen LW. Changes in motor innervation and cholinesterase localization induced by botulinum toxin in skeletal muscle of the mouse: differences between fast and slow muscles. J Neurol Neurosurg Psychiatry. 1970;33:40–54. doi: 10.1136/jnnp.33.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M, Spillantini MG, Davies SW. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol. 1998;8:619–632. doi: 10.1016/s0959-4388(98)80090-1. [DOI] [PubMed] [Google Scholar]
- 12.Haase G, Kennel P, Pettmann B, Vigne E, Akli S, Revah F, Schmalbruch H, Kahn A. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat Med. 1997;3:429–436. doi: 10.1038/nm0497-429. [DOI] [PubMed] [Google Scholar]
- 13.Hassan SM, Jennekens FG, Veldman H, Oestreicher BA. GAP-43 and p75NGFR immunoreactivity in presynaptic cells following neuromuscular blockade by botulinum toxin in rat. J Neurocytol. 1994;23:354–363. doi: 10.1007/BF01666525. [DOI] [PubMed] [Google Scholar]
- 14.Henneman E, Mendell LM. Functional organization of the motoneuron pool and its inputs. Handbook of physiology: the nervous system II (Brooks H, ed), pp 423–507. Williams and Wilkins; Baltimore: 1991. [Google Scholar]
- 15.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsson L, Li X, Tollback A, Grimby L. Contractile properties in single muscle fibers from chronically overused motor units in relation to motoneuron firing properties in prior polio patients. J Neurol Sci. 1995;132:182–192. doi: 10.1016/0022-510x(95)00138-r. [DOI] [PubMed] [Google Scholar]
- 17.Maselli RA, Wollman RL, Leung C, Palombi S, Richman DP, Slalzar-Grueso EF, Roos RP. Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve. 1993;16:1193–1203. doi: 10.1002/mus.880161109. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- 19.Pinter MJ, Waldeck RF, Wallace N, Crok LC. Motor unit behavior in canine motor neuron disease. J Neurosci. 1995;15:3447–3457. doi: 10.1523/JNEUROSCI.15-05-03447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinter MJ, Waldeck RF, Cope TC, Cork LC. Effects of 4-aminopyridine on muscle and motor unit force in canine motor neuron disease. J Neurosci. 1997;17:4500–4507. doi: 10.1523/JNEUROSCI.17-11-04500.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pioro EP, Mitsumoto H. Animal models of ALS. Clin Neurosci. 1995;3:375–385. [PubMed] [Google Scholar]
- 22.Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sagot Y, Tan SA, Hammang JP, Aebischer P, Kato AC. GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmn/pmn mice. J Neurosci. 1996;16:2335–2341. doi: 10.1523/JNEUROSCI.16-07-02335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sagot Y, Rosse T, Vejsada R, Perrelet D, Kato AC. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J Neurosci. 1998;18:1132–1141. doi: 10.1523/JNEUROSCI.18-03-01132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens CF. Neuronal diversity: too many cell types for comfort? Curr Biol. 1998;8:R708–R710. doi: 10.1016/s0960-9822(98)70454-3. [DOI] [PubMed] [Google Scholar]
- 26.Waerhaug O, Lomo T. Factors causing different properties at neuromuscular junctions in fast and slow rat skeletal muscles. Anat Embryol. 1994;190:113–125. doi: 10.1007/BF00193409. [DOI] [PubMed] [Google Scholar]
- 27.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 28.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong PC, Rothstein JD, Price DL. The genetic and molecular mechanisms of motor neuron disease. Curr Opin Neurobiol. 1998;8:791–799. doi: 10.1016/s0959-4388(98)80123-2. [DOI] [PubMed] [Google Scholar]