

Abstract
Recurrent inhibition in olfactory bulb mitral cells is mediated via reciprocal dendrodendritic synapses with granule cells. Although GABAergic granule cells express both NMDA and non-NMDA glutamate receptors, dendrodendritic inhibition (DDI) relies on the activation of NMDA receptors. Using whole-cell recordings from rat olfactory bulb slices, we now show that olfactory NMDA receptors have a dual role; they depolarize granule cell spines, and they provide a source of calcium that can evoke GABA exocytosis. We demonstrate that exogenous NMDA can trigger GABA release after blockade of voltage-dependent calcium channels (VDCCs) with Cd. We also find that postsynaptic depolarization alone can evoke GABA release via a separate mechanism that relies on calcium influx through Cd-sensitive VDCCs. By selectively manipulating postsynaptic responses in granule cells with high-K or low-Na extracellular solutions, we show that endogenous glutamate can elicit GABA release via both NMDA receptor- and VDCC-dependent pathways. Finally, we find that blockade of Na channels in granule cells with tetrodotoxin enhances DDI, presumably by reducing the depolarization of granule cells during DDI and thereby increasing the driving force for Ca entry through NMDA receptors. These results provide evidence of a novel mechanism for evoked transmitter release that depends on Ca influx through ionotropic receptors and provides a new potential site for synaptic plasticity in the olfactory bulb.
Keywords: olfactory bulb, mitral cell, glutamate receptor, EPSP, NMDA receptor, recurrent inhibition
Release of the neurotransmitter contained in presynaptic vesicles is triggered by a rise of intracellular Ca ([Ca2+]i) within the presynaptic terminal (Katz and Miledi, 1967; Augustine et al., 1985; Mintz et al., 1995). Abundant evidence (Adler et al., 1991; Mintz et al., 1995; Borst and Sakmann, 1996; Neher, 1998) indicates that influx through voltage-dependent Ca channels (VDCCs) located near vesicle-docking sites can provide the Ca influx needed to evoke synaptic transmission. However, activation of presynaptic neurotransmitter receptors that are permeable to Ca may also enable a sufficient rise in intracellular Ca to trigger transmitter release. This hypothesis is supported by studies of axon terminals that contain presynaptic nicotinic ACh receptors (nAChRs) that are highly permeable to Ca (Vernino et al., 1992; Rathouz and Berg, 1994; Gray et al., 1996). Application of exogenous nicotine enhanced the frequency of glutamatergic spontaneous EPSC by acting on presynaptic nAChRs in chick medial habenula and interpeduncular nuclei (McGehee et al., 1995). Similar results were found in rat when nicotine applied to mossy fiber synapses in the hippocampus increased the frequency of miniature glutamatergic EPSPs (Gray et al., 1996), an effect that persists after blockade of VDCCs with cadmium.
In the olfactory bulb presynaptic granule cell spines contain functional NMDA receptors (Isaacson and Strowbridge, 1998; Schoppa et al., 1998) that are critically involved in triggering exocytosis of GABA (Isaacson and Strowbridge, 1998). These dendritic spines form reciprocal synapses with the secondary dendrites and somata of glutamatergic mitral cells (Rall et al., 1966; Price and Powell, 1970a–c), the principal cell type in the olfactory bulb. This dendrodendritic microcircuit supports both recurrent feedback (Jahr and Nicoll, 1980, 1982; Nowycky et al., 1981a,b, Isaacson and Strowbridge, 1998; Schoppa et al., 1998) and lateral inhibition of mitral cells (Isaacson and Strowbridge, 1998). The close spatial relationship between the postsynaptic active zone within the spine—presumably containing glutamate receptors—and docked vesicles containing GABA raises the possibility that the influx of Ca through NMDA receptors can directly trigger exocytosis. Alternatively, the depolarization caused by NMDA and AMPA receptor activation could open VDCCs near docked vesicles. In the latter model, the Ca influx through VDCCs triggers exocytosis, whereas influx of Ca through NMDA receptors plays only a modulatory role by elevating residual Ca within the spine. Isaacson and Strowbridge (1998) showed that direct depolarization (with KCl) evoked GABA release from granule cell processes that could be blocked by Cd, supporting the hypothesis that Ca influx through VDCCs can trigger exocytosis. However, it is not clear from these studies whether Ca influx through NMDA receptors present on the same spine also could evoke transmitter release and which mechanism is dominant under physiological conditions (i.e., when receptors on the spine are activated by endogenous glutamate).
To address these questions, we explored the mechanisms controlling GABA release from granule cell spines using whole-cell recordings from rat olfactory bulb slices. We initially confirm that Ca influx through VDCCs can trigger GABA release from granule cells. Unexpectedly, we also find that exogenous NMDA can effectively trigger GABA release from granule cell spines when all VDCCs capable of supporting transmitter release are blocked with Cd. These results support a model in which GABA release can be stimulated by Ca influx through either NMDA receptors or VDCCs. By varying the amount of glutamate released by a mitral cell, we show that Ca influx through VDCCs triggers GABA release in response to relatively small EPSPs, whereas most GABA release in response to large-amplitude EPSPs is triggered directly by Ca influx through NMDA receptors.
MATERIALS AND METHODS
Olfactory bulb slices (300 μm) from 14- to 21-d-old Sprague Dawley rats were made using a Leica (Nussloch, Germany) VT1000S vibratome. Slices were incubated in a 30°C water bath for 30 min and then maintained submerged at room temperature. Whole-cell patch-clamp recordings were made in mitral or granule cells visualized under infrared-differential interference contrast optics (ZeissAxioskop FS) using a Axopatch 1D amplifier (Axon Instruments). Mitral and granule cells were distinguished by their location and the size of their somata. Olfactory bulb slices were superfused with artificial CSF (ACSF) that contained (in mm): NaCl 124, KCl 5, NaH2PO4 1.23, NaHCO3 26, dextrose 10, CaCl2 2.5, and MgSO4 1.2, equilibrated with 95% O2/5% CO2 and warmed to 30°C (flow rate, 1–2 ml/min). As indicated in the figure legends, Mg was omitted from the ACSF in some experiments. Except where noted, tetrodotoxin (TTX; 1 μm) was routinely added to the ACSF to block fast Na currents. In some experiments (indicated in the figure legends)N-methyl-d-glucamine (NMG; 124 mm) was substituted for 124 mm NaCl in the ACSF (pH adjusted to 7.4 by titrating with HCl). This “low-Na” ACSF contained 27 rather than 151 mm Na.
Patch-clamp electrodes used for voltage-clamp recording (2–4 MΩ resistance) typically contained (in mm): CsCl 115, HEPES 10, EGTA 1, TEA-Cl 25, MgATP 4, Na3GTP 0.3, and phosphocreatine 10. For voltage-clamp recordings during focal application of NMDA and KCl, the internal solution contained (in mm): Cs-methanesulfonate 140, NaCl 8, HEPES 10, EGTA 0.2, MgATP 4, Na3GTP 0.3, and phosphocreatine 10. In some experiments (indicated in the figure legends), QX-314 (5 mm) was added to the internal solution to block Na currents in the mitral cell. The internal solution used for current-clamp recordings of granule cells contained (in mm): K-methylsulfate 140, NaCl 8, HEPES 10, EGTA 0.2, MgATP 4, Na3GTP 0.3, and phosphocreatine 10. To evoke glutamate release from mitral cells, steps to +10 mV were used from a holding potential of −70 mV, unless noted otherwise in the figure legends.
Voltage and current records were low-pass filtered at 2 kHz and sampled at 5 kHz using an ITC-18 16-bit analog-to-digital converter (Instrutech). Series resistance, which typically was <10 MΩ, was routinely compensated by >80%. Data were streamed to a hard disk using a custom Visual Basic program or acquired directly into IgorPro (WaveMetrics). IgorPro and Origin 5.0 (Microcal) were used to analyze data. Dendrodendritic inhibition was quantified using custom macros in IgorPro that calculated the postsynaptic current integral (reported in nanoamperes second) over a 2 sec period beginning 50 msec after the end of the presynaptic voltage step. Action potential amplitude was measured from the resting membrane potential. Example traces are averages of five to six responses. In most experiments, drugs were applied by switching the perfusion media. A Picospritzer (General Valve, Fairfield, NJ) was used to focally apply receptor agonists and KCl. TTX was obtained from Calbiochem (La Jolla, CA), the glutamate receptor agonists and antagonists were obtained from Research Biochemicals (Natick, MA), and all other chemicals were obtained from Sigma (St. Louis, MO). Data are shown as the mean ± SEM.
RESULTS
We first examined the postsynaptic glutamate receptors that mediate excitation of granule cells during dendrodendritic inhibition (DDI). Dendrodendritic inhibition in the olfactory bulb is thought to result from glutamate released from the secondary dendrites and somata of mitral cells exciting dendritic spines of granule cells and causing the subsequent release of GABA and feedback inhibition in mitral cells (see Fig. 1A). Previous work (Isaacson and Strowbridge, 1998; Schoppa et al., 1998) suggested that DDI relied on the activation of both NMDA and non-NMDA receptors on granule cells. We investigated the roles of non-NMDA and NMDA receptors in DDI by recording from mitral cells with Mg-free ACSF (to relieve the voltage-dependent Mg block of NMDA receptors.) Mitral cells were recorded under whole-cell patch clamp with a CsCl-based internal solution that resulted in inward GABAAreceptor-mediated currents at a holding potential of −70 mV. We applied a depolarizing voltage step to +10 mV (for 50 msec) to evoke DDI in mitral cells. Under these conditions, the selective non-NMDA receptor antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide disodium (NBQX) did not affect the DDI response (93.0 ± 6.6% of control in 5 μm NBQX; n = 9; 105.2 ± 6.3% of control in 20 μm NBQX;n = 5.) However, the subsequent application of the NMDA receptor antagonist d-2-amino-5-phosphopentanoic acid (d-APV; 50 μm) caused a marked reduction of the dendrodendritic response to 4.9 ± 0.8% of control (n = 9). No further reduction was observed with the combination of NBQX (5 μm) andd-APV (4.5 ± 0.5% of control; 50 μmd-APV;n = 10; see Fig. 1B,C). By contrast, 6,7-dinitroquinoxalone-2,3-dione (DNQX; 20 μm), a less selective AMPA receptor antagonist (Randle et al., 1992), reduced the DDI response to 50.7 ± 6.7% of control (n = 3), consistent with previous work (Isaacson and Strowbridge, 1998). Although we find no requirement for activation of non-NMDA receptors when eliciting DDI in Mg-free ACSF, AMPA receptors may mediate the initial depolarization of granule cells when physiological concentrations of Mg are present (see Isaacson and Strowbridge, 1998; Schoppa et al., 1998, Schoppa and Westbrook, 1999).
Fig. 1.
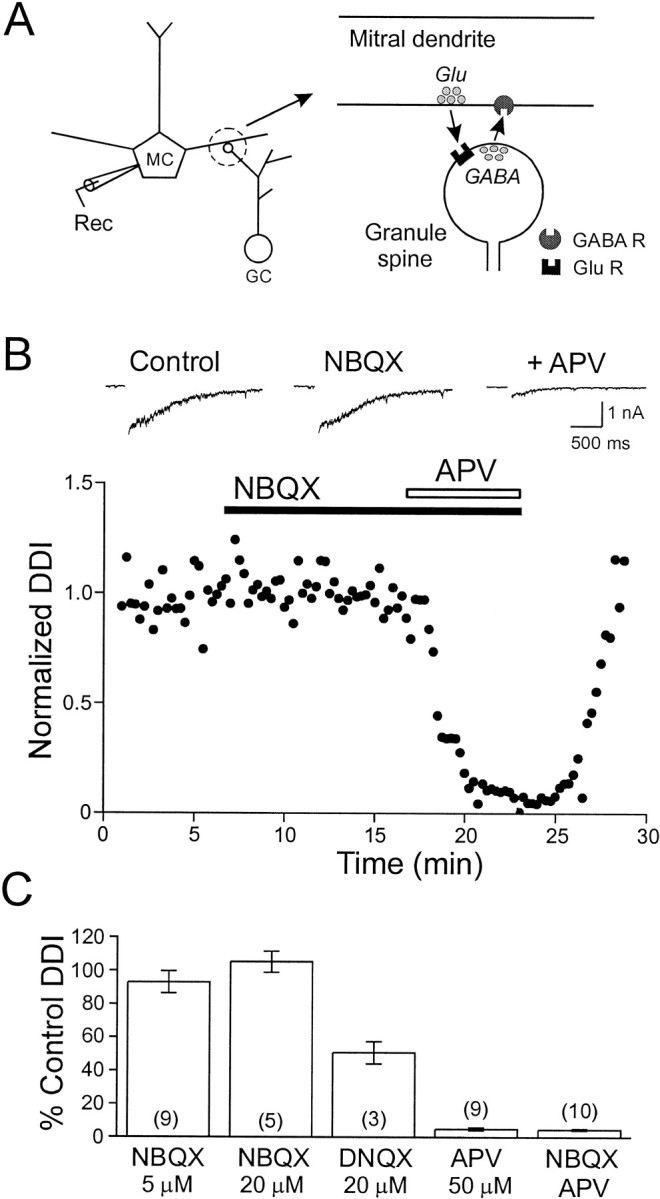
NMDA receptors are required for dendrodendritic inhibition. A, Schematic diagram of the reciprocal synaptic connection between mitral cells and granule cell dendritic spines. B, Bottom, Plot of the reduction in dendrodendritic inhibition by antagonists of NMDA and non-NMDA glutamate receptors. Although the non-NMDA receptor antagonist NBQX (5 μm) did not affect DDI, the subsequent application of d-APV (50 μm) nearly abolished DDI. Top, Sample responses showing control responses (left) and responses in NBQX (middle) and in both NBQX and d-APV (right).C, Summary of the actions of glutamate receptor antagonists. All experiments were performed in Mg-free TTX ACSF using a CsCl-based internal solution. The number of cells tested is shown in parentheses. GC, Granule cell; MC, mitral cell; Rec, recording electrode.
To examine the role of Ca currents through NMDA receptors in triggering dendrodendritic inhibition, we first blocked VDCCs with Cd. We had shown previously (Isaacson and Strowbridge, 1998) that Ca influx through VDCCs in granule cell spines can evoke GABA release. We tested if focal application of NMDA along the secondary dendrites and somata of mitral cells elicits GABAergic IPSCs in mitral cells. In the presence of 100 μm Cd, focal applications of NMDA (0.5 mm; 25–75 msec pressure pulse duration; see Fig.2A) near the cell body region evoked an outward current that persisted for several seconds (Fig. 2B). Most responses to NMDA application in the external plexiform layer (EPL) and mitral cell layer (MCL) were biphasic; an initial IPSC was triggered with very short latency and was followed by a slower, longer-duration IPSC. Both phases were blocked completely by the GABAA receptor antagonist picrotoxin (PTX; 50 μm; Fig.2B–D). Mitral cells were held at +10 mV under voltage clamp, near the reversal potential of glutamate receptors, to minimize currents through NMDA autoreceptors on the mitral cell (Isaacson, 1999; Friedman and Strowbridge, 2000). Synaptic responses to NMDA were not significantly diminished by increasing the concentration of Cd to 200 μm, suggesting that the blockade of VDCCs in granule cells was complete (response integral, 648 ± 218 pA sec in 100 μm Cd and 560 ± 176 pA sec in 200 μm Cd; n = 8;p > 0.05 in paired t test; Fig.2B–D). We also verified that Ca influx through NMDA receptors leads to GABA release in physiological concentrations of Mg by coapplying AMPA and NMDA (both 0.5 mm; 10 msec puff durations) in the EPL in the presence of 1.2 mm Mg. Coapplication of AMPA and NMDA evoked a response in 100 μm Cd that persisted for several seconds (response integral, 1017 ± 384 pA sec;n = 3) and that was abolished with the addition of 50 μm PTX (response integral, −135 ± 20 pA sec; n = 3; Fig. 2E). These findings suggest that the calcium influx through NMDA receptors on dendrites of granule cells can trigger GABA release in the absence of calcium influx through intrinsic voltage-gated calcium channels. Alternatively, it is possible that NMDA receptors are not directly coupled to GABA release but that instead NMDA receptor activation leads to a release of calcium from internal stores in the granule cell, triggering exocytosis. This explanation seems unlikely because depletion of internal stores with thapsigargin (500 nm) had no significant effect on IPSCs evoked by NMDA application near the soma in the presence of 200 μm Cd (115 ± 37% of control DDI response; n = 3; data not shown).
Fig. 2.

Mitral cell IPSCs evoked by Ca influx through NMDA receptors on granule cells. A, Diagram of the experimental setup illustrates the location of the puffer pipette used to activate NMDA receptors on granule cell spines. The pipette was filled with 0.5 mm NMDA and positioned in the EPL, close to the mitral cell body. B, Focal NMDA application in the EPL evokes GABA release in the presence of the Ca channel antagonist Cd. Left, Responses of a mitral cell held at +10 mV to NMDA with 100 μm Cd in the bath (bottomtrace is the average of 5 consecutive responses) are shown. Middle, Increasing the Cd concentration to 200 μm did not reduce the IPSC evoked by NMDA.Right, The response to NMDA was almost completely abolished by the addition of PTX (50 μm).Asterisks indicate onset of focal drug application.C, Plot of NMDA-evoked IPSC versus time in the same mitral cell shown in B is presented.Dashed line indicates steady state level of DDI in 100 μm Cd. D, Summary plot of IPSCs evoked by NMDA in the EPL in 100 μm Cd, 200 μm Cd, and 200 μm Cd + 50 μm PTX is shown. There was no significant difference in the integral of the NMDA-evoked IPSC in 100 and 200 μm Cd. All experiments were performed in Mg-free TTX ACSF using a Cs-methanesulfonate-based internal solution.E, Coapplication of AMPA and NMDA in the EPL evokes GABA release in the presence of Mg (1.2 mm) and Cd (100 μm; left; average of 5 consecutivetraces) that is completely abolished by PTX (50 μm; right).
Although the activation of NMDA receptors on granule cell spines in the EPL leads to a large inhibitory current in mitral cells that is resistant to blockade with Cd, depolarization of granule cell processes outside the EPL and therefore not associated with reciprocal dendrodendritic synapses (Price and Powell, 1970a,b) with either NMDA or KCl always evoked GABA currents that were highly sensitive to Cd. Focal application of NMDA in the granule cell layer (GCL) elicited monophasic outward GABAergic currents in mitral cells (Fig.3A,B) that were markedly slower in onset than were responses to NMDA in the EPL and could be blocked completely by PTX (50 μm; data not shown). Bath application of 100 μm Cd strongly reduced these inhibitory responses (to 14 ± 6% of control;n = 4; Fig. 3F), suggesting that synaptic depolarization of granule cells via NMDA receptors can trigger GABA release through two distinct pathways. When NMDA receptors are activated near sites of reciprocal dendrodendritic synapses with mitral cells (in the EPL or MCL), the Ca influx through these receptors appears to trigger GABA exocytosis directly. However, NMDA application in the GCL, where there are no reciprocal synapses, causes a depolarization of the granule cell that activated VDCCs at GABA release sites in the EPL. These findings suggest that both voltage-gated Ca channels and NMDA receptors are coupled to GABA release from granule cells.
Fig. 3.

Role of Ca channels in granule cells during dendrodendritic inhibition. A, Schematic diagram of the experimental setup is shown. B, Left Column, Responses of a mitral cell held at +10 mV to brief puffs of NMDA under control conditions (bottomtrace is average of 5 responses) are shown. Middle Column, Bath application of Cd (100 μm) nearly abolished the IPSC evoked by focal NMDA application in the GCL. Right Column, The effect of Cd was reversible on washout. C, Diagram of the experimental setup illustrates the location of the puffer pipette filled with KCl (90 mm) in the EPL. D,Left Column, Synaptic responses of a mitral cell to the focal application of KCl under control conditions are shown.MiddleColumns, Bath application of 100 μm Cd completely and reversibly blocked the KCl-evoked IPSC. Right Column, The response to KCl also was blocked by PTX (50 μm). E, Plot of the integral of the KCl-evoked IPSC versus time in the same mitral cell shown inD is presented. Top dashed line indicates basal DDI; bottom dashed line indicates 0 nA sec.F, Summary of the effect of Cd (100 μm) on NMDA-, KCl-, or AMPA + NMDA-evoked IPSCs is shown. All experiments in A–E were performed with Mg-free TTX ACSF using a Cs-methanesulfonate-based internal solution. Experiments with AMPA + NMDA application were performed in TTX ACSF containing 1.2 mm Mg.
Besides blocking VDCCs, this concentration of Cd may also attenuate NMDA responses (Mayer et al., 1989), thereby inhibiting part of the DDI response. We examined this possibility by activating NMDA autoreceptors present on the soma of mitral cells with focal NMDA application in the presence of PTX to block GABAergic inhibition. Cadmium (100 μm) only slightly reduced the EPSC recorded in mitral cells (86.2 ± 10.2% of control; n = 3; data not shown). These results suggest that there is only a minimal reduction of the postsynaptic response to NMDA by the concentration of Cd used to block VDCCs in granule cells.
Although Cd greatly reduced the IPSC evoked by distal NMDA puffs (in the GCL), it is possible that the remaining response was mediated by some Cd-resistant VDCCs. Alternatively, the residual IPSC could be mediated by diffusion of NMDA to the EPL or MCL. To differentiate between these hypotheses, we tested the effect of 100 μmCd on the IPSC evoked by focal application of KCl in the EPL (Fig.3C). Under these conditions, GABA release relies entirely on Ca influx through VDCCs; the KCl-evoked IPSC can be used, therefore, as a sensitive test to determine whether all VDCCs that are coupled to GABA release sites are blocked by 100 μm Cd. Brief KCl puffs evoked a PTX-sensitive response that was rapid in onset and shorter in duration compared with either of the NMDA-evoked responses (Fig. 3D). In three mitral cells, bath application of Cd (100 μm) completely abolished the KCl-evoked IPSC (mean relative response, −33 ± 16% of control;n = 3; Fig. 3D–F), revealing a small inward current deflection that was also observed in PTX. These results demonstrate that GABA can be released from granule cell spines by direct depolarization and activation of VDCCs. Furthermore, VDCCs in granule cells that can trigger GABA release appear to be completely blocked by 100 μm Cd, suggesting that the NMDA-evoked IPSCs we recorded in Cd (Fig. 2B) are elicited by Ca influx through NMDA receptors on granule cell spines.
By using focal depolarization with NMDA and KCl, we have shown that Ca influx through either VDCCs or NMDA receptors can evoke GABA release from granule cells. We next addressed the question of which source of Ca influx is dominant under different physiological conditions by selectively blocking Na or K currents in granule cells. Membrane depolarization in the granule cell spine is likely to activate VDCCs. If GABA release relies primarily on Ca influx through VDCCs, then Na channels may further depolarize granule cells (in addition to the glutamatergic EPSP) and would be expected to enhance GABA release. Blockade of these channels with TTX would be expected to reduce DDI recorded in mitral cells. By contrast, if GABA release normally depends on the Ca influx through NMDA receptors, depolarization provided by nearby Na channels would be expected to reduce the driving force for Ca entry and thereby reduce GABA release. Under these conditions, blockade of Na channels would increase Ca influx through NMDA receptors and enhance DDI.
We examined the effects of blockade of Na channels in granule cells with TTX (1 μm) on the DDI response recorded in mitral cells held at −70 mV and filled with a CsCl-based internal solution containing QX-314 (to block Na channels in the mitral cell; 5 mm). We found that TTX had opposite effects on DDI depending on the amount of glutamate released by the mitral cell. Glutamate release was modulated by altering the duration of the depolarizing voltage step in the mitral cell. Responses to brief steps (2–3 msec; Fig. 4A) were consistently decreased by TTX, with many voltage steps failing to evoke DDI. Even under control conditions, we found that DDI responses evoked by short-duration voltage steps were highly variable. By contrast, TTX enhanced DDI evoked by longer-duration voltage steps (e.g., 25–50 msec; Fig. 4C). Responses to these steps showed less random variability but typically were depressed during stimulus trains, reaching steady-state levels after 10–15 stimuli. Blockade of Na channels in granule cells did not appear to affect this depression (mean change in τ = 8.8 ± 7.5%;n = 8) but rather enhanced the steady-state DDI response. Results from experiments with bath application of TTX are summarized in Figure 4E. We found that TTX significantly decreased DDI responses evoked by voltage steps of 2 or 3 msec duration to 17.9 ± 5.6 and 48.4 ± 2.1% of control, respectively (p < 0.03; n = 9). TTX significantly increased responses to 25 and 50 msec steps to 140 ± 12 and 137 ± 10% of control, respectively (p < 0.03; n = 7 and 9). There was no significant effect of TTX on DDI evoked by 5 msec pulses (90.0 ± 28.0% of control; n = 9). We also confirmed that this concentration of TTX was sufficient to block action potentials in granule cells. When granule cells were recorded under current-clamp conditions, we found that 1 μmTTX completely blocked granule cell action potential firing even after current injection steps that had elicited multiple spikes in control ACSF (n = 2; Fig. 4F).
Fig. 4.

Role of Na channels in granule cells during dendrodendritic inhibition. A, Plot of DDI versus stimulus number before (left) and after (right) bath application of TTX (1 μm). Dendrodendritic IPSCs were recorded in mitral cells and evoked at 0.067 Hz by 3 msec voltage steps from −70 to +10 mV. Short depolarizing pulses often failed to elicit a DDI response in TTX, and the amplitude of successful responses was decreased. Inset, Average successful responses before and after TTX. Calibration: 1 nA, 0.5 sec. B, Schematic diagram of a granule cell spine illustrating the proposed role for Na channels in augmenting the DDI response. AP, Action potential.C, Plot of DDI evoked by longer steps (50 msec) versus stimulus number before (left) and after (right) TTX (different mitral cell from that shown inA). Under control conditions, the DDI response decreases exponentially until a steady-state level is reached (dashedline; τ = 0.038 sec−1). TTX increases the DDI responses to long depolarizing pulses while not significantly affecting the rate of depression during the train (τ = 0.039 sec−1). Inset,Average DDI responses before and after TTX. Calibration: 1 nA, 0.5 sec.D, Schematic diagram illustrating the proposed mechanism by which blockade of Na channels increases the amount of GABA released after NMDA receptor activation. E, Summary plot of the effect of TTX on DDI at different step durations. F,Current-clamp recording of a granule cell response to a brief (100 msec) depolarizing current injection. TTX abolished the action potential firing (right) at a current (60 pA) that evoked multiple action potentials under control conditions (middle). Experiments in A–E were performed in Mg-free ACSF using a CsCl-based internal solution containing QX-314 (5 mm). The experiment shown inF was performed in normal ACSF using a K-methylsulfate-based internal solution.
Although the enhancement of DDI with TTX is likely caused by increased Ca influx through NMDA receptors (attributable to a reduction in the depolarization of the granule cells), DDI may also increase because granule cells become disinhibited. Recordings from mitral cells (Fig.5A) consistently show frequent spontaneous synaptic potentials that are blocked by PTX (50 μm), suggesting that they are mediated by spontaneous spiking in granule cells. We also find that the frequency of spontaneous IPSCs is increased by the bath application of K channel blockers such as Cs (Fig. 5A; 5 mm) and 4-AP (100 μm; data not shown). Because these spontaneous synaptic inputs may modulate DDI, we simultaneously recorded the effect of TTX on DDI and spontaneous synaptic activity (estimated by the current variance in 1 sec epochs) in another population of mitral cells. As shown in Figure 5B, TTX markedly depressed spontaneous synaptic activity (to 23 ± 6% of control; n = 6) at approximately the same time that DDI responses were enhanced (to 140 ± 11% of control; 50 msec voltage-step duration). The two effects of TTX were separable after washout of TTX. Enhanced DDI responses recovered rapidly after washout of TTX, whereas the rate of spontaneous synaptic activity remained depressed for >5 min after DDI responses had returned to control levels. These results suggest that the enhancement of DDI by TTX is not caused by disinhibition of granule cells.
Fig. 5.

Increase in dendrodendritic inhibition with TTX is not caused by a reduction in spontaneous synaptic inputs.A, Bath application of Cs (5 mm) increases the frequency and amplitude of spontaneous IPSCs in voltage-clamped mitral cells. Most spontaneous synaptic inputs are inhibitory and are blocked by PTX (50 μm). Three consecutive sweeps are shown from the same mitral cell held at −70 mV. B, Summary plots of the effects of TTX (1 μm) on dendrodendritic inhibition (top) and spontaneous synaptic activity (current variance; bottom) from six mitral cells are shown. Tetrodotoxin increases dendrodendritic inhibition (to 140 ± 11% of control; n = 6) while reducing spontaneous synaptic activity (current variance decreases to 23 ± 6% of control; n = 6). The two effects of TTX are independent because they recover with different time courses after washout of TTX. Experiments were performed in Mg-free ACSF using a CsCl-based internal solution containing QX-314 (5 mm).
We next examined whether depolarization of granule cells with elevated extracellular K modulates DDI. If DDI responses evoked by long-duration voltage steps depend on Ca influx through NMDA receptors, KCl-induced depolarization should reduce this Ca influx by reducing the driving force for Ca entry. In six mitral cells tested, increasing the extracellular K concentration from 5 to 12 mm consistently decreased the decay time constant to 59.7 ± 5.9% of control. Primarily as a consequence of the shortened response, the integral of the DDI response decreased to 31 ± 4.9% of control (n = 6; Fig.6A). The effects of elevated K were rapidly reversible, suggesting that the modulation of DDI was caused by direct depolarization of granule cell processes. We never observed an initial enhancement of the DDI response after switching to high-K ACSF, as might be expected if a large component of the GABA release was triggered by Ca influx through VDCCs. Instead, these results are consistent with a direct coupling between Ca influx through NMDA receptors and GABA release from granule cell spines.
Fig. 6.
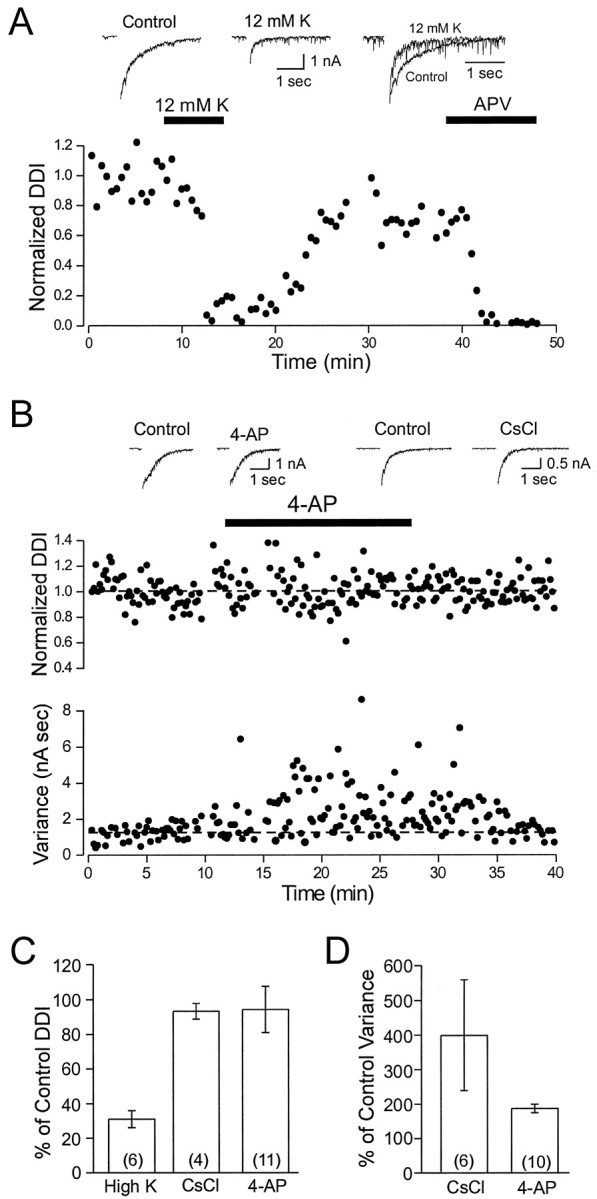
Depolarization of granule cells decreases dendrodendritic inhibition. A, Bottom, Plot of the decrease in DDI (to 10.8% of control) after elevation of extracellular [K+] from 5 to 12 mm is shown. The response in 12 mm[K+]o is reduced nearly to the level seen during subsequent blockade of NMDA receptors with 50 μmd-APV (to 2.4% of control). Top, Left, Middle, Example records show the reduction in peak amplitude and the faster decay rate of the DDI response in elevated [K+]o. Right, Example records are normalized to the peak amplitude. B,Middle, Blockade of K channels by 4-AP (100 μm) does not affect DDI recorded in mitral cells.Bottom, Both 4-AP and Cs (data not shown) increase the spontaneous synaptic activity recorded in mitral cells, quantified by current variance. Dashedlines indicate the control DDI integral and current variance. Top, Example records show typical DDI responses under control conditions and in 4-AP and Cs (5 mm). C, Summary of the effects of increasing [K+]o and blocking K channels on the dendrodendritic response in mitral cells is shown. D, Summary of the effects of K channel blockade on the current variance recorded in mitral cells is shown. Experiments were performed in Mg-free TTX ACSF using a CsCl-based internal solution.
In another test of the NMDA Ca influx model, we examined whether blockade of K channels in granule cells modulated DDI. If GABA release depends on Ca influx through VDCCs, then increasing the excitability of granule cells with 4-aminopyridine (4-AP) or Cs would be expected to enhance DDI. We found, however, that moderate concentrations of 4-AP (100 μm) or CsCl (5 mm) had little effect on DDI evoked by long-duration voltage steps (94.3 ± 13.2%;n = 11; 93.3 ± 4.5; n = 4, respectively; 50 msec voltage steps; Fig. 6B,C). These agents consistently increased spontaneous PTX-sensitive synaptic inputs recorded in mitral cells, suggesting that they were effective in reducing K currents in granule cells. Current variance increased to 187 ± 12.4% of control with 4-AP (n = 10) and to 399 ± 160.0% of control with Cs (n = 6; Fig. 6B,D). Results from experiments with K channel blockers and elevated extracellular K are summarized in Figure 6,C and D. Both the strong inhibition of DDI by high K and the lack of modulation of DDI by 4-AP and Cs are consistent with VDCC-independent GABA release triggered by Ca influx through NMDA receptors on granule cell spines.
Finally, we examined whether substituting NMG, a nonpermeant monovalent cation, for most of the Na normally present in the ACSF would enhance DDI. This manipulation represents a strong test of the hypothesized dependence of GABA release on Ca influx through NMDA receptors. This model predicts that NMG substitution will enhance Ca influx by reducing the postsynaptic depolarization of granule cells during DDI. If GABA release relies instead on activation of VDCCs, the reduction of EPSPs in the granule cell should cause a decrease in DDI. We found that low-Na ACSF consistently increased DDI evoked by long-duration voltage steps (from 0.73 ± 0.17 to 1.32 ± 0.12 nA sec; n = 11; Fig.7A,D). As illustrated in Figure 7A, the enhancement of the DDI integral in low-Na ACSF was caused primarily by a prolongation in the response (DDI decay time constant increased to 183.4 ± 17.8% of control;n = 11). Although the holding current did not change when DDI response increased, we often observed a dramatic increase in holding current after prolonged exposure to low-Na ACSF, presumably reflecting secondary nonspecific effects of the altered extracellular solution. These periods of increased holding current were always accompanied by a dramatic reduction in DDI; the time axis is interrupted, and DDI responses are blanked during this period in Figure 7. Both the initial, presumably direct, effect of low-Na ACSF on DDI and the subsequent increase in holding current were always reversible after switching to normal ACSF (see Fig.7A).
Fig. 7.

Increasing the driving force for Ca through NMDA receptor channels enhances GABA release. A,Middle, Plot of the enhancement of dendrodendritic inhibition after replacement of 124 mm Na with equimolar NMG. Top, Sample records showing dendrodendritic responses before (Control) and after (NMG) switching to low-Na ACSF. Bottom, NMG did not change the holding current during the time that DDI is elevated. The effect of NMG was reversible after washout of NMG. However, after a period of enhanced DDI responses, NMG often caused a pronounced reduction in DDI and a concomitant increase in holding current (indicated by the brokentimeaxis; data not shown). B, Plot of the decrease in granule cell action potential amplitude during NMG substitution. Inset, Representative action potentials before and after NMG substitution. Action potential amplitudes are reduced by NMG substitution at the time that DDI responses were enhanced in the experiment shown in A. C,Bottom, Plot of the elevation of DDI responses by NMG substitution after partial blockade of NMDA receptors on granule cells with 10 μmd-APV. Top, Sample control records, responses in d-APV, and responses after NMG substitution in d-APV (+NMG).D, Summary of the results with NMG substitution. Note the greater enhancement of DDI by NMG after partial blockade of NMDA receptors with d-APV. Experiments in A andC were performed in Mg-free TTX ACSF using a CsCl-based internal solution. The experiment in B was performed in normal ACSF using a K-methylsulfate-based internal solution.
We performed several control experiments with low-Na ACSF. We first determined the time course of the reduction in Na gradient after switching to low-Na ACSF. We recorded action potentials evoked in granule cells by direct current injection (Fig. 7B) in control and low-Na ACSF. The amplitude of these action potentials decreased at approximately the same time that DDI responses were increased (Fig. 7, compare A, B), suggesting that the enhancement of DDI was caused by a reduction in the postsynaptic depolarization of granule cells. Finally, we determined whether NMG substitution also enhanced DDI responses after partial blockade of NMDA receptors. A more prominent role for Ca influx through VDCCs could be masked if the EPSP maximally activated VDCCs in granule cell spines. By using a competitive receptor antagonist to reduce the postsynaptic depolarization, activation of VDCCs should be reduced, and any involvement of Ca influx through VDCCs should be revealed as a decrease in DDI with NMG substitution. We found, however, that even in the presence of 10 μmd-APV, NMG still enhanced DDI responses to long-duration voltage steps (490 ± 142% of DDI response in d-APV,n = 4; Fig. 7C). Results from experiments with NMG substitution are summarized in Figure 7D and are consistent with a direct coupling between Ca influx through NMDA receptors and GABA release from granule cell spines.
DISCUSSION
We find that Ca influx through two distinct pathways can trigger GABA exocytosis from dendrites of olfactory granule cells. In accordance with our previous findings, we show that GABA release can be triggered by Ca influx through VDCCs, mostly likely high-threshold P/Q channels (Isaacson and Strowbridge, 1998). We now report that Ca influx through NMDA receptors on the spines of granule cells can evoke GABA release. This pathway is effective even after blockade of all VDCCs coupled to GABA release by Cd. By selectively altering the postsynaptic depolarization produced by NMDA receptor activation of granule cells, we provide evidence that most of the GABA release evoked by brief EPSPs is caused by Ca influx through VDCCs. However, longer-duration EPSPs evoke additional GABA release, primarily via direct actions of NMDA receptor Ca influx on exocytosis. These data suggest a new role of Ca-permeable ligand-gated channels in directly evoking neurotransmitter release. This pathway also provides a new, potentially important site of synaptic plasticity in the olfactory bulb.
GABA release evoked by Ca influx through NMDA receptors and VDCCs
Dendritic spines of olfactory granule cells function both as postsynaptic elements, with receptors activated by glutamate released from nearby dendritic and somatic processes of mitral cells (Wellis and Kauer, 1993; Isaacson and Strowbridge, 1998; Schoppa et al., 1998), and as presynaptic terminals capable of releasing GABA in a Ca-dependent manner (Isaacson and Strowbridge, 1998). Although granule cells express NMDA and non-NMDA glutamate receptors (Isaacson and Strowbridge 1998;Schoppa et al., 1998; Sassoè-Pognetto and Otterson, 2000), GABA release appears to depend primarily on the activation of postsynaptic NMDA receptors. Electron microscopic studies reveal relatively close spacing between the postsynaptic active zone in granule cell spines, presumably containing most of the postsynaptic glutamate receptors, and sites of docked vesicles containing GABA (Price and Powell, 1970b; Sassoè-Pognetto and Otterson, 2000). The relatively close proximity between presynaptic and postsynaptic sites within the spine raises the possibility that the critical requirement for activation of the NMDA receptor in DDI reflects a direct role of Ca entering through NMDA receptors in triggering GABA exocytosis. Although in some synapses, including the squid giant synapse, VDCCs are thought to be intimatelyassociated with proteins that control exocytosis (Adler et al., 1991), there is evidence from other synapses that transmitter can be released as a consequence of Ca influx from relatively distant sites, potentially including Ca-permeable receptors. For example, the inhibition of glutamate release in the auditory brainstem by the relatively slow-acting Ca chelator EGTA (Borst and Sakmann, 1996) has been interpreted as evidence of distant coupling between sites of Ca entry and vesicle-docking sites. Although we have demonstrated previously (Isaacson and Strowbridge, 1998) that Ca entry through VDCCs can evoke GABA release from granule cells depolarized with KCl, it is unclear from previous studies whether endogenous glutamate released from mitral cells elicits GABA release by activating VDCCs and what role the Ca entering through NMDA receptors plays in exocytosis.
The large Cd-insensitive IPSCs evoked by NMDA observed in this study strongly suggest that Ca entry through postsynaptic NMDA receptors can trigger GABA release from granule cell spines. However, this interpretation depends on the ability of Cd to block all VDCCs that could support GABA release from granule cells. To address this issue we showed that Cd dramatically reduced IPSCs evoked by NMDA application to distal sites on the granule cell. In addition, we showed that Cd completely and reversibly blocked DDI evoked by focal depolarization of spines with KCl. Together, these experiments support a model in which GABA release from granule cell spines can be controlled directly by Ca influx through NMDA receptors. We also observed very different time courses in the responses to NMDA and KCl. Direct activation of NMDA receptors on the granule cell spines evoked biphasic IPSCs. The rapid onset and short duration of the early phase of this response may reflect release from a small, readily releasable pool of vesicles located close to the NMDA receptors. The prolonged second phase may reflect the release of more distant vesicles located closer to VDCCs as well as the repopulation of the proximal release sites. Direct depolarization of the spine evoked an IPSC with very rapid onset kinetics, consistent with relatively close coupling between VDCCs. Although the inability of thapsigargin to modify the biphasic time course of responses to NMDA in the EPL suggests that release of Ca from internal stores does not directly trigger GABA exocytosis, additional experiments will be required to address this issue completely.
Opposite effects of Na channel blockade on dendrodendritic inhibition
Can endogenous glutamate released by mitral cells also evoke GABA release using similar mechanisms? Although this question is difficult to answer directly, we obtained several lines of evidence that suggest endogenous glutamate can trigger recurrent IPSCs via both VDCC- and NMDA receptor-dependent mechanisms. The high degree of response variability when relatively small amounts of glutamate are released is consistent with amplification of the glutamatergic EPSP by intrinsic Na channels in the granule cell. Presumably the glutamate EPSP triggers a variable number of action potentials, thus enhancing the variability normally associated with transmitter release. These granule action potentials, in turn, would provide a larger stimulus for activating VDCCs coupled to GABA release. This model explains why blocking granule cell Na channels with TTX reduces DDI evoked by short-duration voltage steps. Blockade of persistent Na current in granule cells also could contribute to the reduction in DDI. By contrast, when larger amounts of glutamate are released (by using longer-duration voltage steps), TTX has the opposite effect, and DDI responses are enhanced. In addition, control responses evoked by long-duration steps show much less variability than the TTX-sensitive responses elicited by short-duration steps. One potential explanation for the decreased variability is that the larger control EPSP evoked now fully activates VDCCs in granule cells, reducing the variability associated with spike generation. Alternatively, the Ca influx through NMDA receptors activated during long-duration EPSPs causes additional GABA secretion. In both models, activation of Na channels in granule cells functions to decrease Ca entry (and DDI) by reducing the driving force for Ca. A pronounced depression of DDI responses also is apparent after trains of long-duration voltage steps that are not affected by Na channel blockade. Although we did not explore the mechanism underlying this effect, it is possible that the larger concentration of glutamate released by mitral cells activates metabotropic receptors on the mitral or granule cells that function to inhibit DDI. The lack of correlation we found between the time course of enhancement of DDI by TTX and the reduction in spontaneous synaptic activity suggests that the effects of TTX are not caused by disinhibition of granule cells.
Modulation of GABA release by altering the driving force for Ca entry
The results using elevated-K ACSF to depolarize granule cells suggest that GABA release from granule cell spines is limited by the Ca driving force. A dependence on driving force would be expected if Ca entry through NMDA receptors governs GABA release. Postsynaptic depolarization would not be expected to alter the conductance underlying the glutamatergic EPSP—only the inward synaptic current, mediated in part by Ca (Mayer and Westbrook, 1987; Iino et al., 1990). Alternatively, the depolarization provided by the control EPSP may fully activate VDCCs coupled to GABA release; additional depolarization (because of the opening of intrinsic Na channels) would then function to reduce Ca influx through VDCCs. We found that NMG substitution consistently increased DDI in control conditions as well as after the reduction of granule cell EPSPs with 10 μmd-APV. This consistent enhancement of DDI strongly suggests that granule cell depolarization can inhibit GABA release by reducing Ca influx through NMDA receptors. These experiments are complicated by secondary effects of the low-Na ACSF that appear to affect both mitral and granule cell intrinsic properties. We do show, however, that the time course of DDI enhancement is well matched by an indicator of the Na gradient in granule cells (action potential amplitude).
We also found no effect of two K channel blockers on DDI. Such an effect might be expected if GABA release strongly depended on VDCC activation because K channels are likely to regulate granule cell excitability. Modulation of DDI responses by high concentrations of 4-AP was reported recently by Schoppa and Westbrook (1999). The apparent absence of DDI modulation by Cs and 4-AP in our study is consistent with a dominant role for NMDA receptors as the source of the Ca influx triggering GABA release evoked by prolonged granule cell EPSPs.
Our findings support the hypothesis that Ca influx through both NMDA receptors and VDCCs can evoke GABA release from granule cell spines, with VDCCs playing a preferential role when GABA release is triggered by small-amplitude EPSPs and Ca influx through NMDA receptors governing release in response to large EPSPs. The ability of Ca influx through NMDA receptors to evoke transmitter release may account for the critical role of NMDA receptor activation in DDI reported in this and previous studies (Isaacson and Strowbridge, 1998; Schoppa et al., 1998). On the basis of our results, it seems that activation of non-NMDA receptors evokes GABA release exclusively via the VDCC-dependent pathway, whereas NMDA receptor activation seems to trigger GABA release both by activating VDCCs and via direct action of Ca influx through the receptor itself. Although in these experiments glutamate release from the mitral cell was controlled by voltage-clamp command steps, our results suggest that Ca influx through NMDA receptors may govern DDI evoked by high-frequency trains of back-propagating action potentials in mitral cell secondary dendrites. By contrast, we would predict that the feedback inhibition triggered by unitary back-propagating action potentials relies on VDCCs (and perhaps on Na channels) in granule cell spines. The dual role of NMDA receptors on granule cells, both to depolarize the cell and to act as a source of Ca capable of triggering GABA exocytosis, may also lead to new mechanisms for plasticity in the DDI response. Depolarization of granule cells, because of remote EPSPs, for example, may function to shift the mechanisms triggering GABA release and alter the time course of recurrent inhibition.
Our findings provide evidence for direct coupling between Ca entry through presynaptic ligand-gated receptors and evoked transmitter release. This function for NMDA receptors is consistent with the relatively close spacing between presynaptic and postsynaptic sites in granule cell spines in the olfactory bulb (Price and Powell, 1970b;Sassoè-Pognetto and Otterson, 2000). The additional Ca influx provided by NMDA receptors appears to enhance both the amplitude and reliability of reciprocal dendrodendritic inhibition. The dual role of NMDA receptors that have been found in the olfactory bulb may also occur elsewhere in the CNS with other types of Ca-permeable presynaptic receptors. For example, activation of nicotinic ACh receptors leads to Ca accumulation in axon terminals in the spinal cord (McGehee et al., 1995) and increases the frequency of Cd-resistant spontaneous miniature synaptic potentials in both the hippocampus (Gray et al., 1996) and thalamus (Léna and Changeux, 1997).
Footnotes
This study was supported by National Institutes of Health Grant NS 33590. Dr. Strowbridge is a Mount Sinai Health Care Foundation Scholar. We thank Drs. R. Traub and D. Kunze for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Ben W. Strowbridge, Department of Neurosciences, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106-4975. E-mail: bxs48@po.cwru.edu.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol (Lond) 1985;369:163–181. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borst JGG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- 4.Friedman D, Strowbridge BW. Functional role of NMDA autoreceptors in olfactory mitral cells. J Neurophysiol. 2000;84:39–50. doi: 10.1152/jn.2000.84.1.39. [DOI] [PubMed] [Google Scholar]
- 5.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 6.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. J Physiol (Lond) 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 8.Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 9.Jahr CE, Nicoll RA. Dendrodendritic inhibition: demonstration with intracellular recording. Science. 1980;207:1473–1475. doi: 10.1126/science.7361098. [DOI] [PubMed] [Google Scholar]
- 10.Jahr CE, Nicoll RA. An intracellular analysis of dendrodendritic inhibition in the turtle in vitro olfactory bulb. J Physiol (Lond) 1982;326:213–234. doi: 10.1113/jphysiol.1982.sp014187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katz B, Miledi R. A study of synaptic transmission in the absence of nerve impulses. J Physiol (Lond) 1967;192:407–436. doi: 10.1113/jphysiol.1967.sp008307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Léna C, Changeux J-P. Role of Ca2+ ion in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol (Lond) 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayer ML, Vylicky L, Jr, Westbrook GL. Modulation of excitatory amino acid receptors by group IIB metal cations in cultured mouse hippocampal neurones. J Physiol (Lond) 1989;415:329–350. doi: 10.1113/jphysiol.1989.sp017724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGehee DS, Heath MJS, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 16.Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 17.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 18.Nowycky MC, Mori K, Shepherd GM. GABAergic mechanisms of dendrodendritic synapses in isolated turtle olfactory bulb. J Neurophysiol. 1981a;46:639–648. doi: 10.1152/jn.1981.46.3.639. [DOI] [PubMed] [Google Scholar]
- 19.Nowycky MC, Mori K, Shepherd GM. Blockade of synaptic inhibition reveals long-lasting synaptic excitation in isolated turtle olfactory bulb. J Neurophysiol. 1981b;46:649–658. doi: 10.1152/jn.1981.46.3.649. [DOI] [PubMed] [Google Scholar]
- 20.Price JL, Powell TPS. The morphology of the granule cells of the olfactory bulb. J Cell Sci. 1970a;7:91–123. doi: 10.1242/jcs.7.1.91. [DOI] [PubMed] [Google Scholar]
- 21.Price JL, Powell TPS. The synaptology of granule cells in the olfactory bulb. J Cell Sci. 1970b;7:125–155. doi: 10.1242/jcs.7.1.125. [DOI] [PubMed] [Google Scholar]
- 22.Price JL, Powell TPS. The mitral and short axon cells of the olfactory bulb. J Cell Sci. 1970c;7:631–651. doi: 10.1242/jcs.7.3.631. [DOI] [PubMed] [Google Scholar]
- 23.Rall W, Shepherd GM, Reese TS, Brightman MW. Dendrodendritic synaptic pathway for inhibition in the olfactory bulb. Exp Neurol. 1966;14:44–56. doi: 10.1016/0014-4886(66)90023-9. [DOI] [PubMed] [Google Scholar]
- 24.Randle JC, Guet T, Cordi A, Lepagnol JM. Competitive inhibition by NBQX of kainate/AMPA receptor currents and excitatory synaptic potentials: importance of 6-nitro substitution. Eur J Pharmacol. 1992;215:237–244. doi: 10.1016/0014-2999(92)90033-z. [DOI] [PubMed] [Google Scholar]
- 25.Rathouz MM, Berg DK. Synaptic-type acetylcholine receptors raise intracellular calcium levels in neurons by two mechanisms. J Neurosci. 1994;14:6935–6945. doi: 10.1523/JNEUROSCI.14-11-06935.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sassoè-Pognetto M, Otterson OP. Organization of ionotropic glutamate receptors at dendrodendritic synapses in the rat olfactory bulb. J Neurosci. 2000;20:2192–2201. doi: 10.1523/JNEUROSCI.20-06-02192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- 28.Schoppa NE, Kinzie JM, Shara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vernino S, Amador M, Luetje CW, Patrick J, Dani JA. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- 30.Wellis DP, Kauer JS. GABAa and glutamate receptor involvement in dendrodendritic synaptic interactions from salamander olfactory bulb. J Physiol (Lond) 1993;469:315–339. doi: 10.1113/jphysiol.1993.sp019816. [DOI] [PMC free article] [PubMed] [Google Scholar]