

Abstract
Because of discrepancies in previous reports regarding the role of glial cell line-derived neurotrophic factor (GDNF) in motoneuron (MN) development and survival, we have reexamined MNs in GDNF-deficient mice and in mice exposed to increased GDNF after in uterotreatment or in transgenic animals overexpressing GDNF under the control of the muscle-specific promoter myogenin (myo-GDNF). With the exception of oculomotor and abducens MNs, the survival of all other populations of spinal and cranial MNs were reduced in GDNF-deficient embryos and increased in myo-GDNF and in utero treated animals. By contrast, the survival of spinal sensory neurons in the dorsal root ganglion and spinal interneurons were not affected by any of the perturbations of GDNF availability.
In wild-type control embryos, all brachial and lumbar MNs appear to express the GDNF receptors c-ret and GFRα1 and the MN markers ChAT, islet-1, and islet-2, whereas only a small subset express GFRα2. GDNF-dependent MNs that are lost in GDNF-deficient animals express ret/GFRα1/islet-1, whereas many surviving GDNF-independent MNs express ret/GFRα1/GFRα2 and islet-1/islet-2. This indicates that many GDNF-independent MNs are characterized by the presence of GFRα2/islet-2. It seems likely that the GDNF-independent population represent MNs that require other GDNF family members (neurturin, persephin, artemin) for their survival. GDNF-dependent and -independent MNs may reflect subtypes with distinct synaptic targets and afferent inputs.
Keywords: motoneurons, cell death, GDNF, spinal cord, embryo, mouse GDNF receptors, knock-out, transgenic
Glial cell line-derived neurotrophic factor (GDNF) was the first member of the GDNF family of neurotrophic factors to be identified and was originally isolated based on its ability to promote the survival and differentiation of dopaminergic neurons in primary cultures from embryonic ventral midbrain (Lin et al., 1993). Since the initial discovery of GDNF, several other family members with neurotrophic actions have been reported, and receptors for each of these have also now been described (Rosenthal, 1999). The GDNF family of receptors are composed of a complex involving the transmembrane Ret receptor tyrosine kinase and one or more of at least four glycosyl phosphatidyl-inositol (GPI) membrane-anchored ligand-binding components, GFRα1– GFRα4. The four GDNF family ligands all use Ret but have their own preferred co-receptors (GDNF/α1; neurturin/α2; artemin/α3; persephin/α4), although some members may also interact with nonpreferred co-receptors (e.g., GDNF/α2 and neurturin/α1), albeit with lower efficiency.
One neuronal population that is responsive to some of the GDNF family members, including GDNF, are somatic motoneurons (MNs). GDNF and neurturin are expressed in skeletal muscle and Schwann cells of developing and adult mammals (Henderson et al., 1994;Suter-Crazzolara and Unsicker, 1994; Choi-Lundberg and Bohn, 1995; Springer et al., 1995; Sánchez et al., 1996; Wright and Snider, 1996; Nguyen et al., 1998a; Suzuki et al., 1998; Golden et al., 1999), and Ret and GFRα1 are expressed in all populations of spinal and cranial MNs that have been examined so far (Pachnis et al., 1993;Nakamura et al., 1996; Jing et al., 1996; Treanor et al., 1996;Bisgrove et al., 1997; Trupp et al., 1997, 1998; Glazner et al., 1998; Golden et al., 1998; Yu et al., 1998).
These data indicate that GDNF is expressed in cells that closely interact with developing MNs (skeletal muscle, Schwann cells) and that receptors for GDNF are expressed on developing spinal and cranial MNs. GDNF family members can also promote the survival of MNs in vitro and in vivo, and mouse mutants deficient in GDNF or GFRα1 are reported to have reduced MN numbers (Henderson et al., 1994; Soler et al., 1999; Oppenheim et al., 1995; Li et al., 1995; Yan et al., 1995; Moore et al., 1996; Sánchez et al., 1996; Cacalano et al., 1998). Although these various lines of evidence are consistent with the role of GDNF as a neurotrophic survival factor for subpopulations of somatic MNs, several nagging questions remain. For example, muscle-specific overexpression of GDNF in transgenic mice during embryonic development is reported to result in normal MN numbers in neonatal mice (Nguyen et al., 1998b), and there are even discrepancies in the literature as to whether there is any MN loss at all in either GFRα1-deficient mice (cf. Enomoto et al., 1998 withCacalano et al., 1998) or in GDNF-deficient mice (cf. Moore et al., 1996 and Sánchez et al., 1996 with Pichel et al., 1996). It is also not clear whether the apparent loss of MNs in GDNF- or GFRα1-deficient mice involves GDNF regulation of survival versus proliferation, migration, or differentiation of MNs. For these various reasons, we have undertaken a detailed analysis of the role of GDNF in the development of mouse MNs.
MATERIALS AND METHODS
GDNF-deficient embryos. The GDNF−/− mutants were derived from natural or timed mating of GDNF+/− × GDNF+/− mice (from an R29 ES clone on a CD1 background.) The age of the embryos was determined by the presence of vaginal plug in the pregnant mothers and indicated as embryonic day 0.5. Embryos were dissected and immediately immersed in either Bouin's fixative for a few days (for neuronal counting and pyknotic analysis) or 4% paraformaldehyde in 1× PBS for 8–16 hr at 4°C and cryopreserved in 30% sucrose in PBS and sectioned by cryostat at 15 μm. Some of the embryos were freshly frozen on dry ice powder and sectioned by cryostat at 15 μm. All embryos were genotyped by PCR with the following primer sets. To detect the GDNF knock-out allele of a 255 bp PCR product, the upper and lower primers used were 5′-CGGAGCCGGTTGGCGCTACCGG and 5′-ACGACTCGGACCGCCATCGGTG. To detect the GDNF wild-type allele of 337 bp PCR product, we used the following primer set: 5′-GAGAGGAATCGGCAGGCTGCAGCTG and 5′-CAGATACATCCACATCGTTTAGCGG.
Myo-GDNF transgenic mice. To examine the effect of increased target-derived GDNF (and NT-3 or NT-4) on MN survival, we used embryonic and neonatal animals that overexpress these factors under a muscle-specific myogenin promoter that drives transgene expression in mice embryos beginning before the onset of either muscle innervation or the programmed cell death (PCD) of MNs and which continues to be expressed postnatally (Nguyen et al., 1998b). Embryos and neonates from the myo-GDNF transgenic lines examined here express high amounts of GDNF in muscle (Nguyen et al., 1998b). Brain and spinal cord from transgenic and wild-type mice were immersion-fixed in Carnoy's or Bouin's solution, paraffin-embedded, sectioned serially (12 μm), and stained with either thionin or hematoxylin/eosin. Cell counts of healthy surviving and dying pyknotic MNs were performed as described below.
In utero treatment with GDNF. BALB/c ByJ mice (The Jackson Laboratory, Bar Harbor, ME) were bred in Wake Forest Medical School animal facility. On gestation day 14.5 (the morning a vaginal plug was observed is designated E0.5), pregnant females were anesthetized with ether, and partial laparotomy was performed under sterile conditions. One uterus (3–5 embryos) was exposed, and each embryo was injected with 5 μg of GDNF in 5 or 10 μl of saline (0.9% NaCl, pH 7.2) using a modified 10 μl gauge Hamilton microsyringe, as described previously (Houenou et al., 1990, 1994). Injections were made into the amniotic fluid, embryos were replaced in the uterus, and the mother was allowed to recover after the abdomen was sutured. Fetuses from the contralateral uterus were used as controls (saline only). On E18.5, mice were killed with an overdose of ether, and fetuses were collected by cesarean section. Spinal cords from control and trophic factor- or saline-treated embryos were dissected out, fixed in Bouin's solution, and processed as described above.
Neuronal counts. Motoneurons were counted in every fifth or tenth section through each population examined, and the totals were multiplied by five or ten to give an estimate of total cell numbers. Cell counts were done blind as to the treatment condition of the embryo/neonate (control vs experimental) using a well established counting method that effectively eliminates the possibility of counting the same cell (healthy or pyknotic) twice (Clarke and Oppenheim, 1995). Data for pyknotic MNs are expressed as per 1000 surviving MNs.
For assessing sensory neurons, cells in every fifth section of the fourth or fifth lumbar (L4–L5) dorsal root ganglion (DRG) were counted. Spinal interneurons were counted in every 20th section through the entire lumbar enlargement. One lateral half (hemisection) of the spinal cord was used for counts of interneurons, and neurons located in the dorsal horn and ventral horn were excluded from these counts. Cranial MNs in the following motor nuclei were also assessed quantitatively: oculomotor (III), trigeminal (V), abducens (VI), facial (VII), spinal accessory (XI), and hypoglossal (XII). Although a major focus in the spinal cord was on lumbar and brachial MNs, thoracic MNs were also counted in some of the embryos/neonates. Because the rostrocaudal boundaries of the XI motor nucleus was difficult to define precisely, MN counts were confined to the first 1 or 2 cervical segments (C1–C2), and MN numbers were expressed as the number per section for both XI MNs and cervical spinal MNs.
In situ hybridization.α35S-UTP-labeled riboprobes complementary to c-ret, GFRα1, GFRα2, isl-1, isl-2, and ChAT were synthesized according to the supplier's protocol (Stratagene, La Jolla, CA) and used for in situ hybridization as described previously (McMahon et al., 1992). A 0.627 kb antisense riboprobe for a mouse c-ret cDNA at the 3′ untranslated region (3′-UTR) was derived from pMCRET7 cDNA linearized at the Nsi I position (Pachnis et al., 1993). Antisense riboprobe to mouse GFRα1 was complementary to position 720–1415 (GenBank accession number AB000800). Two independent riboprobes were used for detection of GFRα2 transcripts: a 633 bp fragment complementary to rat GFRα2 at amino acid positions 217–247 (Wang, 1998) or a 190 bp mouse GFRα2 riboprobe corresponding to nucleotide position 1–190 (GenBank accession number AF002701). Probes complementary to full-length mouse isl-1 (1.1 kb), rat isl-2 (1.6 kb), and rat ChAT (2.2 kb) cDNAs were obtained from Drs. Sam Pfaff and Terry Rabbitts. In situ hybridization studies were performed on cryosections (transverse or sagittal) of E15.5 and E18.5 embryos. For quantitative analysis of the in situhybridization, adjacent transverse sections on a single slide were hybridized separately with two different riboprobes, and slides of GDNF+/+ and −/− sections were processed simultaneously under identical conditions (e.g., the same stringency of wash and exposure time on emulsion, etc.). Images were imported into the computer directly using a CG-7 frame grabber to avoid distortion of signal intensity using the same exposure for all the sections quantified (Scion Corporation). A stage micrometer was used to calibrate the scale before the data analysis of images. The values for in situsignals are expressed as total pixels (area) in the selected region (ventral horn) and as the average density of pixels in the same region and were quantified using NIH Image 1.60 based on signals from at least five images from each of two or three embryos per region per age. The data are presented as mean pixels or density ± SD. Statistical significance (p value) was calculated by two-tailed Student's t test.
RESULTS
Spinal and cranial MNs are reduced in GDNF-deficient mice
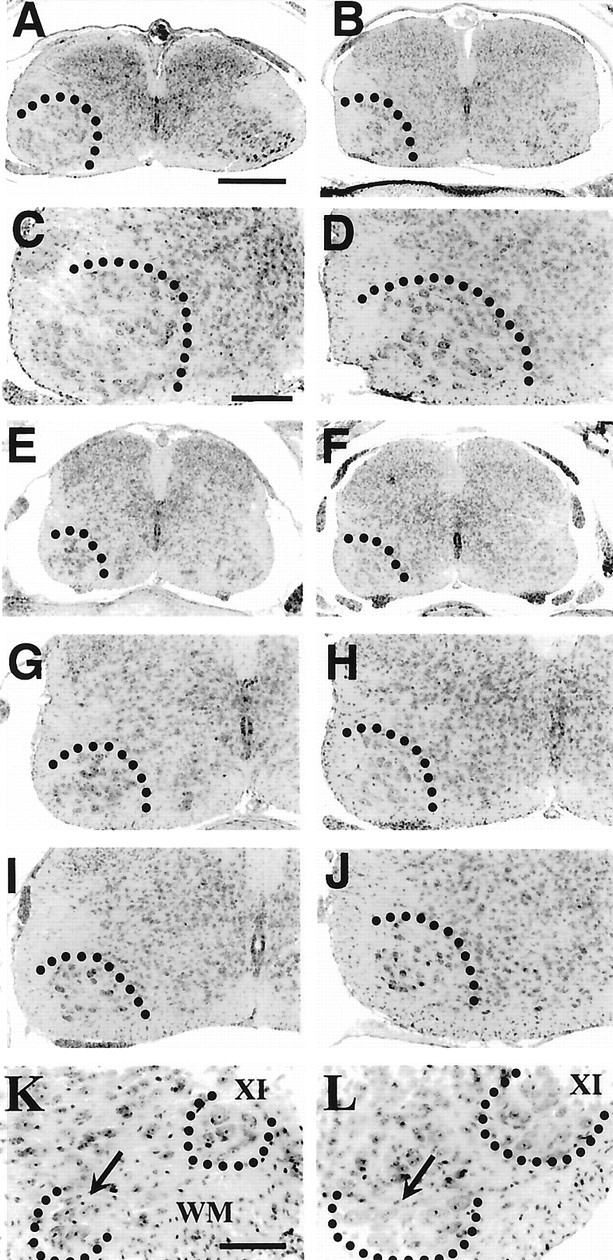
Cell counts of spinal MNs on E18.5 revealed a significant loss in homozygous GDNF-deficient embryos compared to either wild-type controls or heterozygotes (Figs. 1,2A). Brachial, thoracic, and lumbar MNs were reduced by 34, 22, and 37%, respectively. A detailed developmental analysis of lumbar MNs between E12.5 and E18.5 indicates that initially GDNF-deficient embryos have MN numbers comparable to controls but that during the major period of normal cell death (E13.5–E18.5), they lose more MNs than controls, and this loss is associated with increased numbers of degenerating (pyknotic) MNs (Table 1). Although not examined in as much detail, the number of brachial MNs were also comparable in wild-type and GDNF-deficient embryos on E12.5, whereas by E15.5 and E18.5 the GDNF-deficient embryos had 24 and 34% fewer MNs, respectively (data not shown). The number of sensory neurons in the DRG and interneurons in the spinal cord on E18.5 were unaffected in the GDNF-deficient embryos (Fig. 2B).
Fig. 1.
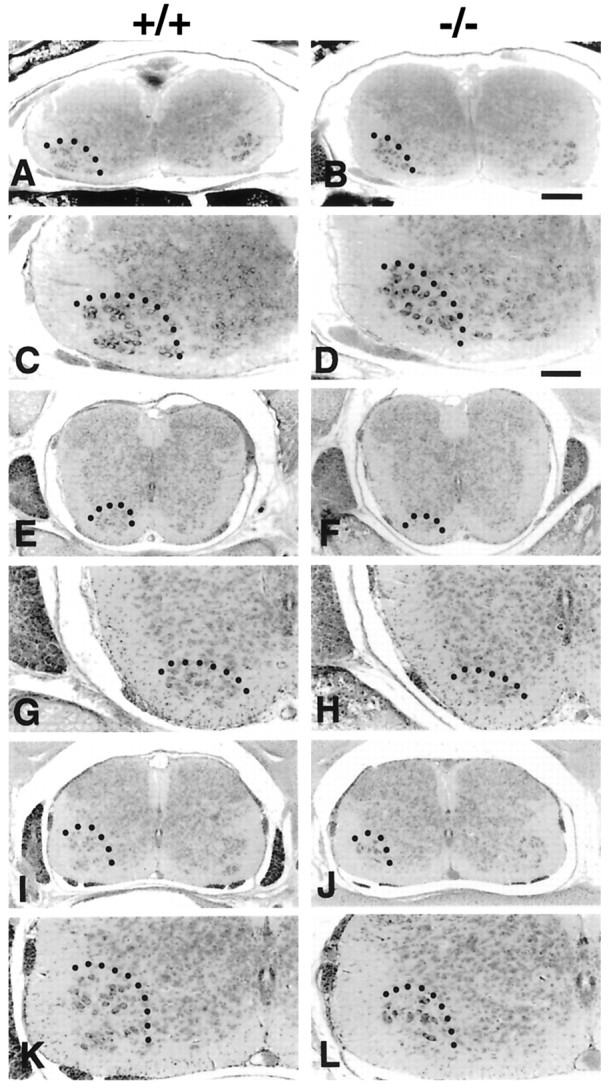
Transverse sections of E18.5 spinal cord of GDNF-deficient (−/−) and wild-type (+/+) control embryos.A–D, Brachial; E–H, thoracic;I–L, lumbar. Dotted lines delineate the ventral horn. Scale bars: B (applies to A, B, E, F, I, J), 200 μm; D (applies toC, D, G, H, K, L), 100 μm.
Fig. 2.
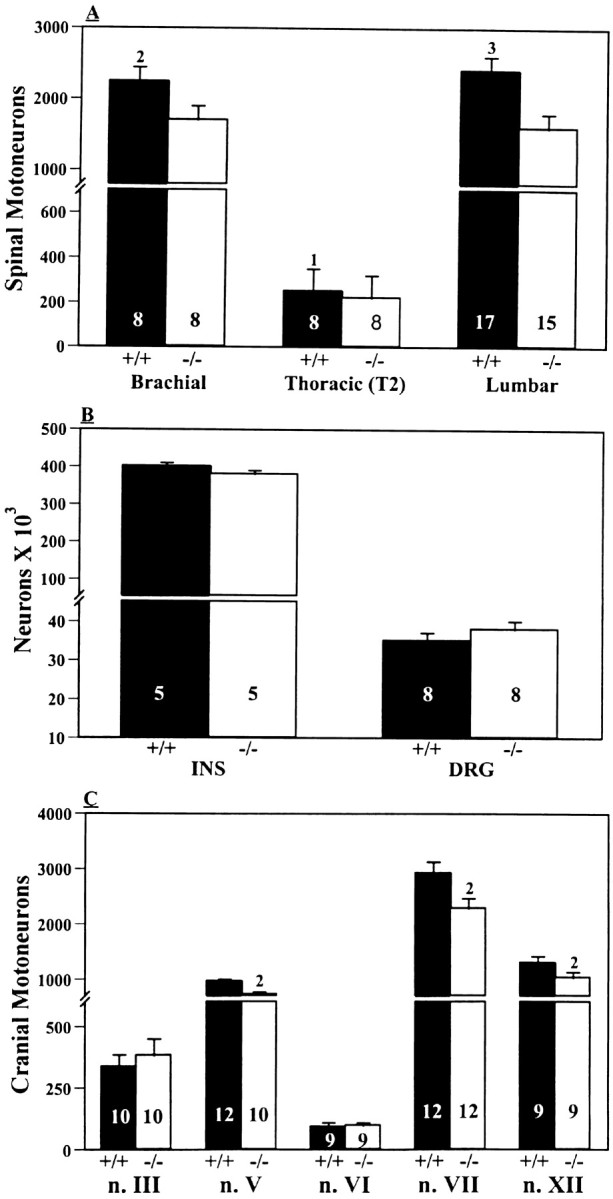
A, Spinal motoneuron numbers (mean ± SD) on E18.5 in GDNF-deficient (−/−) and wild-type control (+/+) embryos. Values in the bars are sample size (embryos).B, The number (mean ± SD) of lumbar spinal interneurons and L4 DRG sensory neurons on E18.5 in GDNF-deficient (−/−) and control (+/+) embryos. C, The number (mean ± SD) of cranial MNs on E18.5 in GDNF-deficient (−/−) and control (+/+) embryos. 1p < 0.05;2p < 0.01;3p < 0.005, ttests.
Table 1.
The number (mean ± SD) of healthy and pyknotic lumbar motoneurons in GDNF-deficient mice
| E12.5 | E13.5 | E14.5 | E15.5 | E16.5 | E18.5 | P2 | |
|---|---|---|---|---|---|---|---|
| Healthy +/+ | 4211 ± 767 | 3936 ± 339 | 3688 ± 226 | 3032 ± 373 | 2870 | 2405 ± 251 | 2290 ± 133 |
| (5) | (5) | (8) | (4) | (1) | (17) | (5) | |
| Healthy −/− | 4480 ± 790 | 3890 ± 271 | 3271 ± 301* | 2595 ± 3281-160 | 2171 | 1506 ± 1511-165 | — |
| (3) | (5) | (7) | (4) | (2) | (15) | — | |
| Pyknotic +/+ | 21 ± 6 | 52 ± 8 | 74 ± 27 | 83 ± 10 | 41 | 9 ± 3 | 2 ± 0.6 |
| (5) | (5) | (8) | (4) | (1) | (12) | (5) | |
| Pyknotic −/− | 16 ± 5 | 54 ± 12 | 128 ± 17* | 112 ± 131-160 | 66 | 13 ± 21-168 | — |
| (3) | (5) | 7 | (4) | (1) | (11) | — |
p < 0.01;
F1-160: p < 0.001;
F1-165: p< 0.002;
F1-168: p<0.05.
An analysis of MN numbers along the rostrocaudal axis of the lumbar spinal cord on E18.5 revealed that most of the MN loss occurred in the rostral three-fifths of the lumbar enlargement (Fig.3A). Because a small amount of normal MN death continues for a few days after E18.5 (Oppenheim et al., 1986) and because most of this loss occurs in caudal lumbar regions (Fig. 3), it seems likely that the apparent restriction of MN loss on E18.5 in GDNF-deficient embryos to the rostral and intermediate (vs caudal) lumbar segments is an artifact of not being able to assess MN numbers postnatally in these embryos because of their embryonic/perinatal death. Although the analysis of MN numbers shows that there is a significant loss in the GDNF-deficient embryos, single Nissl-stained transverse sections of the spinal cord do not always accurately reflect this (Fig. 1) because the loss is distributed along the entire spinal cord (Fig. 3). However, these sections do accurately reflect the normal histology and morphology of the spinal cord in GDNF-deficient embryos.
Fig. 3.
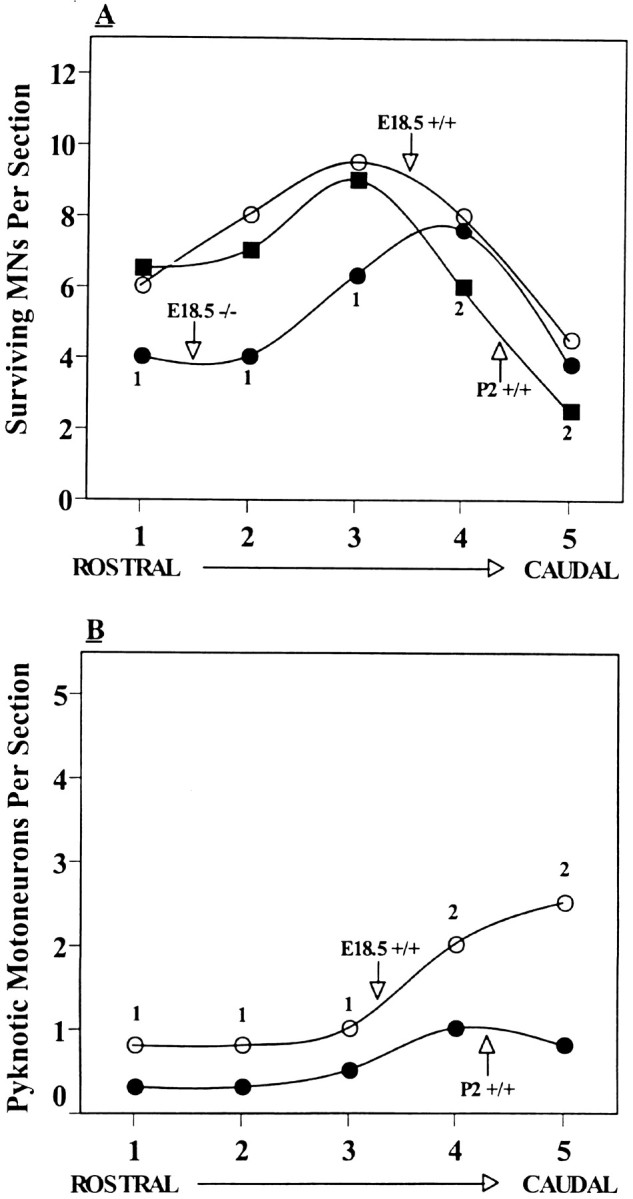
A, The number (mean ± SD, SD not shown for illustrative clarity) of motoneurons per section for five equal lengths of lumbar spinal cord along the rostrocaudal axis on E18.5 (+/+, −/−) and P2 (+/+). B, The number (mean ± SD) of pyknotic motoneurons per section along the rostrocaudal axis of lumbar spinal cord in wild-type control (+/+) animals on E18.5 and P2. In A, E18.5 +/+ versus E18.5 −/−; 1p < 0.005; E18.5 +/+ versus P2 +/+; 2p < 0.01. In B,1p < 0.05;2p < 0.01, ttests.
Because cranial somatic MNs express c-ret and GFRα1, we have also examined MN numbers in several of these nuclei. With the exception of the abducens and oculomotor nuclei, all other cranial motor nuclei examined here exhibit a significant loss of MNs (trigeminal 25%, hypoglossal 21%, and facial 18%; Fig. 2C). Taken together, these data indicate that most populations of spinal and cranial somatic MNs exhibit decreased cell numbers in GDNF-deficient mice and that this loss is attributable to increased naturally occurring cell death.
Reductions in c-ret-, GFRα1-, and GFRα2-expressing cells in GDNF-deficient embryos
Although it is clear from the data on surviving and degenerating MNs that the loss of GDNF results in increased cell death, because not all MNs die, it was of interest to determine the phenotype of surviving versus dying MN subpopulations. Because GDNF is believed to signal primarily via c-ret and GFRα1 (or less effectively via c-ret and GFRα2), we have examined cells expressing these receptor mRNAs usingin situ hybridization. Additionally, to further characterize the surviving MNs in GDNF-deficient embryos, we have examined mRNA expression for ChAT and for the LIM family homeobox nuclear transcription factors islet-1 and islet-2.
The expression of c-ret in presumptive MNs in the ventral horn was reduced in the brachial, thoracic, and lumbar regions of E15.5 GDNF-deficient mice (Fig. 4). Because of the normal rostrocaudal gradient of MN death in the mouse embryo spinal cord (Yamamoto and Henderson, 1999), the decrease in c-ret expression is more apparent in the brachial region at this age. However, by E18.5 all three regions exhibit a clear loss of c-ret expression in ventral cells (Fig. 5), and consistent with the cell counts (Fig. 2), the thoracic region shows less of a loss than brachial or lumbar. Although regions dorsal to the ventral horn that normally express c-ret, GFRα1, and isl-1 (see below) also exhibit reduced mRNA signals, based on our quantification of interneuron numbers at E18.5 (Fig. 2), this reduction is unlikely to reflect cell death.
Fig. 4.
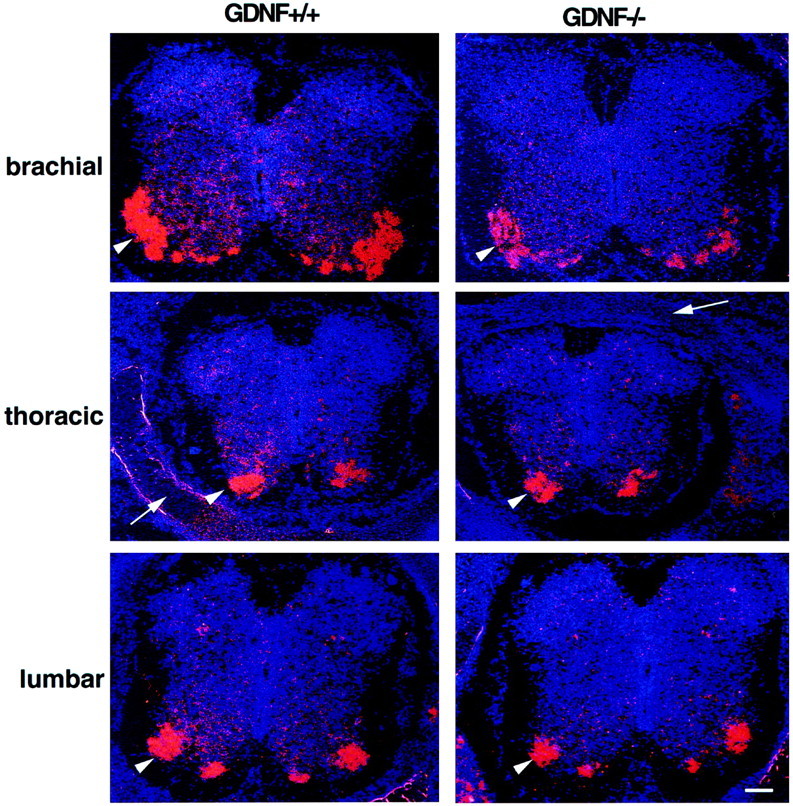
Expression of c-ret in spinal motoneurons of embryos on E15.5. Transverse sections of c-ret in situhybridization at brachial, thoracic, and lumbar levels.Arrowheads indicate ventral horn region;arrows indicate vertebra. Scale bar, 100 μm.
Fig. 5.
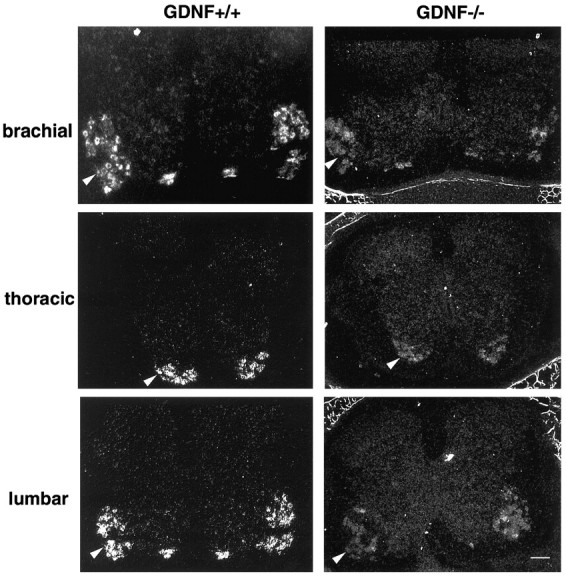
Expression of c-ret in brachial, thoracic, and lumbar spinal cord on E18.5. Scale bar, 200 μm. Note the reduced mRNA signal in surviving MNs compared to E15.5 (Fig. 4).
In an attempt to quantify the loss of c-ret expression, we have averaged the total area (pixels) expressing c-ret in the ventral spinal cord from several embryos in five independent in situ hybridization experiments. The relative reduction in c-ret expression in the ventral horn region of E15.5, GDNF-deficient embryos was 50, 26, and 35% for brachial, thoracic, and lumbar segments, respectively (Table 2). Except for the lumbar region, the density of the c-ret signal was also reduced (Table 3). We have no explanation for the discrepancy in the area versus density data in the lumbar region at this age. However, by E18.5 both measures were also significantly reduced in the lumbar segments (data not shown, but see Fig. 5). These data indicate that the loss of GDNF results in a significant reduction in c-ret expression in ventral neurons in all three spinal regions examined. However, some of the reduction of the c-ret signal in GDNF-deficient embryos appears to reflect a loss of (or reduced) expression in surviving MNs. This is especially obvious on E18.5 (Fig.5). Therefore, the reduced density measure likely represents both cell loss and a reduced in situ signal on the remaining MNs.
Table 2.
Quantification (area) of mRNA signal (mean ± SD) at E15.5
| Probes | Brachial +/+ | Brachial −/− | Thoracic +/+ | Thoracic −/− | Lumbar +/+ | Lumbar −/− |
|---|---|---|---|---|---|---|
| c-ret | 42107.8 ± 10943.4* | 20009.1 ± 4519.8 | 17500.9 ± 3448.8 | 12937.3 ± 4135.2 | 31064.3 ± 8637.9 | 20171.7 ± 4158.7 |
| (6)2-160 | (6) | (7) | (7) | (6) | (6) | |
| p = 0.00352-165 | 50%2-168 | p = 0.04 | 74% | p = 0.02 | ||
| isl-1 | 26216.8 ± 6150.7 | 18276.6 ± 5367.4 | 21476.4 ± 1958.2 | 19031.1 ± 3176.7 | 32472.9 ± 7662.4 | 16417.7 ± 2657.9 |
| (5) | (8) | (5) | (5) | (5) | (4) | |
| p = 0.04 | 70% | p = 0.24 | 88% | p = 0.007 | ||
| GFRα1 | 36664.4 ± 7518.8 | 18368.6 ± 2070.9 | 19778.3 ± 2109.3 | 14578.5 ± 2205.2 | ||
| (5) | (3) | (6) | (7) | NA | NA | |
| p = 0.0037 | 50% | p = 0.001 | 74% | |||
| GFRα2 | 930.4 ± 440.4 | 2730.5 ± 1302.3 | 2037.0 ± 562.8 | 3547.9 ± 447.3 | ||
| (4) | (5) | (6) | (5) | NA | NA | |
| p = 0.03 | 293% | p = 0.0007 | 174% |
pixels ± SD;
F2-160: sample size;
F2-165: p values.
F2-168: The percent change in signal in GDNF null mutants relative to wild-type levels that are expressed as 100%.
Table 3.
Quantification (density) of the c-ret signal (mean ± SEM) at E15.5
| Probe | Brachial | Thoracic | Lumbar | |||
|---|---|---|---|---|---|---|
| +/+ | −/− | +/+ | −/− | +/+ | −/− | |
| c-ret | 0.31 ± 0.02 | 0.23 ± 0.043-150 | 0.34 ± 0.08 | 0.25 ± 0.063-160 | 0.26 ± 0.05 | 0.35 ± 0.10 |
| (6) | (6) | (7) | (7) | (6) | (6) | |
F3-150: p < 0.003;
F3-160: p < 0.03.
In vitro biochemical studies have demonstrated that GDNF binds preferentially to GFRα1 and less efficiently to GFRα2, but that all GDNF family members can signal through c-ret (Baloh et al., 1997;Creedon et al., 1997; Trupp et al., 1997). The retention of many c-ret expressing MNs in GDNF-deficient mice suggests that their survival may be mediated by other GDNF ligands or by GDNF receptor components other than GFRα1. We first examined whether the loss of c-ret-expressing cells was correlated with the loss of GFRα1 or GFRα2 in brachial segments of E15.5 GDNF-deficient embryos. Most MNs in the brachial and lumbar regions appear to express GFRα1, whereas only a subset express GFRα2 (Fig. 6). Although reduced significantly, many GFRα1-expressing MNs remained in the ventral brachial spinal cord of GDNF-deficient animals (Fig. 6). Based on area measurements there was a close correlation between the loss of GFRα1 (50%) and c-ret (50%) expression in GDNF-deficient mice (Table 2). A similar relationship was also observed in the thoracic region (26% loss of c-ret and GFRα1). Although the normal expression of GFRα2 in the brachial ventral horn of wild-type mice is much less than that of GFRα1 (Fig. 6), there is an apparent increased expression of GFRα2 in the ventral horn of GDNF-deficient mice (Fig. 6, Table 2). The low level of GFRα2 expression in wild-type mice is not attributable to poor hybridization of the probe because the weak expression signal was confirmed using two independent riboprobes, and both probes give strong signals in other embryonic tissues (data not shown).
Fig. 6.
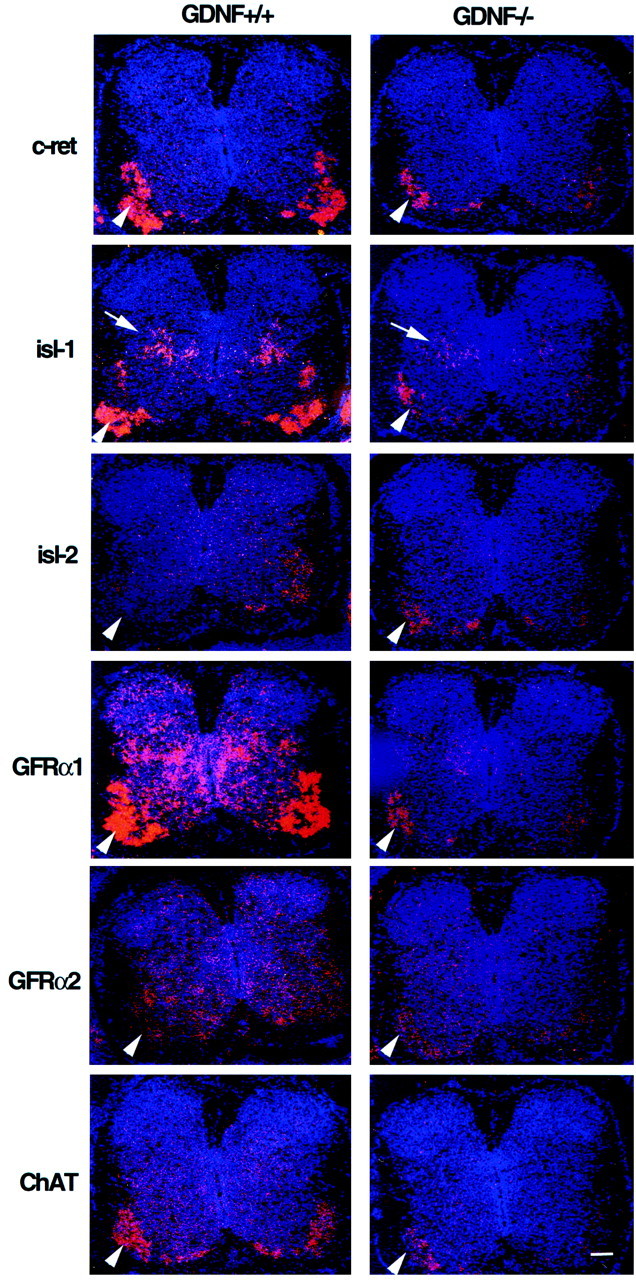
Expression (in situ hybridization using antisense cRNA probes) of multiple markers in transverse sections of E15.5 brachial spinal cord of GDNF-deficient and control embryos.Arrowheads indicate the ventral horn;arrow indicates dorsal population of isl-1-expressing neurons. Scale bar, 100 μm.
To determine whether the surviving neurons in the ventral horn of GDNF-deficient mice were, in fact, differentiating MNs, we examined the expression of two MN-specific markers, ChAT and islet (isl-1 and isl-2), by in situ hybridization. The expression of ChAT appeared to be reduced in tissue sections from E15.5 GDNF-deficient embryos (Fig. 6), and this was confirmed by the area measurements showing a 50 and 37% reduction in brachial and thoracic segments, respectively (Table 4). Because this reduction in ChAT is comparable in magnitude to the reduction in c-ret, this indicates that most, if not all, of the surviving c-ret/ChAT-positive ventral neurons in the GDNF-deficient embryos are differentiating MNs.
Table 4.
Quantification (area) of the ChAT and isl-2 signal (mean ± SEM) at E15.5
| Probe | Brachial | Thoracic | ||
|---|---|---|---|---|
| +/+ | −/− | +/+ | −/− | |
| ChAT | 37,162 ± 1789 | 18,524 ± 39034-150 | 16,670 ± 1825 | 10,468 ± 25554-160 |
| (6) | (6) | (7) | (7) | |
| Isl-2 | 16,805 ± 2131 | 16,150 ± 4729 | ||
| (6) | (6) | N/A | N/A | |
F4-150: p < 0.004;
F4-160: p < 0.025.
To further determine the phenotype of surviving MNs in the GDNF-deficient mice, we examined the expression of isl-1 and isl-2. Whereas isl-2 is MN-specific, isl-1 is strongly expressed in MNs as well as in a subset of dorsal interneurons in the spinal cord. There was a reduction in isl-1 expression in the ventral horn of brachial (30%) and lumbar (50%) segments, but no apparent loss in the thoracic region of GDNF-deficient embryos (Table 2). By contrast, there was no apparent reduction in brachial isl-2 expression on E15.5 (Table 4) or in any segmental region of the ventral horn of E18.5 GDNF-deficient embryos (data not shown); isl-2 area measures in the ventral horn at all spinal levels on E18.5 were also comparable to wild-type embryos (data not shown). Because the area measurements for isl-2, c-ret, isl-1, and ChAT mRNA expression were similar in the brachial ventral spinal cord of GDNF-deficient embryos, this indicates that the surviving GDNF-independent MN population expresses isl-2, whereas the GDNF-dependent MNs are isl-2-negative and isl-1-positive. All surviving c-ret/GFRα1-expressing brachial MNs in the GDNF-deficient mice also appear to coexpress isl-1 and isl-2 as well as show an apparent upregulation of GFRα2 expression (Table 2).
In utero GDNF treatment rescues MNs from cell death
In previous studies, we found that the administration of a single injection of different putative neurotrophic factors to mouse embryosin utero on E14 was sufficient to significantly reduce the normal death of spinal MNs when assessed on E18 (Houenou et al., 1994). In the present study, we observed that GDNF is also able to rescue both spinal and cranial MNs from cell death when administered in utero on E14 (Fig. 7). On average, GDNF treatment increased MN numbers on E18.5 by ∼21%. Except for the abducens and oculomotor nuclei (data not shown), all populations of somatic MNs were rescued from cell death by GDNF. Lumbar spinal interneurons and sensory (L4 DRG) neuron numbers were also unaffected (data not shown). These data suggest that the survival of subpopulations of most somatic MNs is regulated by limiting amounts of endogenous GDNF. As described below, however, MN numbers can be increased even more when endogenous levels of muscle-derived GDNF are constitutively increased throughout the period of normal MN cell death.
Fig. 7.

The number (mean ± SD) of spinal and cranial motoneurons on E18.5 in mouse embryos treated with GDNF in utero on E14. 1p < 0.01;2p < 0.05. E, GDNF-treated; C, Control saline-treated.Numbers in bars are sample size (embryos).
Muscle-specific overexpression of GDNF (myo-GDNF) in transgenic mice promotes MN survival
Neonatal mice from the transgenic lines expressing high levels of GDNF under a muscle-specific (myogenin) promoter (Nguyen et al., 1998b) were found to have a significant increase in spinal and cranial MNs (Table 5, Fig.8). By contrast, transgenic mice that over express NT-3 or NT-4 (Myo-NT-3, myo-NT-4) have MN numbers comparable to controls. Based on cell counts in cervical spinal segments (C1–C2) both cervical spinal MNs (control spinal MNs = 3.6 ± 0.5 per section, n = 5, vs 8.3 ± 2.3 per section, n = 5, for myo-GDNF animals;p < 0.002) and XI cranial MNs (control = 4.7 ± 0.6 per section, n = 5 vs 7.5 ± 0.9 per section, n = 5, for myo-GDNF; p < 0.005) were increased in P1 transgenic mice.
Table 5.
The number of neurons (mean ± SD) in transgenic neonatal mice
| Lumbar | Brachial | DRG | INS | III | V | VI | VII | XII | |
|---|---|---|---|---|---|---|---|---|---|
| Control | 1718 ± 179 | 1781 ± 209 | 4140 ± 330 | 33,000 ± 3500 | 500 ± 121 | 797 ± 27 | 150 ± 24 | 3258 ± 188 | 1007 ± 110 |
| (13) | (4) | (4) | (4) | (3) | (4) | (3) | (4) | (4) | |
| Myo-GDNF | 3142 ± 3365-150 | 3060 ± 2475-150 | 3968 ± 445 | 31,000 ± 3100 | 432 ± 103 | 1146 ± 1265-160 | 158 ± 22 | 3945 ± 2715-160 | 1410 ± 1155-160 |
| (15) | (4) | (4) | (4) | (4) | (4) | (5) | (6) | (6) | |
| Myo-NT-3 | 1852 ± 216 | 1905 ± 366 | 4053 ± 307 | — | — | — | — | — | — |
| (5) | (3) | (3) | — | — | — | — | — | — | |
| Myo-NT-4 | 1694 ± 187 | 1823 ± 265 | 4160 ± 341 | — | — | — | — | — | — |
| (4) | (3) | (3) | — | — | — | — | — | — |
F5-150: p < 0.001;
F5-160: p < 0.01.
Fig. 8.
Transverse sections of P1 spinal cord from myo-GDNF transgenic animals (A, C, E, G, L), controls (B, D, F, H, K), and myo-NT-3 (I) and myo-NT-4 (J) animals. A–D, Brachial; E–J, lumbar;K, L, cervical. Scale bars:A (applies to A, B, E, F), 150 μm; C (applies to C, D, G-J), 80 μm; K (applies to K, L), 70 μm.Dotted lines in A–J delineate the ventral horn, and in K and L, the cervical (arrows) and spinal accessory (XI) MNs.
Consistent with the results from both the GDNF-deficient mice and the mice after in utero administration of GDNF, MN numbers in the abducens and oculomotor nuclei were not affected in the myo-GDNF transgenic mice. Similarly, sensory (DRG) and spinal interneurons were also unaffected in the myo-GDNF mice (Table 5). The increased number of spinal MNs (lumbar 45%, brachial 42%) in the myo-GDNF mice was considerably greater than that seen after in utero GDNF treatment (lumbar 22%, brachial 23%) but was close to the MN loss observed in GDNF-deficient mice (e.g., 37% loss of lumbar MNs vs a 45% increase in myo-GDNF mice). Myo-GDNF embryos examined on E15.5, during the normal cell death period for spinal MNs, had increased numbers of surviving lumbar MNs (3667 ± 194, n = 4 vs 3110 ± 183, n = 4 for controls;p < 0.01) and fewer degenerating (pyknotic) lumbar MNs (48 ± 12, n = 4 vs 67 ± 18,n = 4; p < 0.05). This indicates that the overexpression of GDNF increases MN numbers by preventing programmed cell death.
DISCUSSION
Using three different approaches to perturb GDNF levels in the developing mouse embryo, we have shown that each of the three results in an alteration in the number of MNs that survive during the period of naturally occurring PCD. GDNF-deficient embryos exhibit reductions in brachial, thoracic, and lumbar MNs and in MNs in the III, V, VII, XI, and XII cranial motor nuclei, whereas oculomotor and abducens MNs were unaffected. The overexpression of GDNF in developing muscle (myo-GDNF transgenics) and the injection of exogenous GDNF in uteroincreased MN survival in all of the same MN populations whose numbers were reduced in the GDNF-deficient embryos, whereas again oculomotor and abducen MNs were unaffected. The perturbation of GDNF availability specifically affected MNs, in that the survival of neither sensory (DRG) nor spinal interneurons were altered by any of the three approaches. The effects of overexpression of GDNF in the myo-GDNF transgenics was also trophic factor-specific because MN numbers in myo-NT-3 and myo-NT4/5-transgenic animals were comparable to wild-type control values. When considered together, these results provide compelling evidence for the role of GDNF as a physiologically relevant MN survival factor in the developing murine nervous system. However, because some of these results differ from previous reports they also raise several questions.
Facial MNs
After neonatal axotomy, mammalian facial MNs exhibit a robust survival response to treatment with exogenous GDNF (Henderson et al., 1994; Zurn et al., 1994; Yan et al., 1995; Oppenheim et al., 1995). By contrast, genetic deletion of GDNF or its primary co-receptor GFRα1 are reported to have no effect on the number of surviving facial MNs when examined in embryonic, neonatal, and adult animals (Moore et al., 1996; Sánchez et al., 1996; Cacalano et al., 1998). Using three different approaches to perturb GDNF availability, we have consistently observed an ∼20% increase or decrease (depending on whether GDNF was increased or decreased) in the number of facial MNs of embryonic and neonatal animals. This result is based on the evaluation of >20 experimental and 30 control animals in which cell counts were made blind to treatment. Accordingly, we are confident that the modest but significant alteration of facial MNs observed here is real. Despite this discrepancy, however, there is agreement among all the different studies regarding the role of GDNF in the survival of lumbar and trigeminal MNs, and our results now extend the MN populations that have been shown to respond to GDNF to include cervical, brachial, and thoracic spinal MNs and facial, hypoglossal, and spinal accessory cranial MNs.
Other non-MN populations
None of the three approaches used by us to alter GDNF availability affected the number of surviving sensory DRG neurons, spinal interneurons, or oculomotor or abducens MNs. From this, we conclude that at the embryonic ages examined here GDNF is not likely to be required for the survival of these populations even though all three populations express GDNF receptors. Although a previous study of GDNF-deficient animals reported finding a 20% loss of DRG neurons on P0 (Moore et al., 1996), neonatal animals deficient in the preferred GDNF co-receptor GFRα1 have normal numbers of DRG neurons (Cacalano et al., 1998; Enomoto et al., 1998). Because the excess GDNF in the myo-GDNF transgenic animals is not available to most DRG neurons, the lack of an effect on sensory neuron numbers in these animals is to be expected. One likely explanation for the discrepancy between our results and those of Moore et al. (1996) regarding sensory neurons is the different ages of the GDNF-deficient animals examined in the two studies (E18.5 vs P0).
The failure of both oculomotor and abducens MNs to exhibit a responsiveness to GDNF in any of the three approaches used here to perturb GDNF availability is surprising in light of the fact that these MNs express both c-ret and GFRα receptors in the adult (Trupp et al., 1997; Golden et al., 1998; Glazner et al., 1998). However, receptor expression has not yet been examined during development.
GDNF-dependent and independent MNs
There is considerable evidence supporting the idea that the survival of developing MNs depends on multiple neurotrophic factors produced by different cell types (Oppenheim, 1996; Hanson et al., 1998;Arce et al., 1998). This is perhaps not surprising given that MNs differ phenotypically depending on their location, peripheral targets, and afferent inputs (Tsuchida et al., 1994; Lin et al., 1998). The fact that treatment with specific neurotrophic factors in vitroand in vivo and genetic deletion of specific neurotrophic factors and their receptors only affect a proportion of all MNs in a given population (e.g., lumbar MNs) is consistent with the idea of MN subpopulations that require distinct trophic factors for their survival. A striking example of this is the case of MNs that are responsive to the muscle-derived neurotrophic factor hepatocyte growth factor/scatter factor (HGF/SF). In both chicken and rat, only limb-innervating MNs are rescued from PCD by HGF/SF (Yamamoto et al., 1997; Novak et al., 2000).
The present results are also consistent with this idea in that although the survival of virtually all MN populations examined by us (spinal and cranial) are affected by GDNF availability, only a subpopulation (20–40%) in each region or motor nucleus are dependent on GDNF for their survival. Although alternative explanations of this observation are possible (Oppenheim, 1996), we favor the notion that these represent distinct MN phenotypes that, based on their unique pattern of receptor expression, relative to other MNs, are dependent on GDNF for their survival. This is not meant to exclude the possibility that these same MNs can also respond to or are dependent on other neurotrophic factors (Oppenheim et al., 1993; Oppenheim, 1996; Arce et al., 1998; Hanson et al., 1998).
We find that c-ret and GFRα1 are expressed on most, if not all, spinal MNs in the developing mouse embryo, whereas only a subset of these are lost in GDNF-deficient animals, and only a proportion of the MNs lost during naturally occurring PCD are rescued by increased availability of GDNF (myo-GDNF and in utero treatment). Thus, there are GDNF-dependent and GDNF-independent subpopulations of spinal MNs. Based on our similar findings for cranial MNs (i.e., partial cell losses in each nucleus), these also appear to be composed of GDNF-dependent and independent subpopulations. In a recent study that examined the death of spinal MNs in GFRα1 and GFRα2-deficient mice, a selective vulnerability of subpopulations was also observed (Garcés et al., 2000). Few if any MNs were lost in GFRα2-deficient mice (Rossi et al., 1999), whereas distinct subpopulations were missing in GFRα1-deficient animals. Examination of the spinal cord of our GDNF-deficient embryos revealed a loss of c-ret expression comparable to the amount of cell loss. Although there was a close correlation between the loss of c-ret and GFRα1 expression in spinal MNs, many GFRα1-expressing MNs remained in GDNF-deficient embryos. Additionally, the apparent reduction in c-ret expression in surviving MNs of GDNF-deficient mice on E18.5 suggests that the maintenance of c-ret expression between E15 and E18 may require GDNF. The expression of GFRα2 was considerably less than GFRα1 in spinal MNs of wild-type control embryos (Garcés et al., 2000), and GFRα2 expression was increased in GDNF-deficient embryos. Accordingly, GFRα2-expressing MNs represent only a small subset of all MNs and do not appear dependent on GDNF for their survival.
Our observation that a subpopulation of c-ret/GFRα1-positive MNs are lost in GDNF-deficient embryos is consistent with the reported loss of only subsets of spinal MNs in GFRα1-deficient animals (Cacalano et al., 1998; Garcés et al., 2000) and with previous reports of partial MN losses in GDNF-deficient embryos (Moore et al., 1996;Sánchez et al., 1996). Our failure to observe a loss of GFRα2-expressing MNs in GDNF-deficient embryos is also consistent with GFRα2 being the preferred co-receptor for neurturin (Klein et al., 1997; Heuckeroth et al., 1999). Although the survival of cultured rat MNs is promoted by neurturin (Klein et al., 1997), a quantitative analysis of MNs in Neurturin or GFRα2-deficient animals has not yet been reported (Rossi et al., 1999; Heuckeroth et al., 1999). Interestingly, however, cultured MNs from GFRα2-deficient embryos retain their responsiveness to both GDNF and Neurturin (Garcés et al., 2000), suggesting that mouse spinal MNs may actually respond to Neurturin via receptors other than GFRα2.
To confirm that the surviving cells in the ventral horn of the GDNF-deficient embryos are in fact MNs, we have used two specific MN markers, ChAT and islet. There was a reduction in ChAT expression in the spinal cord of GDNF-deficient embryos consistent with the cell loss and with the reduced expression of c-ret/GFRα1. Using separate probes for isl-1 and isl-2, we found expression of both mRNAs in spinal MNs, whereas isl-1 was also strongly expressed in a subset of dorsal interneurons. Interestingly, there was a reduction of isl-1 expression in GDNF-deficient embryos comparable to the observed MN cell loss and to the reduction in c-ret/GFRα1/isl-1 expression in brachial and lumbar MNs, whereas there was no apparent loss of isl-2 expression. From this, we can conclude that the GDNF-dependent MNs represent a subset of c-ret/GFRα1/isl-1-expressing MNs, whereas the GDNF-independent MNs are an isl-2-positive subpopulation. The fact that all surviving c-ret/GFRα1-expressing MNs also appear to coexpress isl-1 and isl-2 (and show an upregulation of GFRα2) suggests that these represent a subset of MNs that may be dependent on other GDNF family members for their survival. Both neurturin and persephin have been reported to promote the survival of mammalian and avian MNs (Klein et al., 1997; Oppenheim et al., 1998; Soler et al., 1999) and therefore may be trophic factors for these GDNF-independent MNs. We have also found that another GDNF family member, artemin (Baloh et al., 1998), can also promote the survival of subpopulations of avian MNs in vitro and in vivo (R. Oppenheim, unpublished data).
The expression of LIM homeobox genes, including isl-1 and isl-2, by avian embryo MNs is organized topographically in the spinal cord such that the combinatorial expression of these genes define distinct subsets of MNs that reflect their position in the spinal cord and their peripheral targets (Tsuchida et al., 1994). Although the expression of LIM homeobox genes in mammalian MNs is not as well characterized as in the chick, based on the pattern of expression in the chick, one might predict that the differential expression of isl-1 and isl-2 among GDNF-dependent and GDNF-independent mouse MNs may reflect different phenotypes regarding peripheral targets or afferent inputs.
Footnotes
This work was supported by Grants NS20402 and NS31380 from the National Institutes of Health to Ronald W. Oppenheim, a grant from the Muscular Dystrophy Association to Lucien J. Houenou, and by Grant NS37273 from the National Institutes of Health to William D. Snider. We thank Dr. H. Westphal for his advice and support.
Correspondence should be addressed to Dr. Liya Shen, Laboratory of Cellular Carcinogenesis and Tumor Promotion, National Cancer Institute, National Institutes of Health, Building 37, Room 2B15, 9000 Rockville Pike, Bethesda, MD 20892. E-mail: shenl@mail.nih.gov.
REFERENCES
- 1.Arce V, Pollock RA, Philippe J-M, Pennica D, Henderson CE, deLapeyriére D. Synergistic effects of Schwann- and muscle-derived factors on motoneuron survival involved GDNF and cardiotrophin-1 (CT-1). J Neurosci. 1998;18:1440–1448. doi: 10.1523/JNEUROSCI.18-04-01440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baloh RH, Tansey MG, Golden JP, Creedon DJ, Heuckeroth RO, Keck CL, Zimonjic DB, Popescu NC, Johnson EM, Millbrandt J. TrnR2, a novel receptor that mediates neurturin and GDNF signaling through ret. Neuron. 1997;18:793–802. doi: 10.1016/s0896-6273(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 3.Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Millbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-ret receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/s0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- 4.Bisgrove BW, Raibl DW, Walter V, Eisen JS, Grunwald DJ. Expression of c-ret in the zebrafish embryo: Potential roles in motoneuronal development. J Neurobiol. 1997;33:749–768. [PubMed] [Google Scholar]
- 5.Cacalano G, Fariñas I, Want L-C, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi-Lundberg DL, Bohn MC. Ontogeny and distribution of glial cell line-derived neurotrophic factor (GDNF) mRNA in rat. Dev Brain Res. 1995;85:80–88. doi: 10.1016/0165-3806(94)00197-8. [DOI] [PubMed] [Google Scholar]
- 7.Clarke PGH, Oppenheim RW. Neuron death in vertebrate development: in vivo methods. Methods Cell Biol. 1995;46:277–321. [PubMed] [Google Scholar]
- 8.Creedon DJ, Tansey MG, Baloh RH, Osborne PA, Lampe PA, Fahrner TJ, Heuckeroth RO, Millbrandt J, Johnson EM. Neurturin shares receptors and signal transduction pathways with glial cell line-derived neurotrophic factor in sympathetic neurons. Neurobiol. 1997;94:7018–7023. doi: 10.1073/pnas.94.13.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM, Millbrandt J. GFRα1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- 10.Garcés A, Haase G, Airaksinen MS, Livet J, Filippi P, deLapeyriére O. Signaling by GDNF and NTN through GFRα1 and GFRα2 in distinct subpopulations of spinal motoneurons. J Neurosci. 2000;20:4992–5000. doi: 10.1523/JNEUROSCI.20-13-04992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glazner GW, Mu X, Springer JE. Localization of glial cell line-derived neurotrophic factor receptor alpha and c-ret mRNA in rat central nervous system. J Comp Neurol. 1998;391:42–49. doi: 10.1002/(sici)1096-9861(19980202)391:1<42::aid-cne4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 12.Golden JP, Baloh RH, Kotzbauer PT, Lampe PA, Osborne PA, Millbrandt J, Johnson EM. Expression of neurturin, GDNF and their receptors in the adult mouse CNS. J Comp Neurol. 1998;398:139–150. doi: 10.1002/(sici)1096-9861(19980817)398:1<139::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Golden JP, DeMaro JA, Osborne PA, Millbrandt J, Johnson EM. Expression of neurturin, GDNF and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol. 1999;158:504–528. doi: 10.1006/exnr.1999.7127. [DOI] [PubMed] [Google Scholar]
- 14.Hanson MG, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 16.Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, Jackman A, Molliver DC, Bardgett ME, Snider WD, Johnson EM, Millbrandt J. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory and parasympathetic neurons. Neuron. 1999;22:253–263. doi: 10.1016/s0896-6273(00)81087-9. [DOI] [PubMed] [Google Scholar]
- 17.Houenou LJ, Pincon-Raymond M, Garcia L, Harris AJ, Rieger F. Neuromuscular development following tetrodotoxin-induced inactivity in mouse embryos. J Neurobiol. 1990;21:1249–1261. doi: 10.1002/neu.480210809. [DOI] [PubMed] [Google Scholar]
- 18.Houenou LJ, Li L, Lo AC, Yan Q, Oppenheim RW. Naturally occurring and axotomy-induced motoneuron death and its prevention by neurotrophic agents: a comparison between chick and mouse. Prog Brain Res. 1994;102:217–226. doi: 10.1016/S0079-6123(08)60542-7. [DOI] [PubMed] [Google Scholar]
- 19.Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis J-C, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNF-α, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 20.Klein RD, Sherman D, Ho WH, Stone D, Bennett GL, Moffat B, Vandlen R, Simmons L, Gu Q, Hongo J-A, Devaux B, Poulsen K, Armanini M, Nozaki C, Asai N, Goddard A, Phillips H, Henderson CE, Takahashi M, Rosenthal A. A GPI-linked protein that interacts with ret to form a candidate neurturin receptor. Nature. 1997;387:717–721. doi: 10.1038/42722. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Wu W, Lin L-F, Lei M, Oppenheim RW, Houenou LJ. Rescue of adult mouse motoneurons form injury-induced cell death by glial cell line-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:9771–9775. doi: 10.1073/pnas.92.21.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin LH, Doherty DH, Lile JD, Becktesh S, Collins F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 23.Lin JH, Saito T, Anderson DJ, Lance-Jones C, Jessell TM, Arber S. Functionally related motor neuron pool and muscle sensory afferent subtypes defined by coordinate ETS gene expression. Cell. 1998;95:393–407. doi: 10.1016/s0092-8674(00)81770-5. [DOI] [PubMed] [Google Scholar]
- 24.McMahon A, Joyner A, Bradley A, McMahon J. The midbrain-hindbrain phenotype of Wnt-1-/Wnt-1- mice results from stepwise deletion of engrailed-expression cells by 9.5 days post coitum. Cell. 1992;69:581–595. doi: 10.1016/0092-8674(92)90222-x. [DOI] [PubMed] [Google Scholar]
- 25.Moore MW, Klein RD, Fariñas, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Ohta K, Hirokawa K, Fukushima M, Uchino M, Ando M, Tanaka H. Developmental and denervation changes in c-ret proto-oncogene expression in chick motoneurons. Mol Brain Res. 1996;39:1–11. doi: 10.1016/0169-328x(95)00347-u. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen QT, Keller-Peck CR, Lichtman JW, Silos-Santiago I, Snider WD. GDNF expression is restricted to a subset of muscles and muscle fibers. Soc Neurosci Abstr. 1998a;24:2002. [Google Scholar]
- 28.Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscles. Science. 1998b;279:1725–1730. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- 29.Novak KD, Prevette D, Wang SW, Gould T, Oppenheim RW. Hepatocyte growth factor/scatter factor (HGF/SF) is a neurotrophic survival factor for lumbar but not for other somatic motoneurons in the chick embryo. J Neurosci. 2000;20:326–337. doi: 10.1523/JNEUROSCI.20-01-00326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oppenheim RW. Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron. 1996;17:195–197. doi: 10.1016/s0896-6273(00)80151-8. [DOI] [PubMed] [Google Scholar]
- 31.Oppenheim RW, Houenou LJ, Rieger F, Pincon-Raymond M, Powell JA, Standish JL. The development of motoneurons in the embryonic spinal cord of the mouse mutant, muscular dysgenesis (mdg/mdg). Dev Biol. 1986;114:426–436. doi: 10.1016/0012-1606(86)90207-1. [DOI] [PubMed] [Google Scholar]
- 32.Oppenheim RW, Prevette D, Haverkamp LJ, Houenou L, Yin QW, McManaman J. Biological studies of a putative avian muscle-derived neurotrophic factor that prevents naturally occurring motoneuron death in vivo. J Neurobiol. 1993;24:1065–1079. doi: 10.1002/neu.480240806. [DOI] [PubMed] [Google Scholar]
- 33.Oppenheim RW, Houenou LJ, Johnson JE, Lin L-FH, Li L, Lo AC, Newsome AL, Prevette DM, Wang SW. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 34.Oppenheim RW, Wang SW, Prevette D. A comparison between GDNF, neurturin and persephin in promoting motoneuron survival. Soc Neurosci Abstr. 1998;24:807. [Google Scholar]
- 35.Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- 36.Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 37.Rosenthal A. The GDNF protein family: gene ablation studies reveal what they really do and how. Neuron. 1999;22:201–207. doi: 10.1016/s0896-6273(00)81077-6. [DOI] [PubMed] [Google Scholar]
- 38.Rossi J, Luukko K, Poteryaev D, Laurikainen A, Sun YF, Laakso T, Eerikäinen S, Tuominen R, Lakso M, Rauvala H, Arumäe U, Pasternack M, Saarma M, Airaksinen MS. Retarded growth and deficits in the enteric and parasympathetic nervous system in mice lacking GFRα2, a functional neurturin receptor. Neuron. 1999;22:243–252. doi: 10.1016/s0896-6273(00)81086-7. [DOI] [PubMed] [Google Scholar]
- 39.Sánchez MP, Silos-Santiago I, Frisén J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 40.Soler RM, Dolcet X, Encinas M, Eges J, Bayascas JR, Comella JX. Receptors of the glial cell line derived neurotrophic factor family of trophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci. 1999;19:9160–9169. doi: 10.1523/JNEUROSCI.19-21-09160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Springer JE, Seeburger JF, He J, Gabrea A, Blankenhorn EP, Bergman LW. cDNA sequence and differential mRNA regulation of two forms of glial cell line-derived neurotrophic factor in Schwann cells and rat skeletal muscle. Exp Neurol. 1995;131:47–52. doi: 10.1016/0014-4886(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 42.Suter-Crazzolara C, Unsicker K. GDNF is expressed in two forms in many tissues outside the CNS. Mol Neurosci. 1994;5:2486–2488. doi: 10.1097/00001756-199412000-00020. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki H, Hase A, Miyata Y, Arahata K, Akazawa C. Prominent expression of glial cell line-derived neurotrophic factor in human skeletal muscle. J Comp Neurol. 1998;402:303–312. [PubMed] [Google Scholar]
- 44.Treanor JS, Goodman L, deSauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–84. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 45.Trupp M, Belluardo N, Funakoshi H, Ibáñez CF. Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene and GDNF receptor-α indicates multiple mechanisms of trophic actions in the adult rat CNS. J Neurosci. 1997;17:3554–3567. doi: 10.1523/JNEUROSCI.17-10-03554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trupp M, Raynoschek C, Belluardo N, Ibanez CF. Multiple GPI-anchored receptors control GDNF-dependent and independent activation of the c-Ret receptor tyrosine kinase. Mol Cell Neurosci. 1998;11:47–63. doi: 10.1006/mcne.1998.0667. [DOI] [PubMed] [Google Scholar]
- 47.Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 1994;79:957–970. doi: 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 48.Wang CY, Ni J, Jiang H, Hsu TA, Dugich-Djordjevic M, Feng L, Zhang M, Mei L, Gentz R, Lu B. Cloning and characterization of glial cell line-derived neurotrophic factor receptor-B: a novel receptor for members of glial cell line-derived neurotrophic factor family of neurotrophic factors. Neuroscience. 1998;83:7–14. doi: 10.1016/s0306-4522(97)00462-4. [DOI] [PubMed] [Google Scholar]
- 49.Wright DE, Snider WD. Focal expression of glial cell line-derived neurotrophic factor in developing mouse limb bud. Cell Tissue Res. 1996;286:209–217. doi: 10.1007/s004410050689. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto Y, Henderson CE. Patterns of programmed cell death in populations of developing spinal motoneurons in chicken, mouse and rat. Dev Biol. 1999;214:60–71. doi: 10.1006/dbio.1999.9413. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto Y, Livit J, Pollock RA, Garces A, Arce V, deLapeyrière O, Henderson CE. Hepatocyte growth factor (HGF/SF) is a muscle-derived survival factor for a subpopulation of embryonic motoneurons. Development. 1997;124:2903–2913. doi: 10.1242/dev.124.15.2903. [DOI] [PubMed] [Google Scholar]
- 52.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 53.Yu T, Scully S, Yu Y, Fox GM, Jing S, Zhou R. Expression of GDNF family receptor components during development: implications in the mechanisms of interaction. J Neurosci. 1998;18:4684–4696. doi: 10.1523/JNEUROSCI.18-12-04684.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zurn AD, Baetge EE, Hammang JP, Tan SA, Aebischer P. Glial cell line-derived neurotrophic factor (GDNF), a new neurotrophic factor for motoneurons. NeuroReport. 1994;6:113–118. doi: 10.1097/00001756-199412300-00030. [DOI] [PubMed] [Google Scholar]