

Abstract
Protein aggregates containing intermediate filaments (IFs) are a hallmark of degenerating spinal motor neurons in amyotrophic lateral sclerosis (ALS). Recently, we reported that a deficiency in neurofilament light subunit (NF-L), a phenomenon associated with ALS, promoted the formation of IF inclusions with ensuing motor neuron death in transgenic mice overproducing peripherin, a type III IF protein detected in axonal inclusions of ALS patients. To further assess the role of NF-L in the formation of abnormal IF inclusions, we generated transgenic mice overexpressing human neurofilament heavy subunits (hNF-H) in a context of targeted disruption of the NF-L gene (hH;L−/− mice). The hH;L−/− mice exhibited motor dysfunction, and they developed nonfilamentous protein aggregates containing NF-H and peripherin proteins in the perikarya of spinal motor neurons. However, the perikaryal protein aggregates in the hH;L−/− mice did not provoke motor neuron death, unlike toxic IF inclusions induced by peripherin overexpression in NF-L null mice (Per;L−/− mice). Our results indicate that different types of IF protein aggregates with distinct properties may occur in a context of NF-L deficiency and that an axonal localization of such aggregates may be an important factor of toxicity.
Keywords: neurofilament, peripherin, intermediate filament, transgenic mouse, ALS, amyotrophic lateral sclerosis, motor neuron disease, neurodegeneration
Amyotrophic lateral sclerosis (ALS) is a late onset motor neuron disease characterized by the degeneration of spinal motor neurons, leading to paralysis and death. Although ∼3% of ALS patients have inherited mutations in the Cu/Zn superoxide dismutase (SOD1) gene (Rosen et al., 1993), the causes of disease for the vast majority of ALS patients remain unknown. Inclusion bodies, also called spheroids, containing neuronal intermediate filaments (IFs) are a common feature of degenerating motor neurons in ALS (Carpenter, 1968; Corbo and Hays, 1992; Migheli et al., 1993). Such IF inclusions also occur in transgenic mice expressing ALS-linked SOD1 mutant proteins, suggesting a role for these inclusions in the pathology of ALS (Tu et al., 1996; Beaulieu et al., 1999a). Adult motor neurons can express up to five different IF proteins detectable in IF inclusions of ALS patients (Tu et al., 1996; Corbo and Hays, 1992; Migheli et al., 1993). These proteins are the neurofilament (NF) light (NF-L), medium (NF-M), and heavy subunits (NF-H), α-internexin and peripherin (Hoffman and Lasek, 1975; Pachter and Liem, 1985; Parysek and Goldman, 1988). Multiple cell transfection and transgenic mouse studies have demonstrated that NFs are formed by the heteropolymerization of NF-L with NF-M and/or NF-H (Lee et al., 1993; Ching and Liem, 1993; Zhu et al., 1997; Jacomy et al., 1999). On the other hand, α-internexin and peripherin can either self-polymerize into IFs or interact with NF proteins according to modalities that are not yet well understood (Parysek et al., 1991; Ching and Liem, 1993; Athlan and Mushynski, 1997; Beaulieu et al., 1999a,b).
A 60% decrease in mRNA levels for NF-L has been reported in motor neurons of ALS patients (Bergeron et al., 1994). Although the NF-L knock-out genotype (NF-L−/−) is not pathogenic for mice (Zhu et al., 1997), our recent studies showed that NF-L levels can affect the progression of motor neuron pathologies caused by an upregulation of other IF proteins. The overexpression of wild-type peripherin led to the formation of IF inclusions with ensuing death of motor neurons in 2-year-old mice (Beaulieu et al., 1999a). Remarkably, the motor neuron disease in peripherin transgenic mice was precipitated by a deficiency of NF-L (Beaulieu et al., 1999a). Transgenic mice expressing a human NF-H protein (hNF-H) also develop abnormal IF accumulations in spinal motor neurons, with ensuing motor dysfunction during aging (Côté et al., 1993). Again, in this mouse model, the pathogenesis was altered by changes in NF-L levels. Thus, the overexpression of a human NF-L transgene in hNF-H mice was able to reduce the perikaryal NF swellings and rescue the motor neuronopathy (Meier et al., 1999).
To further analyze the role of NF-L in the formation of neuronal IF inclusions and in motor neuron disease, we generated transgenic mice overexpressing hNF-H proteins in a context of NF-L deficiency. In absence of NF-L subunits, the hNF-H transgenic mice developed nonfilamentous protein aggregates containing NF-H and peripherin proteins in the perikarya of spinal motor neurons. However, the protein aggregates in the hH;L−/− mice did not provoke motor neuron death, unlike IF inclusion bodies resulting from the overexpression of peripherin proteins in NF-L null mice. Our results suggest that axonal localization is a factor that may contribute to the toxicity of IF protein aggregates in motor neurons.
MATERIALS AND METHODS
Transgenic mice. The hH;L−/− mice were obtained by breeding the previously described hNF-H transgenic mice (line 200) (Côté et al., 1993) with NF-L−/− mice (Zhu et al., 1997). The genotypes of the resulting mice were determined by Southern blot analysis of mouse tail DNA according to previously described procedures (Côté et al., 1993; Zhu et al., 1997). All hNF-H transgenic mice used in this study were homozygous for the hNF-H transgene and in a C57BL6 enriched background. The peripherin transgenic mice overexpressing the wild-type mouse peripherin protein under the control of the peripherin promoter (Per mice) and Per;L−/− mice were described previously (Beaulieu et al., 1999a). The use and maintenance of mice described in this article were performed according to the guidelines of Care and Use of Experimental Animals of the Canadian Council on Animal Care.
Antibodies. The anti-human NF-H (OC95) and anti-mouse NF-H (OC59) rat monoclonal antibodies were kind gifts from Dr. Virginia Lee (University of Pennsylvania, Philadelphia, PA). The anti-NF-H hypophosphorylated (Smi-32) and hyperphosphorylated (Smi-31) monoclonal antibodies were from Sternberger Monoclonals (Lutherville, MD). The anti-NF-H polyclonal antibody (N-4142) was from Sigma (Oakville, Canada). The anti-NF-M monoclonal antibody (NN-18), the anti-NF-L monoclonal antibody (NR-4), and the anti-actin clone c4 monoclonal antibody were from Roche Molecular Biochemical (Laval, Canada). The anti-peripherin monoclonal antibody (MAB1527), the anti-peripherin polyclonal antibody (AB1530), and the anti α-internexin polyclonal antibody (AB1515) were from Chemicon (Mississauga, Canada). The anti-MG160 polyclonal antibody (Croul et al., 1990) was a kind gift from Dr. Nicolas K. Gonatas (University of Pennsylvania, Philadelphia, PA).
Western blot analysis. For total protein extraction, tissues were homogenized in SDS–urea buffer (5 mg/ml SDS, 8m urea). For isolation of the cytoskeletal insoluble proteins, tissues were first homogenized at 4°C in a Triton buffer containing 10 mm tris, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, and a cocktail of protease inhibitors (2 mm PMSF, 2 mg/ml leupeptin, 1 mg/ml pepstatin, and 10 mg/ml aprotinin). Homogenates were centrifuged at 14,000 ×g for 15 min at 4°C using a Sorvall MC-12V centrifuge (Dupont, Guelph, Canada). The supernatants (soluble fraction) were collected and kept for further use. The pellets (insoluble fraction) were resuspended in a volume of SDS–urea buffer equivalent to the volume of the soluble fraction. For both types of extracts, the protein concentration was measured using a DC-protein assay (Bio-Rad, Mississauga, Canada). Proteins were separated on 10% SDS-PAGE and transferred to nitrocellulose membranes. To ensure that equivalent amounts of proteins were used for each sample, duplicate gels were routinely stained with Coomassie blue-R250. Proteins were detected using primary antibodies and peroxidase-conjugated anti-mouse IgG, anti-rat IgG, or anti-rabbit IgG secondary antibodies from Jackson ImmunoResearch (Mississauga, Canada). Immune complexes were revealed using the Renaissance chemiluminescence reagents (NEN, Boston, MA). Densitometric analysis were performed from autoradiograms using NIH Image software version 1.62 for Power Macintosh (Apple Computers, Cupertino, CA).
Partial proteolysis of NF-H. Partial proteolytic digestion was performed following a modification of the method described byJulien and Mushynski (1983). The spinal cord samples were homogenized in Triton buffer in the absence of protease inhibitor. Soluble and insoluble protein fractions were prepared by centrifugation as described above. The pellets were resuspended at a concentration of 3 mg/ml protein in 0.05 m2(N-morpholino) ethanesulphonic acid, pH 6.5, and the supernatants were diluted at the same protein concentration in the same buffer. Both fractions were then digested separately with α-chymotrypsin (Roche Molecular Biochemical, Laval, Canada) at 30°C for 0, 8, or 15 min with an enzyme-to-protein ratio of 1:3000. The digestion was stopped by the addition of PMSF at a final concentration of 2 mm. Each digestion product was then analyzed by Western blotting.
Immunohistological analysis. Mice were anesthetized by injection of chloral hydrate. For immunohistochemistry, mice were perfused with a 16 gm/l sodium cacodylate buffer, pH 7.5, followed by fixative (3% glutaraldehyde in sodium cacodylate buffer). Floating 50-μm-thick vibratome sections were rinsed in PBS, treated for 30 min with a 10 mg/ml sodium borohydride solution, and then blocked for 1 hr in a buffer containing 30 mg/ml BSA, 0.5% Triton X-100, and 0.3 mg/ml hydrogen peroxide in PBS. Incubation with antibodies was performed overnight at room temperature in a buffer containing 30 mg/ml BSA and 0.05% Triton X-100 in PBS. Labeling was done using a Vector ABC kit (Vector Laboratories, Burlingame, CA) and Sigmafast tablets (Sigma). For double indirect immunofluorescence, mice were perfused with PBS, pH 7.5, followed by 40 mg/ml paraformaldehyde in PBS. Tissues were post-fixed for 2 hr in paraformaldehyde, rinsed in PBS, and incubated overnight in a 200 mg/ml phosphate-buffered sucrose solution. Sections of 10 μm were prepared using a cryostat. Sections were blocked for 30 min in PBS blocking buffer and incubated overnight with primary antibodies. Immune complexes were revealed with anti-mouse FITC and anti-rabbit rhodamine-conjugated antibodies (Jackson ImmunoResearch) at a dilution of 1:200. Samples were mounted in Prolong (Molecular Probes, Eugene, OR) and examined under a fluorescence microscope. Composite overlaid pictures of double immunofluorescence were derived from digitalized color films using Photoshop 4.0 for Macintosh computers (Adobe Systems, San Jose, CA).
In situ detergent solubility assay. The spinal cords were rapidly dissected out from unfixed mice, frozen at −80°C in Tissue Tek (Miles, Elkhart, IN), and cut in 10 μm sections using a cryostat. Sections were then incubated on slides at room temperature for 15 min in Triton buffer (10 mm Tris, 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100) or in PBS, rinsed three times in PBS, and fixed for 30 min with a 4% paraformaldehyde phosphate-buffered solution. The sections were then processed for double indirect immunofluorescence in parallel with untreated sections obtained from mice fixed with a 4% paraformaldehyde solution.
Light and electron microscopy. Tissues were prepared for embedding in Epon as described by Zhu et al. (1997). Thin sections were stained with toluidine blue and examined under a light microscope. Counting and morphological analysis of L5 ventral root axons was performed using an Image-1 analysis software (Universal Imaging, West Chester, PA). For electron microscopy, sections were stained with a lead citrate solution and examined with a Phillips CM10 transmission electron microscope.
RESULTS
Absence of NF-L does not alleviate neuronopathy caused by overexpression of hNF-H
The hH;L−/− mice were derived by breeding procedures with hNF-H transgenic mice (line 200) (Côté et al., 1993) and NF-L−/− mice (Zhu et al., 1997) to obtain mice that were homozygous for both hNF-H transgene and NF-L disrupted gene (hNF-H+/+;NF-L−/−). Western blots revealed reduced levels of mouse and human NF-H proteins in the spinal cord of hH;L−/− mice when compared with homozygous hNF-H transgenic mice having a normal NF-L genotype (Fig. 1A). Similarly, a decrease of endogenous NF-H protein content also occurred in NF-L−/− mice. The reductions in levels of NF-H, NF-M, and to a lesser extent in peripherin have been reported in NF-L−/− mice and are probably the result of an enhanced proteolytic turnover of unassembled or disorganized IF proteins in the absence of NF-L (Zhu et al., 1997; Williamson et al., 1998; Beaulieu et al., 1999a; Levavasseur et al., 1999).
Fig. 1.

Motor dysfunction despite reduced levels of hNF-H in hH;L−/− mice. A, Western blot analysis of total protein extracts (10 μg) from the spinal cord of 3-month-old control (W), NF-L−/− (L−/−), hNF-H transgenic (hH), and hH;L−/− mice. Protein immunodetection was performed using the following antibodies: hNF-H (OC95, 1:200 dilution), mouse NF-H (mNF-H) (OC59, 1:200 dilution), NF-L (NR-4, 1:1000 dilution), and actin (clone c4, 1:5000 dilution). B, Severe motor dysfunction in a 2-year-old hH;L−/− mouse.
Despite overall reduction in levels of hNF-H protein, mice with the hH;L−/− genotype showed neuropathological dysfunctions similar to those occurring in the hNF-H transgenic mice (Côté et al., 1993). Both hNF-H transgenic mice and hH;L−/− mice developed normally and had no overt phenotypes within their first month of life. After this period, they progressively developed tremors and muscle weakness culminating in a severe impairment of hind limb functions at ∼2 years of age (Fig. 1B).
Nonfilamentous NF-H inclusions in the absence of NF-L
Light microscopy of thin sections from the lumbar spinal cord stained with toluidine blue did not reveal inclusions in motor neurons of adult normal mice (Fig.2A). However, small inclusions were observed in perikarya of some motor neurons from NF-L−/− mice (Fig. 2B). Large inclusions filling nearly all of the perikarya were observed in most spinal motor neurons of both hNF-H transgenic and hH;L−/− mice (Fig.2C,D). These inclusions were formed before 3 weeks of age and persisted throughout life. Except for their smaller size, the inclusions of hH;L−/− mice did not differ from those of hNF-H transgenic mice when observed at light microscopy. Immunohistochemical staining with the OC95 antibody confirmed that the inclusions in hNF-H transgenic mice and hH;L−/− mice contained the hNF-H protein (Fig. 2E). These inclusions were also detected with the Smi-31 and Smi-32 antibodies, indicating the presence of both hyperphosphorylated (Smi-31) and hypophosphorylated (Smi-32) NF-H epitopes (Fig. 2E; see Fig.4A). Electron microscopy revealed that the inclusions in hNF-H transgenic mice consisted of disorganized aggregates of 10 nm filaments (Fig. 3A). In contrast, the inclusions in hH;L−/− mice were devoid of any filaments (Fig. 3B,C). Despite this lack of filaments, the inclusions in hH;L−/− mice excluded other cytoplasmic organelles, such as mitochondria and microtubules, suggesting that the inclusions are not composed of freely diffusing proteins (Fig.3C).
Fig. 2.
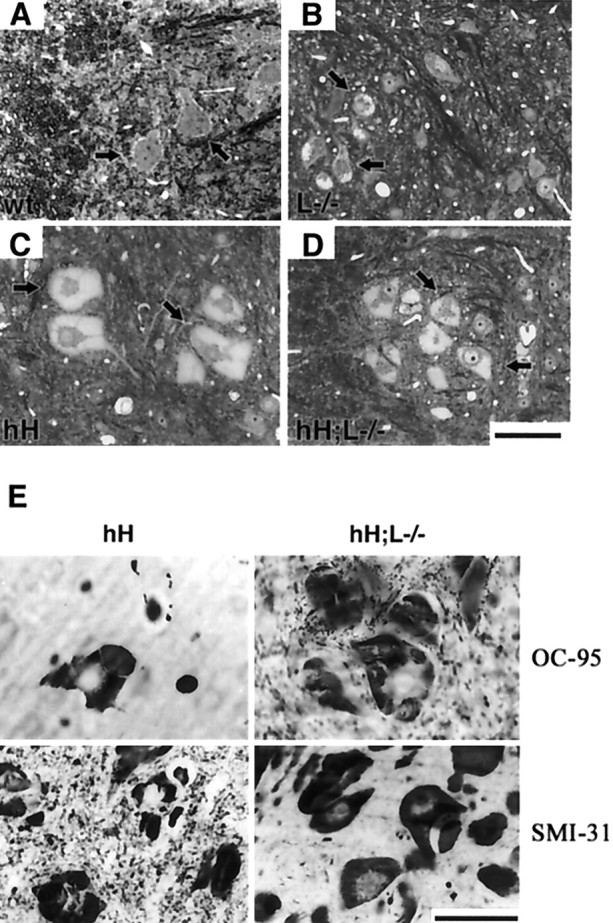
Perikaryal inclusions in the absence of NF-L (A–D). Spinal cord sections stained with toluidine blue show the presence of hyaline inclusions in the perikarya of motor neurons (black arrowheads) from NF-L−/− (B), hNF-H transgenic (C), and hH;L−/− (D) mice at 2 years of age. Such inclusions were absent in motor neurons of normal (wt) mice (A). E, Immunohistochemistry revealing the presence of hNF-H (OC-95) and phosphorylated NF-H epitopes (SMI-31) in inclusions of both hH and hH;L−/− mice. The scale bar in D applies toA–D. Scale bars, 50 μm.
Fig. 4.

The inclusions in hH;L−/− mice are resistant to Triton extraction. Cryostat sections from unperfused 3-month-old hNF-H transgenic and hH;L−/− mice were incubated for 15 min in either PBS or 1% Triton buffer. A double immunofluorescence analysis demonstrates that the NF-H inclusions stained by Smi-32 immunoreactivity (arrowheads) are not extracted by this treatment, whereas the Golgi apparatus membrane-associated protein MG160 is extracted. Scale bar, 50 μm. B, Western blot of soluble (S) and insoluble (I) fractions (2.5 μg) of spinal cord homogenates from 3-month-old hNF-H transgenic (hH) and hH;L−/− mice. Protein detection was performed using the following antibodies: OC95 (1:200), hNF-H; OC59 (1:200), mouse NF-H and NF-M; NN-18 (1:1000), NF-L; NR-4 (1:1000), peripherin (Per); MAB1527 (1:1000), actin (Act); and clone c4 (1:5000) and α-internexin (α-Int) AB1515 (1:2000).
Fig. 3.
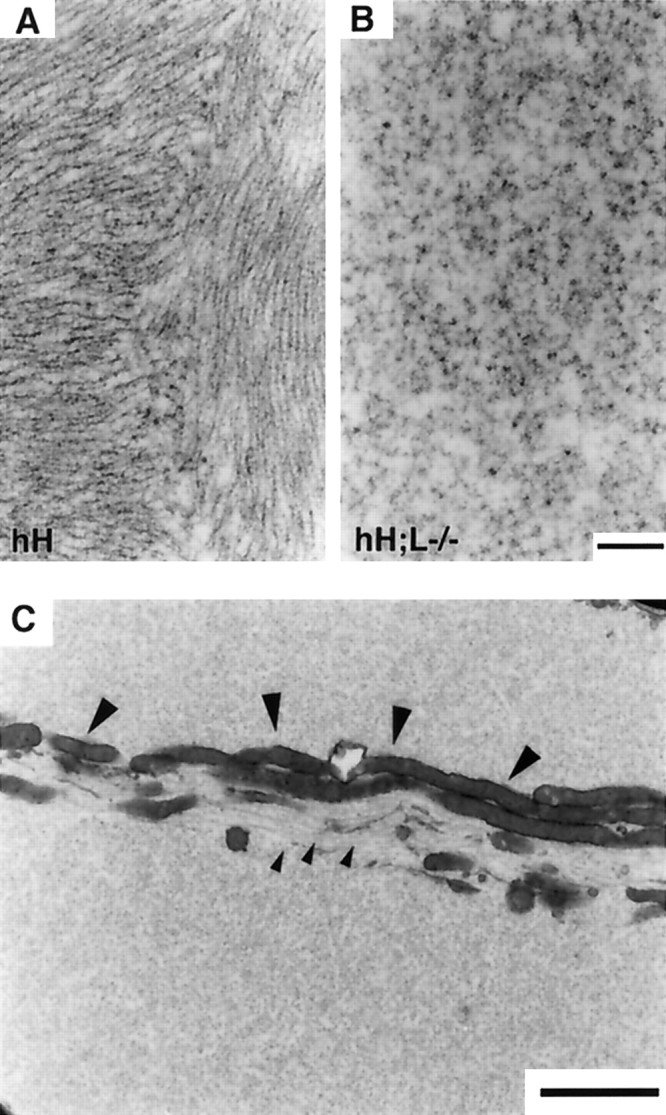
Absence of IF structures in perikaryal inclusions of hH;L−/− mice. A, B, Electron micrographs show IF structures in inclusions of hNF-H transgenic mice (A) and their absence in the inclusions of hH;L−/− mice (B). C, Electron micrograph showing the segregation of microtubules (small arrowheads) and mitochondria (large arrowheads) in an inclusion of hH;L−/− mouse. Scale bars: (in B)A, B, 0.3 μm; C, 2 μm.
The inclusions in hH;L−/− mice are resistant to Triton extraction
To further characterize the interactions of IF proteins in the inclusions of hH;L−/− mice, an in situ Triton solubilization assay was performed on unfixed cryostat sections from the spinal cord. The NF-H protein was detected using the monoclonal antibody Smi-32, whereas a polyclonal antibody against the Golgi apparatus-associated protein MG160 (Croul et al., 1990) was used as a control for protein extraction. The Triton treatment successfully abolished MG160 immunoreactivity, although it failed to remove NF-H inclusions in sections prepared from the hNF-H transgenic and hH;L−/− mice (Fig. 4A). This suggested that some interactions still occur between IF proteins in the inclusions of hH;L−/− mice. Western blots of soluble and insoluble spinal cord extracts from hNF-H transgenic and hH;L−/− mice confirmed that 30% of mouse NF-H and 16% of human NF-H protein existed in an insoluble form in the hH;L−/− mice (Fig. 4B). The other neuronal IF proteins, NF-M, α-internexin, and peripherin, were mostly recovered in the insoluble fractions in hNF-H transgenic and hH;L−/− mice (Fig. 4B).
Assembly of the NF-H rod domain in hH;L−/− mice
Because the first step of IF assembly involves the formation of coiled-coil dimers between the rod domains of IF proteins (for review, see Fuchs and Weber, 1994), we have performed partial α-chymotrypsin digestion to examine whether the rod domain of NF-H is involved in protein–protein interactions in hH;L−/− mice. Potential α-chymotrypsin cleavage sites (Fig.5A) were identified from the published sequence of the mouse (GenBank accession numberP19246) and human (GenBank accession number P12036) NF-H proteins. Multiple potential α-chymotrypsin digestion sites exist in the head and rod domain of both mouse and human NF-H proteins. It has been demonstrated previously that α-chymotrypsin treatment of unassembled NF-H proteins leads to the formation of a single 160 kDa fragment containing the tail domain of NF-H, whereas digestion of assembled NF-H proteins generates additional fragments in the range of 40 kDa corresponding to segments of the head and rod domain of NF-H, which are protected from α-chymotrypsin digestion because of assembly into IF structures (Julien and Mushynski, 1983). Partial α-chymotrypsin digestions were performed on soluble and insoluble fractions of spinal cord homogenates from either the hNF-H transgenic or hH;L−/− mice. Similar results were obtained with mice of these two genotypes (Fig. 5B). Digestion of proteins from the soluble fractions led to the formation of an NF-H fragment of ∼160 kDa detectable by Western blotting with either a polyclonal anti-NF-H antibody or the anti-tail domain antibody Smi-32 (Sternberger and Sternberger, 1983). Thus, this large 160 kDa fragment contained the non-digested tail domain of NF-H proteins. The same large fragment was also obtained after α-chymotrypsin digestion of the insoluble fraction of homogenates. However, staining of Western blots from the insoluble protein digestion products with the anti-NF-H polyclonal antibody revealed additional NF-H immunoreactive fragments in the range of 40–50 kDa. These fragments were not detected by Smi-32 staining, indicating that they are not derived from the tail domain of NF-H and that they correspond to protected fragments from the head and rod domains. These results further suggest that the NF-H rod domain is involved in the formation of insoluble protein aggregates.
Fig. 5.

Partial α-chymotrypsin digestion of insoluble NF-H. A, Potential α-chymotrypsin digestion sites (black arrowheads) along the head and rod domains of both mouse (mNF-H) and human (hNF-H) NF-H proteins. Black boxesindicate the α-helical segments of NF-H rod domain. B, Western blot analysis of the α-chymotrypsin digestion products. Soluble and insoluble fractions from spinal cord homogenates of hNF-H transgenic or hH;L−/− mice were treated with α-chymotrypsin for 0, 8, and 15 min as described in Materials and Methods. The immunodetection of NF-H digestion fragments was performed using an anti-NF-H polyclonal antibody (N-4147, 1:2000 dilution) or the Smi-32 (1:500 dilution) monoclonal antibody, which is specific to unphosphorylated (Lys/Ser/Pro) residues present in the tail domains of both human and mouse NF-H proteins. Digestion of soluble NF-H proteins from both mouse models led to the formation of a single large fragment immunoreactive for both Smi-32 and NF-H polyclonal antibody, whereas digestion of insoluble NF-H proteins led to the formation of additional NF-H fragments (black arrowheads) that were only immunoreactive with the polyclonal antibody. Numbers on the left of gels in B correspond to molecular weight markers in kilodaltons.
NF-H colocalizes with peripherin in inclusion bodies of hH;L−/− mice
We have searched for potential interaction partners of NF-H in inclusions bodies from the hH;L−/− mice. In the absence of NF-L, the NF-H protein can form heterodimers or interact in vivo with α-internexin and peripherin, although the interaction of peripherin with NF-H does not lead to the formation of a normal IF network (Ching and Liem, 1993; Athlan and Mushynski, 1997; Beaulieu et al., 1999a,b). Double immunofluorescence staining was performed using the Smi-32 monoclonal antibody and polyclonal antibodies directed against α-internexin or peripherin. The results demonstrate the presence of peripherin in most inclusions of hH;L−/− mice, whereas α-internexin was only rarely detected in these same inclusions (Fig.6).
Fig. 6.

Colocalization of NF-H with peripherin but not with α-internexin in hH;L−/− mice. The NF-H protein was detected (green) using the mouse monoclonal antibody Smi-32 (1:500). Polyclonal antibodies (red) were used to detect peripherin (Per; AB1530, 1:5000) and α-internexin (α-Inter; AB1515, 1:100). Composite images were obtained by overlaying the green and redimages. Scale bar, 100 μm.
Disparate effects of NF-L deficiency in hNF-H and peripherin transgenic mice
Light microscopy of thin sections from the L5 ventral roots revealed a reduction of axonal caliber for the hNF-H transgenic, NF-L−/−, and hH;L−/− mice (Fig.7A–E). However, the extent of axonal atrophy differs for each genotype. The hNF-H transgenic mice were the least affected by the reduction of axonal caliber, followed by the NF-L−/− and hH;L−/− mice, which exhibited the most severe atrophy of axons (Fig. 7E). Despite robust motor dysfunction and axonal atrophy in hNF-H transgenic and hH;L−/− mice, no massive degeneration of axons was observed in 2-year-old mice of either genotype (Fig. 7A–D). In agreement with our previous observations with NF-L−/− mice (Zhu et al., 1997; Beaulieu et al., 1999a), there was a similar ∼20% loss of L5 ventral roots axons in 2-year-old mice from the NF-L−/− and hH;L−/− genotypes (Fig.7F). In contrast, 6-month-old Per;L−/− mice suffered a 46% loss of their motor neurons, whereas the peripherin transgenic mice having normal NF-L levels did not suffer axonal loss at this age (Fig. 7F). These results show that an NF-L deficiency enhances the detrimental effects of peripherin overexpression but not of hNF-H overexpression.
Fig. 7.
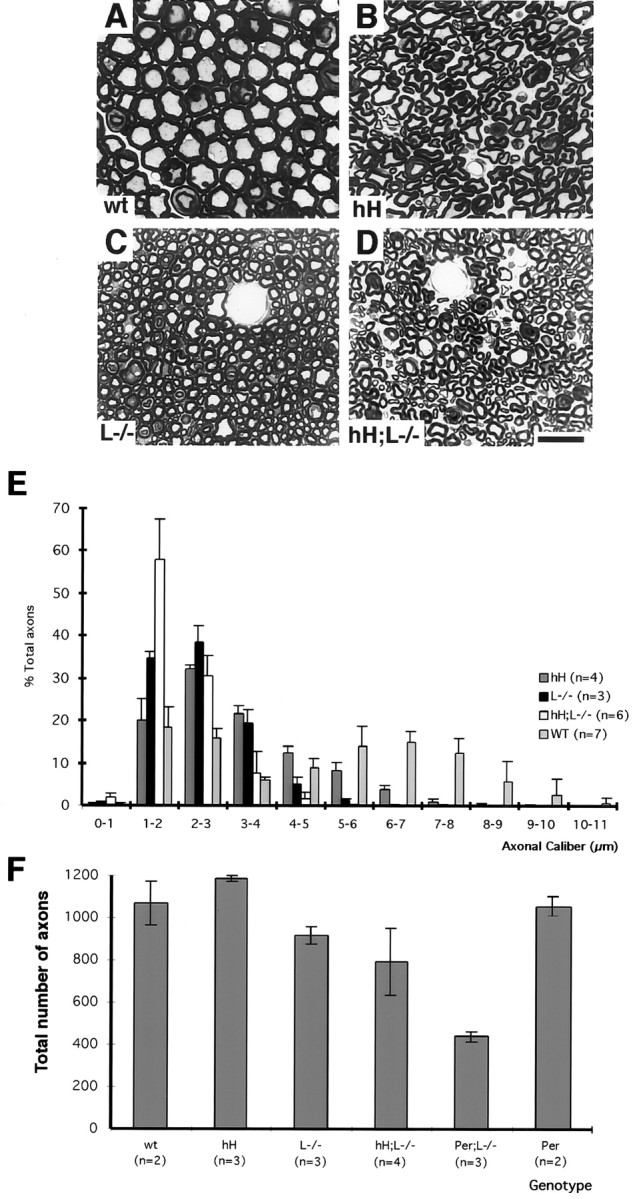
hNF-H overexpression exacerbates axonal atrophy but not axonal loss in NF-L null mice. A–D, Toluidine blue-stained sections of the L5 ventral roots from control (A), hNF-H transgenic (B), NF-L−/− (C), and hH;L−/− (D) mice. Note that little axonal degeneration occurred in the hNF-H transgenic and hH;L−/− mice. E, Caliber distribution of L5 ventral root axons in normal (WT), hNF-H transgenic (hH), NF-L−/− (L−/−), and hH;L−/− mice. F, Average number of axons in L5 ventral roots of mice. These counts were performed on 2-year-old normal (wt), hNF-H transgenic (hH), NF-L−/− (L−/−), and hH;L−/− mice and on 6-month-old Per;L−/− and peripherin transgenic mice (Per). Scale bar, 5 μm. Error bars indicate SD.
Axonal but not perikaryal IF inclusions correlate with motor neuron death
Both the hH;L−/− and Per;L−/− mice develop IF inclusions containing peripherin and NF-H (Fig. 6) (Beaulieu et al., 1999a). However, only the Per;L−/− mice suffer motor neuron death. Immunohistochemistry and light microscopy of spinal cord sections from 3-month-old hH;L−/− and Per;L−/− mice revealed major differences in the size and distribution of peripherin-containing inclusions between mice of these two genotypes (Fig.8B–E). Motor neurons of hH;L−/− mice were characterized by the formation of a single large accumulation that filled most of the perikaryon with occasional filling of the proximal axons. In contrast, motor neurons of Per;L−/− mice contained multiple small inclusions that were found in both perikarya and axons. Western blots of soluble and insoluble protein preparations from the spinal cord of 3-month-old NF-L−/−, hH;L−/− and Per;L−/− mice revealed that peripherin was totally insoluble in these three types of mice. However, peripherin was much more abundant in the Per;L−/− mice (Fig. 8A).
Fig. 8.
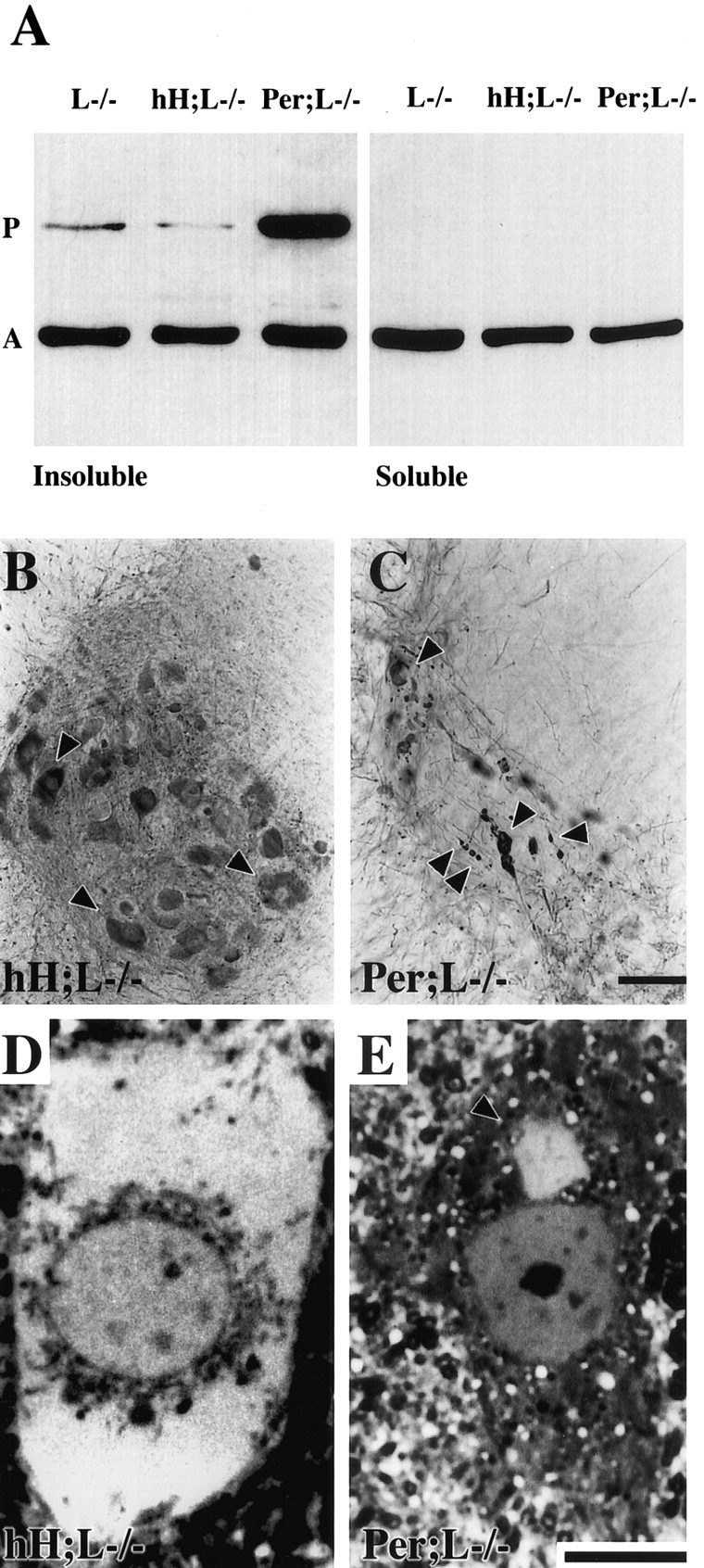
Peripherin detection in protein aggregates of hH;L−/− and Per;L−/− mice. A, Western blot of soluble and insoluble fractions of spinal cord homogenates from NF-L−/− (L−/−), hH;L−/−, and Per;L−/− mice. The insoluble protein fraction (2.5 μ g) and an equal volume of the soluble protein fraction were loaded on gel. Protein detection was performed using the following antibodies: peripherin (P); MAB1527 (1:1000), actin (A); and clone c4 (1:5000). B,C, Immunohistochemical detection of peripherin in the spinal cord with a polyclonal antibody (AB1530, 1:5,000) showing the difference in the size and distribution of peripherin-containing inclusions (arrowheads) between hH;L−/− (B) and Per;L−/− (C) mice. D, E, Thin sections stained with toluidine blue showing a large hyaline inclusion filling the perikaryon of a motor neuron in an hH;L−/− mouse (D) and a typical small hyaline inclusion (arrowhead) in the perikaryon of a motor neuron in a Per;L−/− mouse (E). Scale bars: C, 100 μm;E, 10 μm.
DISCUSSION
The presence of IF protein aggregates in motor neurons is a common pathological finding in human ALS (Carpenter, 1968) (for review, seeChou, 1995), but it appears paradoxical that the levels of NF-L mRNA are reduced in this disease (Bergeron et al., 1994). The results presented here demonstrate that the formation of IF protein aggregates in motor neurons may occur in a context of NF-L deficiency. However, in the absence of NF-L, different types of IF protein aggregates with distinct properties were formed by the overexpression of different IF transgenes.
The overexpression of hNF-H proteins in NF-L null mice led to formation of protein aggregates in the perikarya of motor neurons. These perikaryal aggregates in hH;L−/− mice were not as large as those occurring in hNF-H transgenic mice having normal NF-L levels, and they were not composed of 10 nm filaments. The lack of NF-L caused a reduction in total NF-H content. The decreased NF-H levels are likely the result of increased proteolytic degradation of unassembled NF-H proteins in the absence of NF structures (Zhu et al., 1997). Yet, a fraction of NF-H proteins escaped degradation and formed insoluble protein aggregates in the hH;L−/− mice. Although IF structures were not detected in these aggregates, our results suggest that the NF-H protein was present in the form of oligomeric structures. Thus, the NF-H rod domain in the insoluble protein fraction was protected from partial digestion by α-chymotrypsin (Fig. 5), indicating the formation of coiled-coil structures involving the rod domain of NF-H. Moreover, the colocalization of peripherin with NF-H in the protein aggregates of hH;L−/− mice suggests the existence of oligomeric structures made up of NF-H together with peripherin. This view is in agreement with reports that peripherin can form heterodimers with NF-H (Athlan et al., 1997) and that NF-H and NF-M can prevent the formation of a normal peripherin IF network in transfected cultured cells and transgenic mice (Beaulieu et al., 1999a,b).
The overexpression of peripherin in NF-L null mice also resulted in the development of IF protein aggregates. However, the aggregates in Per;L−/− mice are composed of disorganized 10 nm filaments, unlike aggregates in hH;L−/− mice that are nonfilamentous. The different types of IF protein inclusions and their distinct localization in these two mouse models are basically the result of different ratios of peripherin to NF-H proteins. At a low peripherin-to-NF-H ratio occurring in the hH;L−/− mice, the assembly of peripherin into IF structures was impeded by excess NF-H, preventing the formation of a 10 nm filament network. Such destabilization of peripherin by NF-H has been observed previously in SW13vim(−) cells cotransfected with peripherin and NF-H expression vectors (Beaulieu et al., 1999b). On the other hand, at a high peripherin-to-NF-H ratio as in the Per;L−/− mice, peripherin proteins were able to form disorganized filaments with NF-H and move into motor axons (Beaulieu et al., 1999a).
The mouse models described here provide good examples of distinct phenotypes produced by different types of IF protein aggregates. The small IF aggregates in the Per;L−/− mice are very toxic and provoke progressive motor neuron death. In contrast, the large perikaryal swellings in motor neurons of hNF-H;L+/+ or hNF-H;L−/− transgenic mice are relatively well tolerated. These hNF-H transgenic mice develop motor dysfunction, but they do not suffer motor neuron death. Alterations in the electrophysiological properties of peripheral nerves detected in the hNF-H transgenic mice are likely responsible for such motor dysfunction (Kriz et al., 2000).
The toxic effects of IF aggregates in the Per;L−/− mice, which have morphological features similar to inclusion bodies in human ALS, may be related in part to the sequestering of organelles such as mitochondria (Beaulieu et al., 1999a). On the contrary, cellular organelles were segregated from the perikaryal protein aggregates of the hNF-H;L−/− mice (Fig. 3). It is also possible that the location of IF protein aggregates in the axon constitutes an important determinant of toxicity. As shown in Table 1, which summarizes the various transgenic mouse models with IF abnormalities, there is one example of partial motor neuron loss associated with massive, strictly perikaryal, IF inclusions. The NF-H/β-galactosidase transgene resulted in large perikaryal NF accumulations, a scarcity of axonal IF structures comparable with the one observed in NF-L−/− mice, and in a 20% motor neuron loss (Eyer and Peterson, 1994; Tu et al., 1997). However, it is noteworthy that the massive death of spinal motor neurons occurred only in mouse models with multiple IF inclusions in motor axons, i.e., in mice expressing a mutant NF-L protein (Lee et al., 1994) or peripherin transgenes (Beaulieu et al., 1999a). Such axonal IF inclusions could block axonal transport, a mechanism of neurodegeneration referred as the “strangulation model” (for review, see Julien, 1997; Cleveland, 1999).
Table 1.
Effects of intermediate filament alterations on spinal motor neurons
| Transgene | Perikaryal IF inclusions | Axonal IF inclusions | Scarcity of axonal IF structures | Overt motor dysfunction | Motor neuron loss | Reference |
|---|---|---|---|---|---|---|
| Mouse NF-H | Massive | Some | No | No | No | Marszalek et al., 1996 |
| hNF-H | Massive | Some | No | Yes | No | Côté et al 1993; this paper |
| NF-H/β-galactosidase | Massive | No | Yes | No1-a | 20% | Eyer and Peterson, 1994; Tu et al., 1997 |
| MSV-NF-M1-b | No | No | No | No | No | Wong et al., 1995 |
| MSV-NF-M mutant | Massive | Some | No | No | No | Wong et al., 1995 |
| hNF-L | No | No | No | No | No | Julien et al., 1987 |
| MSV-NF-L1-b | Massive | Some | No | Yes | No | Xu et al., 1993 |
| MSV-NF-L mutant | Massive | Multiple | No | Yes | 40% (2 months)1-c | Lee et al., 1994;Houseweart and Cleveland, 1999 |
| α-Internexin | No | No | No | No1-a | No | Ching et al., 1999 |
| Thy1-Peripherin1-b | No | Yes | No | No at 14 months | No at 14 months/35% loss at 28 months | Beaulieu et al., 1999a |
| Peripherin | No | Yes | No | No at 14 months | No at 6 months/35% loss at 28 months | Beaulieu et al., 1999a; this paper |
| Thy1-Peripherin; L−/−1-b | Small | Multiple | Yes | Yes | 47% at 6 months/59% at 14 months | Beaulieu et al., 1999a |
| Per;L−/− | Small | Multiple | Yes | Yes | 46% at 6 months | Beaulieu et al., 1999a; this paper |
| hNF-H;hNF-L | Few | Few | No | No | No | Meier et al., 1999 |
| hH;L−/− | Massive | Some | Yes | Yes | 20% (developmental) | This paper |
| NF-L−/− | Yes | No | Yes | No | 20% (developmental) | Zhu et al., 1997; this paper |
Although these mice do not develop lower motor neuron dysfunction, they do suffer motor dysfunction at the level of the cerebellum during aging.
MSV and Thy-1 designate the human Thy-1 gene promoter and the murine sarcoma virus long-term repeat promoter.
Motor neuron loss stops after this period because of a lack of transgene expression.
The results presented here demonstrate that an NF-L deficiency may influence differently the formation and morphology of IF protein aggregates in neurons depending on the levels of other IF proteins. The absence of NF-L reduced the size of perikaryal swellings in transgenic mice overproducing human NF-H proteins (Fig. 2), whereas it precipitated the formation of IF aggregates and motor neuron death in mice overexpressing peripherin (Beaulieu et al., 1999a). Distinct neuronal cell populations may express various levels of each neuronal IF protein and therefore may develop IF protein aggregates with different characteristics in a situation of NF-L deficiency. Our findings may be of importance for neurodegenerative diseases because decreased levels of NF-L mRNA are associated with ALS (Bergeron et al., 1994), Alzheimer's disease (Crapper McLachlan et al., 1988), and to a lower extent, aging (Parhad et al., 1995).
Footnotes
This research was supported by the Medical Research Council of Canada (MRC), the Amyotrophic Lateral Sclerosis (ALS) Association (USA), and the ALS Society of Canada. J-M.B. is a recipient of studentships from the Natural Science and Engineering Research Council of Canada, Les Fonds de la Recherche en Santé du Québec, and the McDonald Stewart Foundation. J-P.J. has an MRC Senior Scholarship. The technical assistance of Pascale Hince, Daniel Houle, and Gaetan Gagnon is gratefully acknowledged.
Correspondence should be addressed to Dr. Jean-Pierre Julien, The Montreal General Hospital Research Institute, 1650 Cedar Avenue, Montreal, Quebec, Canada, H3G 1A4. E-mail: mdju@musica.mcgill.ca.
Dr. Jacomy's present address: Institut National de la Recherche Scientifique–Institut Armand-Frappier, Laval, Quebec, Canada H7V 1B7.
REFERENCES
- 1.Athlan ES, Mushynski WE. Heterodimeric associations between neuronal intermediate filament proteins. J Biol Chem. 1997;272:31073–31078. doi: 10.1074/jbc.272.49.31073. [DOI] [PubMed] [Google Scholar]
- 2.Beaulieu JM, Nguyen MD, Julien JP. Late onset death of motor neurons in mice overexpressing wild-type peripherin. J Cell Biol. 1999a;147:531–544. doi: 10.1083/jcb.147.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaulieu JM, Robertson J, Julien JP. Interactions between peripherin and neurofilaments in cultured cells: disruption of peripherin assembly by the NF-M and NF-H subunits. Biochem Cell Biol. 1999b;77:41–45. [PubMed] [Google Scholar]
- 4.Bergeron C, Beric-Maskarel K, Muntasser S, Weyer L, Somerville MJ, Percy ME. Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J Neuropathol Exp Neurol. 1994;53:221–230. doi: 10.1097/00005072-199405000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology. 1968;18:841–851. doi: 10.1212/wnl.18.9.841. [DOI] [PubMed] [Google Scholar]
- 6.Ching GY, Liem RKH. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ching GY, Chien CL, Flores R, Liem RKH. Overexpression of a-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. J Neurosci. 1999;19:2974–2986. doi: 10.1523/JNEUROSCI.19-08-02974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou SM. Pathology of motor system disorder. In: Leigh PN, Swash M, editors. Motor neuron disease: biology and management. Springer; London: 1995. pp. 53–92. [Google Scholar]
- 9.Cleveland DW. From Charcot to SOD1: mechanisms of selective motor neuron death in ALS. Neuron. 1999;24:515–520. doi: 10.1016/s0896-6273(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 10.Corbo M, Hays AP. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J Neuropathol Exp Neurol. 1992;51:531–537. doi: 10.1097/00005072-199209000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Côté F, Collard JF, Julien JP. Progressive neuropathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 12.Crapper McLachlan DR, Lukiw WJ, Wong L, Bergeron C, Bech-Hansen NT. Selective messenger RNA reduction in Alzheimer's disease. Mol Brain Res. 1988;3:255–262. doi: 10.1016/0169-328x(88)90048-4. [DOI] [PubMed] [Google Scholar]
- 13.Croul S, Mezitis SG, Stieber A, Chen YJ, Gonatas JO, Goud B, Gonatas NK. Immunocytochemical visualization of the Golgi apparatus in several species, including human, and tissues with an antiserum against MG-160, a sialoglycoprotein of rat Golgi apparatus. Histochem Cytochem. 1990;38:957–963. doi: 10.1177/38.7.2355176. [DOI] [PubMed] [Google Scholar]
- 14.Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-β-galactosidase fusion protein. Neuron. 1994;12:389–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman PN, Lasek RJ. The slow component of axonal transport. J Cell Biol. 1975;642:316–322. doi: 10.1083/jcb.66.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houseweart MK, Cleveland DW. Bcl-2 overexpression does not protect neurons from mutant neurofilament-mediated motor neuron degeneration. J Neurosci. 1999;19:6446–6456. doi: 10.1523/JNEUROSCI.19-15-06446.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacomy H, Zhu Q, Couillard-Després S, Beaulieu JM, Julien JP. Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J Neurochem. 1999;73:972–984. doi: 10.1046/j.1471-4159.1999.0730972.x. [DOI] [PubMed] [Google Scholar]
- 19.Julien J. Neurofilaments and motor neuron disease. Trends Cell Biol. 1997;7:243–249. doi: 10.1016/S0962-8924(97)01049-0. [DOI] [PubMed] [Google Scholar]
- 20.Julien JP, Mushynski WE. The distribution of phosphorylation sites among identified proteolytic fragments of mammalian neurofilaments. J Biol Chem. 1983;258:4019–4025. [PubMed] [Google Scholar]
- 21.Julien JP, Trerjakoff I, Beaudet L, Peterson A. Expression and assembly of a human neurofilament protein in transgenic mice provide a novel neuronal marking system. Genes Dev. 1987;1:1085–1095. doi: 10.1101/gad.1.10.1085. [DOI] [PubMed] [Google Scholar]
- 22.Kriz J, Meier J, Julien JP, Padjen AL (2000) Altered ionic conductances in axons of transgenic mouse expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Exp Neurol, in press. [DOI] [PubMed]
- 23.Lee MK, Xu Z, Wong PC, Cleveland DW. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee MK, Marszalek JR, Cleveland DW. A mutant neurofilament subunit causes massive, selective motor neuron death: Implication for the pathogenesis of human motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 25.Levavasseur F, Zhu Q, Julien JP. No requirement of α-internexin for nervous system development and for radial growth of axons. Mol Brain Res. 1999;69:104–112. doi: 10.1016/s0169-328x(99)00104-7. [DOI] [PubMed] [Google Scholar]
- 26.Marszalek JR, Williamson TL, Lee MK, Xu Z, Hoffman PN, Becher MW, Crawford TO, Cleveland DW. Neurofilament subunit NF-H modulates axonal diameter by selectively slowing neurofilament transport. J Cell Biol. 1996;135:711–724. doi: 10.1083/jcb.135.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier J, Couillard Despres S, Jacomy H, Gravel C, Julien JP. Extra neurofilament NF-L subunits rescue motor neuron disease caused by overexpression of the human NF-H gene in mice. J Neuropathol Exp Neurol. 1999;58:1099–1110. [PubMed] [Google Scholar]
- 28.Migheli A, Pezzulo T, Attanasio A, Schiffer D. Peripherin immunoreactive structures in amyotrophic lateral sclerosis. Lab Invest. 1993;68:185–191. [PubMed] [Google Scholar]
- 29.Pachter JS, Liem RK. alpha-Internexin, a 66-kD intermediate filament-binding protein from mammalian central nervous tissues. J Cell Biol. 1985;101:1316–1322. doi: 10.1083/jcb.101.4.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parhad IM, Scott JN, Cellars LA, Bains JS, Krekoski CA, Clark AW. Axonal atrophy in aging is associated with a decline in neurofilament gene expression. J Neurosci Res. 1995;41:355–366. doi: 10.1002/jnr.490410308. [DOI] [PubMed] [Google Scholar]
- 31.Parysek LM, Goldman RD. Distribution of a novel 57 kDa intermediate filament (IF) protein in the nervous system. J Neurosci. 1988;8:555–563. doi: 10.1523/JNEUROSCI.08-02-00555.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parysek LM, McReynolds MA, Goldman RD, Ley CA. Some neural intermediate filaments contain both peripherin and the neurofilament proteins. J Neurosci Res. 1991;30:80–91. doi: 10.1002/jnr.490300110. [DOI] [PubMed] [Google Scholar]
- 33.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Percak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH, Jr, Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee M K, Valentine JS, Bredesen DE. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 34.Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu PT, Robinson KA, de Snoo F, Eyer J, Peterson A, Lee VMY, Trojanowski JQ. Selective degeneration of Purkinje cells with lewy body-like inclusions in aged NFHLACZ transgenic mice. J Neurosci. 1997;17:1064–1074. doi: 10.1523/JNEUROSCI.17-03-01064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong PC, Marszalek J, Crawford TO, Xu Z, Hsieh ST, Griffin JW, Cleveland DW. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol. 1995;130:1413–1422. doi: 10.1083/jcb.130.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Z, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 40.Zhu Q, Couillard-Despres S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]