

Abstract
Many cases of early-onset inherited Alzheimer's disease (AD) are caused by mutations in the presenilin-1 (PS1) gene. Studies of cultured neural cells suggest that PS1 mutations result in perturbed cellular calcium homeostasis and may thereby render neurons vulnerable to apoptosis. In light of evidence that metabolic impairment plays a role in AD, that cerebral ischemia may be a risk factor for AD, and that individuals with AD have increased morbidity and mortality after stroke, we examined the impact of a PS1 mutation on neuronal vulnerability to ischemic injury. We report that the extent of brain injury after focal cerebral ischemia reperfusion is increased, and behavioral outcome is worsened, in PS1 mutant knock-in mice compared to wild-type mice. Cultured cortical neurons from PS1 mutant mice exhibit increased vulnerability to glucose deprivation and chemical hypoxia compared to their wild-type counterparts. Calcium imaging studies demonstrated enhanced elevation of intracellular calcium levels after glucose deprivation and chemical hypoxia in neurons from PS1 mutant mice. Agents that block calcium release from IP3- and ryanodine-sensitive stores (xestospongin and dantrolene, respectively) protected against the endangering action of the PS1 mutation. Our data suggest that presenilin mutations may promote neuronal degeneration in AD by increasing the sensitivity of neurons to age-related ischemia-like conditions. The data further suggest that drugs that stabilize endoplasmic reticulum calcium homeostasis may prove effective in suppressing the neurodegenerative process in AD patients.
Keywords: Alzheimer's disease, dantrolene, endoplasmic reticulum, knock-in, stroke, transgenic
Stroke and Alzheimer's disease (AD) are two age-related neurological disorders that are leading causes of death and disability worldwide (Small et al., 1997; Elkind and Sacco, 1998). Mechanisms of neuronal injury and death after stroke have been well characterized and involve energy depletion, overactivation of glutamate receptors, excessive calcium influx, and oxidative stress (for review, see Choi, 1996; Mattson and Mark, 1996). In the case of AD, the mechanisms of neuronal degeneration are less well understood but appear to include metabolic compromise, oxidative stress, and disruption of cellular calcium homeostasis (for review, see Mattson, 1997). Therefore, although the time course of the disease process may differ in stroke and AD, the neuronal cell death mechanism in each disorder appears to involve energy deficit, oxidative stress, and perturbed calcium homeostasis. In support of the latter statement, an overlapping set of neurons (e.g., hippocampal CA1 neurons and cortical pyramidal neurons) are vulnerable in both stroke and AD (Katzman, 1986;Ginsberg and Butso, 1989). In addition, history of ischemic stroke increases risk for AD (Kokmen et al., 1996), and recent studies have shown that the extent of AD pathology (plaques and tangles) is increased in AD patients with coexisting evidence of cerebral ischemia (Nagy et al., 1997; Snowdon et al., 1997).
Whereas most cases of AD are not caused by a specific genetic defect and have a late age of onset, some cases are characterized by an early age of onset and a dominant inheritance pattern. Mutations in the gene encoding presenilin-1 (PS1) on chromosome 14 are responsible for many such cases of inherited AD (for review, see Hardy, 1997; Mattson et al., 1998). PS1 is an integral membrane protein that is expressed in neurons throughout the brain wherein it is localized primarily in the endoplasmic reticulum (ER). Two pathogenic mechanisms for PS1 mutations have been proposed. One mechanism involves altered proteolytic processing of the amyloid precursor protein (APP), resulting in increased production of neurotoxic forms of amyloid β-peptide and decreased levels of the neuroprotective secreted form of amyloid precursor protein (Mattson et al., 1993a; Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996; Ancolio et al., 1997; Guo et al., 1998). Altered APP processing may result in increased oxidative stress and neuronal apoptosis (Mattson, 1997). A second mechanism involves perturbed calcium regulation, which results in enhanced elevations of intracellular calcium levels under conditions of oxidative and excitotoxic stress (Guo et al., 1996, 1997, 1999b). We recently produced and characterized knock-in mice expressing the M146V PS1 mutation (Guo et al., 1999a). We now report data from studies of these mice showing that PS1 mutations render neurons vulnerable to ischemic injury, by a mechanism linked to calcium release from ER.
MATERIALS AND METHODS
PS1 mutant knock-in mice and protocol for focal cerebral ischemia. The targeting strategy used to generate PS1 mutant knock-in mice is detailed elsewhere (Guo et al., 1999a, 1999b). PS1 mutant knock-in mice and wild-type mice were bred in house and were maintained on the same genetic background (BL6/129Sv). Previous studies have characterized these mice, showing that the knock-in mice express mutant PS1 at normal levels, that they exhibit no overt developmental abnormalities, and that they do exhibit increased levels of Aβ1–42 in brain tissue and increased vulnerability of hippocampal neurons to apoptosis and excitotoxicity (Guo et al., 1999a, 1999b). The protocol for performing focal cerebral ischemia using a middle cerebral artery occlusion (MCAO) reperfusion method was similar to that used in our previous studies (Bruce et al., 1996; Keller et al., 1998), which were adapted from Huang et al. (1994) Briefly, mice were anesthetized with 350 mg/kg chloral hydrate. In all mice, the left femoral artery was cannulated for arterial blood pressure and blood gas determination. Arterial blood samples (50 μl) were analyzed for pH, arterial oxygen pressure, and partial pressure of carbon dioxide using a blood gas/pH analyzer (Corning 178; CIBA-Corning Diagnostics, Medford, MA). Samples were taken immediately before, 10 min after MCA occlusion, and 10 min after the start of reperfusion. Rectal and temporalis muscle temperature was maintained at 37.0 + 0.5°C with a temperature control unit (FHC, Brunswick, ME). The left common carotid artery was exposed, and the occipital, superior thyroidal, maxillary, and lingual branches of the external carotid artery were coagulated; the pterygopalatine artery was ligated. An incision was made in the external carotid artery, and a polylysine-coated suture was advanced into the middle cerebral artery. The thread was left in place for 60 min and then removed to allow reperfusion. Cerebral blood flow was determined in anesthetized animals under resting conditions, 30 min into the ischemia, and 10 min after reperfusion. Measurements were made using a laser Doppler flowmeter (Laserflo BPM; Vasomedic). Two flexible probe tips were secured 2 mm posterior and 3 mm lateral to bregma and 2 mm posterior (peri-infarct region) and 6 mm lateral to bregma on the ischemic hemisphere. After surgery, mice were returned to their cages and given ad libitum access to food and water. Twenty four hours after MCAO, the mice were euthanized by anesthesia overdose, and coronal brain sections (1-mm-thick) were stained with 1% triphenyltetrazolium chloride (TTC). After killing, cerebral infarct sizes were determined by staining 2 mm brain sections with 2,3,5-triphenyltetrazolium chloride (TTC), capturing images of stained sections using a Hitachi KPD50 color digital camera (Medical Video System, Winston-Salem, NC), and quantifying infarct size/section and calculating volume using a Macintosh G3 computer and image analysis software (Scion Image NIH version 1.59, Frederick, MD) as described (Keller et al., 1998). Neurological deficits were assessed 22 hr after ischemia based on a scale from 0 (no deficits) to 3 (severe deficits) as described previously (Huang et al., 1994; Endres et al., 1999).
Data analysis. Differences between the wild-type and PS-1 mutant mice were compared by paired and unpaired two-tailed Student'st test or by ANOVA followed by Scheffe's test (physiological measurements) or Bonferroni test (cerebral blood flow). For neurological assessment, the Mann–Whitney rank sum Utest was used. p values of <0.05 were considered significant.
Neocortical cell culture methods and quantification of neuron survival. Cultures of dissociated neocortical cells were prepared from embryonic day 18 wild-type and homozygous PS-1 m146VKI mouse pups using methods similar to those described previously (Guo et al., 1999a). Briefly, cerebral hemispheres were removed and incubated for 15 min in Ca2+- and Mg2+-free HBSS (Life Technologies, Gaithersburg, MD) containing 0.2% trypsin. Cells were dissociated by trituration and plated into polyethyleneimine-coated plastic or glass-bottomed culture dishes containing Minimum Essential Medium with Earle's salts supplemented with 10% heat-inactivated fetal bovine serum and (in mm): 2l-glutamine, 1 pyruvate, 20 KCl, 10 sodium bicarbonate, and 1 HEPES, pH 7.2. After cell attachment (3–6 hr after plating), the culture medium was replaced with Neurobasal Medium with B27 supplements (Life Technologies). Experiments were performed in 7- to 9-d-old cultures; >90% of the cells in the cultures were neurons, and the remaining cells were astrocytes, as judged by cell morphology and immunostaining with antibodies against neurofilaments, and glial fibrillary acidic protein. Glucose deprivation was performed as described previously (Cheng and Mattson, 1991), and involved replacing the culture maintenance medium with glucose-free Locke's buffer (in mm: NaCl, 154; KCl, 5.6; CaCl2, 2.3; MgCl2, 1.0; NaHCO3, 5; and HEPES, 5 mm; pH 7.2); control cultures were incubated in Locke's buffer containing 3.6 mm glucose. Concentrated stocks of NaCN (Sigma, St. Louis, MO) were prepared in Locke's buffer and were added directly to the culture medium to achieve the final concentration. The method for quantification of neuron survival in cortical cell cultures was described previously (Cheng and Mattson, 1991). Briefly, undamaged neurons in premarked microscope fields were counted before and at indicated time points after exposure to experimental treatments. Neurons with intact neurites and a cell body that was smooth and round or oval in shape were considered viable. Neurons with beaded or fragmented neurites and a cell body that was shrunken and rough in appearance were considered nonviable. Assessments of neuronal survival were performed in a blind manner without knowledge of the source of the cells or their treatment history.
Measurement of intracellular free calcium levels.Intracellular free calcium levels ([Ca2+]i) were quantified by fluorescence ratio imaging of the calcium indicator dye fura-2 using methods described previously (Mattson et al., 1995; Guo et al., 1999a). Briefly, cells were loaded with fura-2 AM (30 min incubation in the presence of 10 μm fura-2) and imaged using a Zeiss AttoFluor system with a 40× oil objective. The average [Ca2+]i in individual neuronal cell bodies was determined from the ratio of the fluorescence emissions obtained using two different excitation wavelengths (334 and 380 nm). The system was calibrated with solutions containing either no Ca2+ or a saturating level of Ca2+ (1 mm) using the formula: [Ca2+]i =Kd[(R −Rmin)/(Rmax−R)](Fo/Fs).
RESULTS
Infarct size is increased after focal cerebral ischemia in PS1 mutant mice
Wild-type (n = 12) and PS1 mutant knock-in mice (n = 12) were subjected to middle cerebral artery occlusion and reperfusion. There were no significant differences in physiological parameters (cerebral blood flow, mean arterial pressure, blood pO2, pCO2, pH, and glucose concentration) between wild-type and PS1 mutant mice at baseline, during ischemia, or after reperfusion (Table1). Mice were killed 24 hr after the onset of reperfusion, and infarct sizes were quantified (see Materials and Methods). Infarct size was significantly increased, by an average of 32%, in PS1 mutant mice compared to wild-type mice (Fig.1A). Behavioral deficits were also significantly enhanced in PS1 mutant mice compared to wild-type mice (Fig. 1B).
Table 1.
Physiological parameters in wild-type and PS1 mutant knock-in mice at baseline, during ischemia, and after reperfusion
| Pre-ischemia | Ischemia | Post-ischemia | |
|---|---|---|---|
| Cerebral blood flow (% baseline) | |||
| Wild-type | 100 ± 4 | 22 ± 5 | 94 ± 5 |
| PS1Mut | 100 ± 5 | 25 ± 4 | 97 ± 4 |
| Blood pressure (mmHg) | |||
| Wild-type | 89.8 ± 9.2 | 88.7 ± 10.1 | 91.5 ± 10.8 |
| PS1Mut | 91.0 ± 9.5 | 89.4 ± 7.8 | 94.2 ± 11.5 |
| Heart rate (beats per minute) | |||
| Wild-type | 417 ± 28 | 425 ± 34 | 452 ± 21 |
| PS1Mut | 424 ± 21 | 433 ± 31 | 461 ± 23 |
| pO2(mmHg) | |||
| Wild-type | 91.6 ± 4.3 | 92.4 ± 6.2 | 94.6 ± 5.0 |
| PS1Mut | 88.4 ± 5.1 | 91.3 ± 4.4 | 92.6 ± 4.5 |
| pCO2(mmHg) | |||
| Wild-type | 47.6 ± 4.5 | 48.3 ± 4.7 | 50.5 ± 3.1 |
| PS1Mut | 49.7 ± 3.6 | 47.5 ± 5.6 | 49.3 ± 4.7 |
| pH | |||
| Wild-type | 7.33 ± 0.05 | 7.35 ± 0.03 | 7.34 ± 0.04 |
| PS1Mut | 7.36 ± 0.04 | 7.34 ± 0.02 | 7.33 ± 0.04 |
Values are the mean and SD of measurements made in 12 wild-type and 12 PS1 mutant knock-in mice. Measurements were made 10 min before occlusion, during ischemia (10 min after occlusion), and 10 min after reperfusion (except for cerebral blood flow, which was measured 30 min after reperfusion).
Fig. 1.

PS1 mutant knock-in mice exhibit increased brain damage and behavioral deficits after focal ischemia-reperfusion. Wild-type and PS1 mutant mice were subjected to middle cerebral artery occlusion for 1 hr. Twenty two hours later neurological deficits were assessed (A), and at the 24 hr time point, the mice were killed, brain sections were stained with TTC, and infarct volume was quantified (B). Values are the mean and SE of analyses performed in 12 wild-type and 12 PS1 mutant mice. *p < 0.001 (infarct volume) andp < 0.01 (neurological deficit) compared to value for wild-type mice (paired t tests).
Cortical neurons from PS1 mutant mice exhibit increased vulnerability to glucose deprivation and chemical hypoxia
To more directly examine the vulnerability of neurons from PS1 mutant mice to ischemia-related insults and to ascertain the underlying mechanisms, we established cortical cell cultures from wild-type and PS1 mutant mice. Cultures were exposed to glucose deprivation and NaCN (chemical hypoxia), two insults relevant to focal ischemic brain injury that have previously been shown to damage and kill cultured cortical and hippocampal neurons by a mechanism involving energy depletion, overactivation of glutamate receptors, and disruption of cellular calcium homeostasis (Rothman, 1984; Cheng and Mattson, 1991; Goldberg and Choi, 1993; Mattson et al., 1993b). Glucose deprivation resulted in a progressive decrease in neuronal survival in cultures from wild-type mice during a 48 hr exposure period (Fig.2A). The extent of neuronal loss was significantly greater in cultures from PS1 mutant mice at each time point, with the rate of neuronal death during the first 24 hr of glucose deprivation being markedly accelerated. Neuronal death induced by NaCN was also significantly enhanced at each post-insult time point in cultures from PS1 mutant mice compared to cultures from wild-type mice (Fig. 2A). The concentration–effect curve for NaCN-induced neuronal death was shifted to the left such that more neurons from PS1 mutant mice were killed at each NaCN concentration (0.5, 1, and 2 mm) examined (Fig. 2B). In an additional experiment, we quantified Trypan blue-negative neurons in cultures of wild-type and PS1 mutant cells 24 hr after glucose deprivation or exposure to 5 mm NaCN (Fig. 2C). The latter results confirm that neurons expressing mutant PS1 exhibit increased vulnerability to cell death induced by glucose deprivation and chemical hypoxia.
Fig. 2.
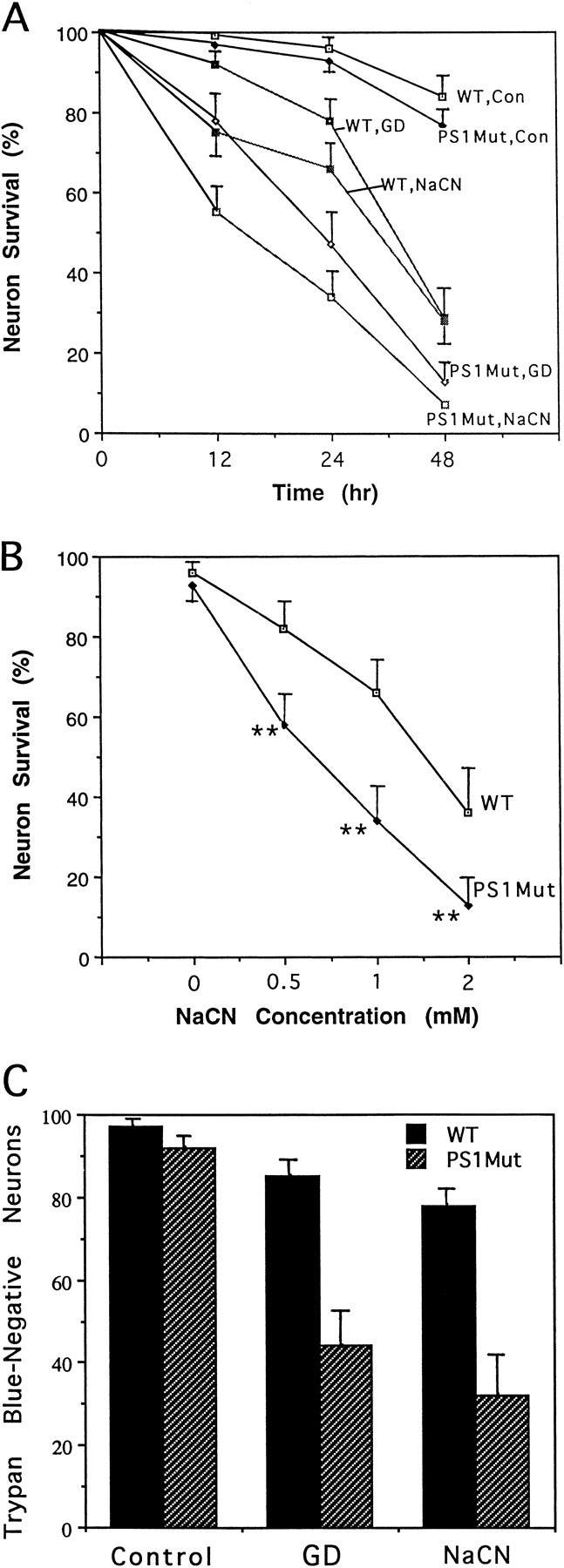
Cortical neurons from PS1 mutant mice exhibit increased vulnerability to cell death induced by glucose deprivation and chemical hypoxia. A, Cultures from wild-type and PS1 mutant mice were exposed to saline (control), glucose deprivation (GD), or 1 mm NaCN, and neuronal survival was quantified at the indicated time points. Values are the mean and SD of determinations made in four to six cultures. Values for neurons from PS1 mutant mice were significantly different than values for wild-type mice at each time point (GD: 12 hr, p< 0.01; 24 hr, p < 0.001; 48 hr,p < 0.01; NaCN: 12 hr, p < 0.01; 24 hr, p < 0.001; 48 hr,p < 0.001); ANOVA with Scheffe's post hoc tests. B, Cultures from wild-type and PS1 mutant mice were exposed for 24 hr to saline (0) or the indicated concentrations of NaCN, and neuronal survival was quantified. Values are the mean and SD of determinations made in four to six cultures. **p < 0.01 compared to corresponding value for cultures from wild-type mice (ANOVA with Scheffe's post hoc tests). C, Cultures from wild-type and PS1 mutant mice were exposed for 24 hr to saline, glucose deprivation (GD), or 5 mm NaCN. The percentage of Trypan blue-negative neurons in each culture was determined. Values are the mean and SD of determinations made in four cultures. The values for cultures from PS1Mut mice exposed to GD or NaCN were significantly less than the corresponding value for cultures from wild-type mice (p < 0.01).
Perturbed calcium homeostasis contributes to increased vulnerability of neurons expressing mutant PS1 to ischemic injury
Previous studies of cultured PC12 cells overexpressing mutant PS1 suggested that PS1 mutations perturb endoplasmic reticulum calcium homeostasis such that calcium release is enhanced after stimulation of IP3 receptors and ryanodine receptors (Guo et al., 1996, 1997). Because loss of cellular calcium homeostasis is believed to contribute to ischemic neuronal injury (Choi, 1996; Mattson and Mark, 1996), we measured intracellular free calcium levels ([Ca2+]i) after exposure of cortical neurons to glucose deprivation and chemical hypoxia in cultures from wild-type and PS1 mutant knock-in mice. Basal levels of [Ca2+]iwere similar in cortical neurons from wild-type mice (79 ± 4 nm) and PS1 mutant mice (89 ± 5 nm). After glucose deprivation there was a progressive increase in the average [Ca2+]i in neurons from both wild-type and PS1 mutant mice, but the magnitude of the increase was significantly greater in neurons from PS1 mutant mice at each time point (6, 12, and 24 hr) examined (Fig.3A). The [Ca2+]i increase after exposure of neurons to NaCN was also significantly greater in neurons from PS1 mutant mice compared to neurons from wild-type mice (Fig. 3B). Pretreatment of cultures with xestospongin, a specific inhibitor of calcium release from IP3-sensitive ER stores (Gafni et al., 1997), significantly attenuated cell death induced by glucose deprivation (GD) and chemical hypoxia in both wild-type and PS1 mutant neurons (Fig. 4). Pretreatment of cultures with dantrolene, an inhibitor of calcium release from ryanodine-sensitive ER stores (Usachev et al., 1993; Tasker et al., 1998), also markedly attenuated cell death induced by GD and chemical hypoxia in both wild-type and PS1 mutant neurons (Figs. 4,5). Xestospongin and dantrolene rescued up to 80% of neurons in cultures from PS1 mutant mice, indicating that calcium release from ER stores plays a critical role in the neuronal death that occurs after exposure to these ischemia-related insults.
Fig. 3.

PS1 mutation exacerbates disruption of calcium homeostasis after glucose deprivation and chemical hypoxia in cultured cortical neurons. A, Cultures from wild-type (WT) and PS1 mutant mice were left untreated (basal) or were subjected to glucose deprivation (GD) for the indicated time points. The [Ca2+]i was then measured in neurons by imaging of the calcium indicator dye fura-2. Measurements were made in 15–22 neurons per culture, and values are the mean and SD of data obtained from four separate cultures per condition. **p < 0.01 compared to corresponding value for neurons from WT mice. B, Cultures from wild-type (WT) and PS1 mutant mice were left untreated (basal) or were exposed to 1 mm NaCN for the indicated time points. The [Ca2+]i was then measured in neurons by imaging of the calcium indicator dye fura-2. Measurements were made in 12–20 neurons per culture, and values are the mean and SD of data obtained from four separate cultures per condition. *p < 0.05; **p < 0.01 compared to corresponding value for neurons from wild-type mice.
Fig. 4.
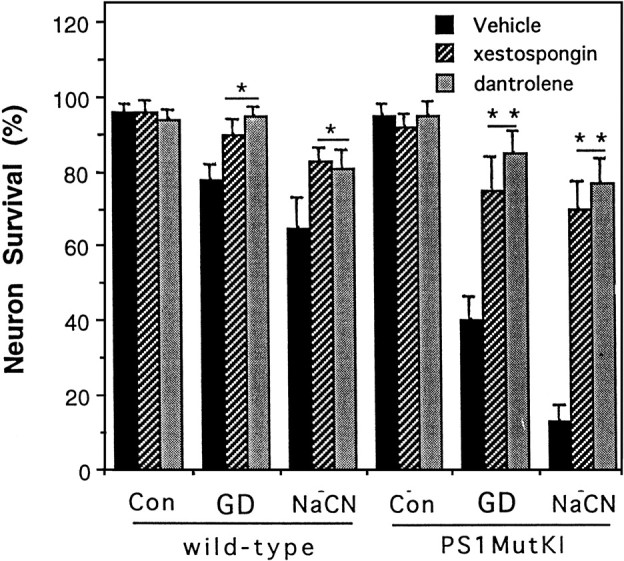
Evidence that calcium release from endoplasmic reticulum is necessary for the cell death-enhancing effect of mutant PS1 in cortical neurons. Cultures were pretreated for 1 hr with 0.2% dimethylsulfoxide (vehicle), 10 μm dantrolene, or 1 μm xestospongin. Cultures were then exposed to saline (Con), glucose deprivation (GD), or 1 mm NaCN for 24 hr, and neuronal survival was quantified. Values are the mean and SD of determinations made in four to six cultures. *p < 0.01, **p < 0.001 compared to value for vehicle-treated cultures (ANOVA with Scheffe's post hoc tests).
Fig. 5.

Dantrolene protects neurons expressing mutant PS1 against hypoxic injury. Phase-contrast micrographs showing cortical neurons in cultures from PS1 mutant knock-in mice 24 hr after exposure to saline (Control), 1 mm NaCN, or 10 μm dantrolene + 1 mm NaCN.
DISCUSSION
The present findings suggest that PS1 mutations render neurons vulnerable to ischemic injury by a mechanism involving enhanced calcium release from ER. The increase in infarct size after focal ischemia-reperfusion in PS1 mutant knock-in mice was apparently not the result of systemic alterations in these mice because physiological parameters were essentially identical in both wild-type and mutant mice. A direct adverse effect of the PS1 mutation in neurons is suggested by several observations. Neurons in cortical cell cultures, a preparation highly enriched in neurons, exhibited increased vulnerability to glucose deprivation and chemical hypoxia, two insults relevant to ischemic neuronal injury. In addition, immunohistochemical analyses of rodent brain tissue have shown levels of expression of PS1 are considerably greater in neurons than in non-neuronal cells (Cribbs et al., 1996; Elder et al., 1996; Lah et al., 1997). Moreover, the magnitude of elevation of [Ca2+]i after glucose deprivation and exposure to NaCN was increased in neurons from PS1 mutant mice.
Previous studies of cultured PC12 cells (Guo et al., 1996, 1997) and synaptosomes from transgenic mice (Begley et al., 1999) overexpressing PS1 mutations suggested that altered PS1 function disrupts cellular calcium homeostasis. However, interpretation of the latter findings was tempered by several potential caveats of the systems used. Although PC12 cells have some properties of neurons, they do not exhibit several important characteristics (e.g., expression of ionotropic glutamate receptors and synaptic connections) of neurons vulnerable in AD. Moreover, the PC12 cell lines used overexpressed mutant PS1 at high levels, making it uncertain whether the altered ER calcium homeostasis was the specific result of the PS1 mutations. Caveats with PS1 mutant transgenic mice (Duff et al., 1996; Begley et al., 1999) include overexpression of the transgene and expression of endogenous wild-type PS1, and (because the transgene is randomly integrated into the genome) the possibility that the phenotype results from disruption of an endogenous gene cannot be ruled out. PS1 mutant knock-in mice overcome many of these pitfalls such that mutant PS1 is expressed at physiological levels, endogenous mouse PS1 is not expressed, and the mutant PS1 is targeted to the endogenous mouse PS1 locus (Guo et al., 1999a, 1999b) The cell survival and calcium imaging data obtained in the present study of PS1 mutant knock-in mice therefore strongly suggest that PS1 mutations disrupt neuronal calcium homeostasis and render neurons vulnerable to metabolic insults.
Because PS1 is localized to ER (Guo et al., 1996; Kovacs et al., 1996;Walter et al., 1996) and disrupts ER calcium regulation (Guo et al., 1996, 1997; present study), it is likely that this effect of PS1 is responsible for the increased neuronal vulnerability to ischemic injury documented in the present study. Consistent with the latter interpretation, previous studies have shown that drugs that block calcium release from ER, including dantrolene, can protect cultured neurons against metabolic and excitotoxic insults (Frandsen and Schousboe, 1991). Administration of dantrolene to neonatal brain slice immediately after an ischemia-like insult significantly enhanced cellular recovery, as indicated by reduced energy depletion and suppression of poly-ADP-ribose polymerase activation (Tasker et al., 1998). Wei and Perry (1996) showed that intravenous administration of dantrolene to gerbils immediately after transient global forebrain ischemia resulted in a significant decrease in loss of CA1 hippocampal neurons. In the same model, Zhang et al. (1993) reported a significant decrease in damage to CA1 neurons in gerbils receiving an intraventricular bolus of dantrolene 30 min after reperfusion. Calcium release from IP3-sensitive stores may also play a role in ischemic neuronal death in vivo, as suggested by studies showing correlations between IP3 receptor levels and selective neuronal vulnerability in hippocampus (Araki et al., 1991; Sharp et al., 1993).
Both ryanodine receptors and IP3 receptors are expressed at high levels in cortical pyramidal neurons, but their subcellular distributions appear to differ, with ryanodine receptors being present mainly in apical dendrites and IP3receptors being concentrated in proximal dendrites and cell bodies (Sharp et al., 1993). PS1 appears to be present in ER throughout neurons (cell body, axons, and dendrites) (Cook et al., 1996; Cribbs et al., 1996; Elder et al., 1996). It is therefore likely that PS1 is present in both ryanodine- and IP3-sensitive stores. Although the mechanism whereby PS1 mutations perturb ER calcium signaling is not known, recent findings suggest that PS1 interacts with ryanodine receptors and/or associated proteins (Kim et al., 1998;Mattson et al., 1999). Another potential mechanism whereby PS1 mutations might increase the amount of calcium released from ER stores is by increasing the total pool of calcium. Support for the latter possibility comes from studies showing that PC12 cells overexpressing mutant PS1 exhibit enhanced calcium release after exposure to thapsigargin, a selective inhibitor of the ER calcium ATPase (Guo et al., 1996).
Although our data strongly suggest a primary role for altered ER calcium regulation in the endangering action of PS1 mutations, additional mechanisms might play a role. One well-documented effect of PS1 mutations is that they result in altered proteolytic processing of APP in a manner that increases production of Aβ (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996; Guo et al., 1999b) and decreases production of the secreted form of APP (sAPPα; Ancolio et al., 1997). Aβ has been shown to increase neuronal vulnerability to excitotoxic and metabolic insults by a mechanism that involves, in part, disruption of calcium homeostasis (Mattson et al., 1992; Keller et al., 1998). However, the amino acid sequence of rodent Aβ is different from that of human Aβ, and this difference appears to greatly decrease the neurotoxicity of rodent Aβ (Otvos et al., 1993), making it unlikely that increased production of Aβ accounts for the present findings. On the other hand, sAPPα stabilizes cellular calcium homeostasis and protects neurons against metabolic, excitotoxic, and oxidative insults (Mattson et al., 1993a; Goodman and Mattson, 1994; Furukawa et al., 1996). We previously reported that intraventricular administration of sAPPα reduces damage to hippocampal neurons after transient global forebrain ischemia in rats (Smith-Swintosky et al., 1994). Thus, a decrease in levels of sAPPα (Ancolio et al., 1997) could contribute to increased vulnerability of PS1 mutant knock-in mice to focal ischemic injury.
Finally, our data have implications for interventions that may prevent or attenuate the neurodegenerative process in AD patients. We have shown that drugs that block calcium release from ER can protect neurons expressing mutant PS1 against metabolic insults. Because reduced energy availability to neurons is believed to play a role in the neurodegenerative process in AD (for review, see Mattson et al., 1999), such drugs may prove useful in inhibiting the neurodegenerative cascade.
Footnotes
This work was supported by the National Institutes of Health (National Institute on Aging and National Institute of Neurological Disorders and Stroke). We thank J. Partin and L. Yang for technical assistance.
Correspondence should be addressed to Mark P. Mattson, Chief of the Laboratory of Neurosciences, National Institute on Aging, GRC 4F01, 5600 Nathan Shock Drive, Baltimore, MD 21224. E-mail:mattsonm@grc.nia.nih.gov.
REFERENCES
- 1.Ancolio K, Marambaud P, Dauch P, Checler F. α-secretase-derived product of β-amyloid precursor protein is decreased by presenilin-1 mutations linked to familial Alzheimer's disease. J Neurochem. 1997;69:2494–2499. doi: 10.1046/j.1471-4159.1997.69062494.x. [DOI] [PubMed] [Google Scholar]
- 2.Araki T, Kato H, Kogure K. Alteration of second messenger systems after transient cerebral ischemia in gerbils: protective effect of pentobarbital and an autoradiographic analysis. Neurosci Lett. 1991;130:57–60. doi: 10.1016/0304-3940(91)90226-j. [DOI] [PubMed] [Google Scholar]
- 3.Begley JG, Duan W, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ25–35/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 5.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 6.Cheng B, Mattson MP. NGF and bFGF protect rat and human central neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron. 1991;7:1031–1041. doi: 10.1016/0896-6273(91)90347-3. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 8.Cook DG, Sung JC, Golde TE, Felsenstein KM, Wojczyk BS, Tanzi RE, Trojanowski JQ, Lee VM, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cribbs DH, Chen L, Bendle SM, La Ferla FM. Widespread neuronal expression of the presenilin-1 early-onset Alzheimer's disease in the murine brain. Am J Pathol. 1996;148:1797–1806. [PMC free article] [PubMed] [Google Scholar]
- 10.Duff K, Eckman C, Zehr C, Yu X, Prada C-M, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 11.Elder GA, Tezapsidis N, Carter J, Shioi J, Bouras C, Li D, Johnston JM, Efthimiopoulos S, Friedrich VL, Robakis NK. Identification and neuron specific expression of the S182/presenilin 1 protein in human and rodent brains. J Neurosci Res. 1996;45:308–320. doi: 10.1002/(SICI)1097-4547(19960801)45:3<308::AID-JNR13>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Elkind MS, Sacco RL. Stroke risk factors and stroke prevention. Semin Neurol. 1998;18:429–440. doi: 10.1055/s-2008-1040896. [DOI] [PubMed] [Google Scholar]
- 13.Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP, Kwiatkowski DJ, Moskowitz MA. Neuroprotective effects of gelsolin during murine stroke. J Clin Invest. 1999;103:347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frandsen A, Schousboe A. Dantrolene prevents glutamate cytotoxicity and calcium release from intracellular stores in cultured cerebral cortical neurons. J Neurochem. 1991;56:1075–1078. doi: 10.1111/j.1471-4159.1991.tb02031.x. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa K, Barger SW, Blalock E, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted β-amyloid precursor protein. Nature. 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 16.Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- 17.Ginsberg M, Butso R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman Y, Mattson MP. Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 20.Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid β-peptide. NeuroReport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 21.Guo G, Sopher BL, Pham DG, Furukawa K, Robinson N, Martin GM, Mattson MP. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Q, Robinson N, Mattson MP. Secreted APPα counteracts the pro-apoptotic action of mutant presenilin-1 by activation of NF-κB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 23.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999a;5:101–107. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 24.Guo Q, Sebastian L, Sopher BL, Miller MW, Glazner GW, Ware CB, Martin GM, Mattson MP. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a presenilin-1 mutation. Proc Natl Acad Sci USA. 1999b;96:4125–4130. doi: 10.1073/pnas.96.7.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 27.Katzman R. Alzheimer's disease. N Engl J Med. 1986;314:964–973. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- 28.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen H-C, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial MnSOD prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kokmen E, Whisnant JP, O'Fallon WM, Chu CP, Beard CM. Dementia after ischemic stroke: a population-based study in Rochester, Minnesota (1960–1984). Neurology. 1996;46:154–159. doi: 10.1212/wnl.46.1.154. [DOI] [PubMed] [Google Scholar]
- 30.Kovacs DM, Fausett HJ, Page KJ, Kim T-W, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 31.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattson MP. Cellular actions of β-amyloid precursor protein and it's soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 33.Mattson MP, Mark RJ. Excitotoxicity and excitoprotection in vitro. Adv Neurol. 1996;71:1–37. [PubMed] [Google Scholar]
- 34.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattson MP, Cheng B, Culwell A, Esch F, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of β-amyloid precursor protein. Neuron. 1993a;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 36.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993b;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 37.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Guo Q, Furukawa K, Pedersen WA. Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer's disease. J Neurochem. 1998;70:1–14. doi: 10.1046/j.1471-4159.1998.70010001.x. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann NY Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- 40.Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B, Joachim C, Litchfield S, Barnetson L, Smith AD. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:165–170. doi: 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Otvos L, Szendrei GI, Lee VM, Mantsch HH. Human and rodent Alzheimer β-amyloid peptides acquire distinct conformations in membrane-mimicking solvents. Eur J Biochem. 1993;211:249–257. doi: 10.1111/j.1432-1033.1993.tb19893.x. [DOI] [PubMed] [Google Scholar]
- 42.Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4:1884–1891. doi: 10.1523/JNEUROSCI.04-07-01884.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. The amyloid β protein deposited in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 44.Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST, Ferris SH, Finkel SI, Gwyther LP, Khachaturian ZS, Lebowitz BD, McRae TD, Morris JC, Oakley F, Schneider LS, Streim JE, Sunderland T, Teri LA, Tune LE. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer's Association, and the American Geriatrics Society. JAMA. 1997;278:1363–1371. [PubMed] [Google Scholar]
- 46.Smith-Swintosky VL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP. Secreted forms of β-amyloid precursor protein protect against ischemic brain injury. J Neurochem. 1994;63:781–784. doi: 10.1046/j.1471-4159.1994.63020781.x. [DOI] [PubMed] [Google Scholar]
- 47.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 48.Tasker RC, Sahota SK, Cotter FE, Williams SR. Early postischemic dantrolene-induced amelioration of poly(ADP-ribose) polymerase-related bioenergetic failure in neonatal rat brain slices. J Cereb Blood Flow Metab. 1998;18:1346–1356. doi: 10.1097/00004647-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 49.Usachev Y, Shmigol A, Pronchuk N, Kostyuk P, Verkhratsky A. Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- 50.Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlinsy MB, Fraser P, St George-Hyslop P, Selkoe DJ, Haass C. The Alzheimer's disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med. 1996;2:673–691. [PMC free article] [PubMed] [Google Scholar]
- 51.Wei H, Perry DC. Dantrolene is cytoprotective in two models of neuronal cell death. J Neurochem. 1996;67:2390–2398. doi: 10.1046/j.1471-4159.1996.67062390.x. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Andou Y, Masuda S, Mitani A, Kataoka K. Dantrolene protects against ischemic, delayed neuronal death in gerbil brain. Neurosci Lett. 1993;158:105–108. doi: 10.1016/0304-3940(93)90623-s. [DOI] [PubMed] [Google Scholar]