

Abstract
The selective degeneration of an axon, without the death of the parent neuron, can occur in response to injury, in a variety of metabolic, toxic, and inflammatory disorders, and during normal development. Recent evidence suggests that some forms of axon degeneration involve an active and regulated program of self-destruction rather than a passive “wasting away” and in this respect and others resemble apoptosis. Here we investigate whether selective axon degeneration depends on some of the molecular machinery that mediates apoptosis, namely, the caspase family of cysteine proteases. We focus on two models of selective axon degeneration: Wallerian degeneration of transected axons and localized axon degeneration induced by local deprivation of neurotrophin. We show that caspase-3 is not activated in the axon during either form of degeneration, although it is activated in the dying cell body of the same neurons. Moreover, caspase inhibitors do not inhibit or retard either form of axon degeneration, although they inhibit apoptosis of the same neurons. Finally, we cannot detect cleaved substrates of caspase-3 and its close relatives immunocytochemically or caspase activity biochemically in axons undergoing Wallerian degeneration. Our results suggest that a neuron contains at least two molecularly distinct self-destruction programs, one for caspase-dependent apoptosis and another for selective axon degeneration.
Keywords: neuron, apoptosis, nerve growth factor, optic nerve, sciatic nerve, retina, dorsal root ganglia
The axon of a nerve cell usually degenerates when the cell dies. Part of the axon can also degenerate without the death of the parent neuron in response to local neurotrophin deprivation (Campenot, 1982) or injury and in many metabolic, toxic, and inflammatory disorders (Griffin et al., 1996). The selective degeneration of unwanted axon branches is also important in establishing topographic maps during neural development (O'Leary and Koester, 1993). Despite its significance, little is known about the molecular mechanisms of axon degeneration.
Studies of the breakdown of the distal portion of a transected axon, a process called Wallerian degeneration (Waller, 1850), suggest that axon degeneration can involve an active and regulated self-destruction program rather than a passive “wasting away” and in this respect is similar to apoptosis. In vertebrates, the distal portion of an axon can remain viable and able to conduct action potentials in vivofor up to a few days after axotomy, but then there is a period of rapid destruction in which the axolemma and axonal cytoskeleton are dismantled. Neurofilament breakdown, which depends on Ca2+ and the Ca2+-activated protease calpain (George et al., 1995), is a prominent feature of this degenerative phase and is often used as a marker for it.
Because axotomy interrupts the supply of proteins and organelles to the axon from the cell body, it was once thought that Wallerian degeneration results from axonal starvation. The recent discovery of the naturally occurring Wlds mutant strain of mice in which Wallerian degeneration is greatly slowed (Lunn et al., 1989) suggests that Wallerian degeneration is an active process. Unlike those from wild-type mice, the distal portion of a transected Wlds axon remains viable and able to conduct action potentials for up to 2 weeks (Lunn et al., 1989). This remarkable property is intrinsic to the axon and does not depend on macrophages or glial cells (Perry et al., 1990b; Glass et al., 1993).
Here we focus on Wallerian degeneration and localized axon degeneration induced by local deprivation of neurotrophin in a compartmented cell-culture system (Campenot, 1982), which may serve as an in vitro model for some forms of axon degeneration during development (Campenot, 1982; Cowan et al., 1984). We explore whether these two forms of axon degeneration depend on some of the molecular machinery that mediates apoptosis, namely, the caspase family of cysteine proteases. As mediators of an intracellular proteolytic cascade during apoptosis, caspases cleave one another and various other intracellular proteins following specific aspartic acids to kill the cell (Nicholson and Thornberry, 1997; Cryns and Yuan, 1998). The caspases required for apoptosis differ according to cell type. Caspase-3, for example, seems to be especially important for many types of neuronal apoptosis (Friedlander and Yuan, 1998; Porter and Jänicke, 1999), including apoptosis in the developing CNS. Mice in which the caspase-3 gene has been inactivated die perinatally with a vast excess of cells in their CNS, apparently because of decreased apoptosis in neuroepithelial cells (Kuida et al., 1996; Woo et al., 1998).
MATERIALS AND METHODS
Animals and reagents. Sprague Dawley rats, C57Bl/6 mice, and anesthetics were purchased from the University College London Animal Facility. The day of birth was designated as postnatal day 0 (P0). All reagents were purchased from Sigma (Poole, UK) unless otherwise stated. The peptide caspase inhibitors benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoromethylketone [z-Val-Ala-Asp(O-Me)-CH2F (zVAD-fmk)] and Boc-Asp(O-Me)-CH2F (BocD-fmk), the cathepsin B inhibitor z-Phe-Ala-CH2F (zFA-fmk), and the fluorogenic peptide caspase substrates z-Asp-Glu-Val-Asp-aminotrifluoromethylcoumarin (zDEVD-AFC), z-Val-Glu-Ile-Asp-AFC (zVEID-AFC), and z-Tyr-Val-Ala-Asp-AFC (zYVAD-AFC) were purchased from Enzyme Systems Products (Livermore, CA). Stock solutions of the inhibitors (50 mm) and substrates (20 mm) were made up in dry DMSO and stored in aliquots at −80°C. In all experiments involving inhibitors, parallel treatment of nerves, cells, or distal axons with DMSO alone had no effect. Murine nerve growth factor (NGF; 7S) was purchased from TCS Biologicals (Buckingham, UK) and stored in aliquots at −80°C until needed; it was then added directly to the culture medium without filtration.
Nerve transection. For optic nerve transections, P4 rats were anesthetized by hypothermia. The left eye was retracted to allow access to the optic nerve, which was cut with ophthalmology scissors just behind the globe. For sciatic nerve transections, adult C57Bl/6 mice were anesthetized using a mixture of Hypnorm and Diazepam. An incision was made near the left hip, and the sciatic nerve was cut with scissors just posterior to the spinal cord. After surgery the incisions were sutured, and the animals were returned to their cages. After 1–2 d the animals were anesthetized with a lethal injection of Sagatal. They were perfused through the heart with PBS to remove blood cells, followed by 4% paraformaldehyde in PBS (PFA-PBS). Successful transection was confirmed by visual inspection. The nerves were removed and immersed in PFA-PBS for 2–4 hr.
Explant cultures. For retinal explant cultures, eyes from P7 rats were placed in PBS, and the neural retinae were removed. The whole retina was placed (pigment layer down) on a 1% agarose disk (∼1.5 cm in diameter) in a 35 mm bacteriological dish. Before the tissue was added, the agarose disks were preincubated at 37°C in 5% CO2 in 0.5 ml of Neuralbasal medium (NBM; Life Technologies, Gaithersburg, MD) with 5% fetal calf serum (FCS), 100 ng/ml NGF, and 50 U/ml penicillin–streptomycin; in some experiments the appropriate concentrations of peptide caspase inhibitors or staurosporine (STS; 2 μm) and cycloheximide (CHX; 10 μg/ml) were added. Four small radial slits were made in the retina using surgical scissors, and the edges of the retina were smoothed using a fine paintbrush. The medium was replaced after 24 hr.
Optic and sciatic nerves in P7 rats were cut with surgical scissors, freed of connective tissue with forceps, washed in NBM, placed on agarose disks, and cultured as described above.
Whole dorsal root ganglia (DRG) were dissected from P0 rats and placed in NBM. The nerve roots and connective tissue were removed using fine forceps. Ganglia were cultured on glass coverslips coated with poly-d-lysine (PDL; 10 μg/ml) and laminin (10 μg/ml) in individual wells of a 24-well tissue culture plate (Falcon) in 0.5 ml of growth medium, which consisted of NBM supplemented with 100 μg/ml transferrin, 16 μg/ml putrescine, 5 μg/ml insulin, 39 ng/ml sodium selenite, and 100 μg/ml crystallized BSA [modified from Bottenstein and Sato (1979)], as well as with 60 μg/mlN-acetyl-l-cysteine and 100 ng/ml NGF. After 7–10 d in culture, most ganglia exhibited extensive neurite outgrowth. The neurites were cut close to the body of the ganglion using an 18 gauge, unbeveled needle (Becton Dickinson, Rutherford, NJ), and the ganglion bodies were removed; in some experiments a peptide caspase inhibitor (zVAD-fmk or BocD-fmk; 100 μm), the cathepsin B inhibitor zFA-fmk (100 μm), or the calpain inhibitorN-acetyl-Leu-Leu-norleucinal (ALLN; 200 μm) was added 1 hr before cutting. Axon degeneration was assessed repeatedly after transection with inverted phase microscopy.
Dissociated-cell culture. Whole DRG were dissected from three to four P0 rats and collected in NBM containing 10% FCS (NBM/FCS) and 50 U/ml penicillin–streptomycin. They were washed once, resuspended in 3 ml of NBM/FCS containing 0.1% collagenase, transferred to a 35 mm dish, and incubated at 37°C in 5% CO2 for 30 min. They were then washed with NBM, resuspended in NBM containing 0.25% trypsin and 0.8 U/ml DNase, and incubated at 37°C for 30 min. A single-cell suspension was prepared by triturating the ganglia with a Pasteur pipette and then passing the cells through 60 μm nylon mesh to remove debris. The cells were centrifuged at 1000 rpm for 6 min and resuspended in growth medium containing 10 μm cytosine arabinoside (AraC). Many of the non-neural cells were removed by preplating the suspension at 37°C in NBM/FCS for 1.5 hr on 35 mm tissue culture dishes (Nunc). The cells were then either plated at a density of ∼5000 neurons/well on glass coverslips coated with laminin and PDL in 24-well dishes or plated in compartmented culture dishes (see below). After 2 d, the medium was replaced with fresh growth medium and changed every 3–4 d thereafter.
Compartmented culture system. Compartmented culture dishes were prepared as described previously (Campenot, 1997) with minor modifications. Briefly, 35 mm tissue culture dishes (Falcon) were treated with poly-d,l-ornithine (5 μg/ml) and then laminin (5 μg/ml; Life Technologies). A series of shallow parallel scratches were made across the bottom of the dish, and a drop of 1% methylcellulose in F14 medium (Imperial Laboratories, Hampshire, UK) was applied on top of the scratches. Teflon dividers (Tyler Research Instruments, Edmonton, Alberta, Canada) were coated with silicon grease (Dow Corning) and then gently pressed onto the dish, forming fluid-tight seals between the three compartments. All three compartments were filled with growth medium, and DRG neurons, dissociated from PO rats as described above, were added to the central compartment. The medium was changed every 2–4 d, with 10 μm AraC present in the distal compartments from days 2 to 3 and 5 to 6 to eliminate glial cells. After 7 d, axons had extended through the silicon grease into the two distal compartments. Medium in the right-hand compartment was replaced with growth medium lacking NGF and containing either anti-NGF antibodies (0.2 μg/ml; Boehringer Mannheim, Mannheim, Germany) or anti-NGF antibodies and 100 μm zVAD-fmk or zFA-fmk or with growth medium containing STS (1 μm) and CHX (10 μg/ml). To prevent bulk flow of the medium containing NGF into the right-hand compartment, the level of the medium in the right-hand compartment was kept several millimeters above the level in the other compartments. zVAD-fmk and zFA-fmk were refreshed after 48 hr. Treatment of dissociated DRG neurons with 100 μm zVAD-fmk for 4 d indicated that this inhibitor was nontoxic. Axon degeneration was repeatedly assessed during NGF deprivation by inverted phase-contrast microscopy.
Fixation and frozen sections. All cultures were fixed by removing most of the culture medium and then adding PFA-PBS. After 0.5–1 hr at room temperature, the coverslips were washed and stored in PBS at 4°C until use.
Retinae, optic nerves, and sciatic nerves were fixed by immersion in PFA-PBS for 2–4 hr at 4°C. They were then washed in PBS, transferred to 1 m sucrose in PBS overnight, transferred to a solution of 50% ornithine carbamyl transferase (OCT) compound (Miles, Elkhart, IN) in 1 m sucrose for 2–4 hr, embedded in OCT, and frozen in liquid nitrogen. Frozen sections were cut at 10 or 15 μm and collected onto glass slides that had been coated with 1% gelatin. Sections were allowed to dry for several hours at room temperature and then stored at −20°C until use.
Immunocytochemistry. To detect activated caspase-3, we used two preparations of rabbit polyclonal antibodies—CM1 and Ab206. The CM1 antibodies were produced against (and affinity-purified on) a 13 amino acid peptide corresponding to the C terminal of the large (p20) subunit of activated human and mouse caspase-3 (Armstrong et al., 1997). In Western blots, these antibodies recognize the activated form of caspase-3 but not the inactive proenzyme (Armstrong et al., 1997;Srinivasan et al., 1998). These antibodies have been used to detect activated caspase-3 in mouse brain during development (Srinivasan et al., 1998) and after ischemic injury (Namura et al., 1998), as well as in mouse sperm and chicken erythrocytes (Weil et al., 1998) and rat oligodendrocytes (Gu et al., 1999). The Ab206 antibodies were produced against the peptide sequence GIETD, corresponding to the C terminal of the large (p17) subunit of activated human and rodent caspase-3. The peptide was conjugated via an N-terminal cysteine residue to keyhole limpet hemocyanin, and the conjugate was used to immunize rabbits. In Western blots, Ab206 antibodies label a single 17 kDa polypeptide in extracts of apoptotic, but not healthy, cultured neurons (R. Siman, unpublished observations).
To detect proteins cleaved by caspase-3 and (to a lesser extent) its close relatives such as caspase-2 and caspase-7, we used rabbit polyclonal antibodies (Ab127) that were produced against the peptide sequence KGDEVD, a caspase recognition domain that is found at the C terminal of many caspase-cleaved protein fragments. The Ab127 antibodies have been characterized extensively as a biochemical and immunohistochemical marker for caspase-mediated proteolysis associated with apoptosis in vitro and in vivo (Siman et al., 1999).
Slides, coverslips, and compartmented culture dishes were washed three times in PBS and incubated for 30 min in blocking solution (10% goat serum and 0.4% Triton X-100 diluted in Tris-buffered saline containing 1% BSA and 10 mml-lysine) at room temperature and then for 1–2 hr in CM1 antibodies (1.2 μg/ml in blocking solution without goat serum), Ab206 antibodies (1:2000), or Ab127 antibodies (1:2000). After three washes in PBS, the bound antibodies were detected with biotinylated goat anti-rabbit Ig antibodies (diluted 1:200; Amersham), followed by streptavidin-Texas Red (diluted 1:200; Amersham), both at room temperature for 45 min. In parallel with all experiments involving the CM1, Ab206, and Ab127 antibodies, we stained dissociated DRG neurons treated with STS and CHX for 4–6 hr or deprived of NGF and treated with anti-NGF antibodies for 24 hr as a positive control for the staining procedure.
To detect neurofilaments, we mainly used the N52 monoclonal antibody (ascites fluid; diluted 1:400) (Shaw et al., 1986), which recognizes the largest neurofilament protein. We also used (data not shown) the NA1297 cocktail of affinity-purified rabbit antibodies (diluted 1:100; Affiniti) that together recognize all three neurofilament proteins. Although the N52 antibodies seemed to label a subset of axons, the results with the two antibody preparations were equivalent.
We also detected axons with a monoclonal antibody against Thy-1.1 (OX7; hybridoma supernatant; diluted 1:1; originally provided by A. Williams, Oxford University) or affinity-purified rabbit antibodies against protein gene product 9.5 (PGP 9.5; ascites fluid; diluted 1:500; Ultraclone). We used the same antibodies to identify retinal ganglion cells (RGCs) in sections of retinal explants. The anti-PGP 9.5 antibodies recognize horizontal cells, as well as RGCs, in the mammalian retina. The N52 and OX7 antibodies were detected with fluorescein-conjugated goat anti-mouse Ig (diluted 1:200; Jackson ImmunoResearch, West Grove, PA); the NA1297 and anti-PGP 9.5 antibodies were detected using fluorescein-conjugated goat anti-rabbit Ig (diluted 1:200; Jackson ImmunoResearch).
Apoptotic cells were identified by their nuclear morphology, which was assessed by staining for 30 min with propidium iodide (PI; 0.8 μg/ml) and RNase (0.1 mg/ml) or a cell-permeable form of bisbenzimide (Hoechst 33342; 4 μg/ml). Slides and coverslips were mounted in Citifluor (Citifluor Products, Canterbury, UK) and examined with a Bio-Rad MRC-1000 confocal laser-scanning microscope or a Zeiss Axioplan 2 fluorescence microscope.
Cytosolic extracts. We prepared cytosolic extracts of sciatic nerve explants from P7 rats and of thymocytes from P0 rats that were cultured for various time periods with or without STS (5 μm) and CHX (10 μg/ml). The cells and explants were washed twice with PBS and twice with PBS containing 0.5 mmEDTA (PBS-EDTA). The nerves were then placed in PBS-EDTA, finely chopped using surgical scissors, and washed by centrifugation. S100 cytosolic extracts were prepared by sonication on ice, as described byLiu et al. (1996), with some modifications; the thymocytes and nerve segments were resuspended (without sucrose) in ice-cold buffer A (20 mm HEPES-KOH, 10 mm KCl, 1.5 mmMgCl2, 1 mm Na-EDTA, 1 mmNa-EGTA, and 1 mm DTT, pH 7.5) supplemented with protease inhibitors (0.1 mm phenylmethylsulfonyl fluoride, 5 μg/ml pepstatin A, 10 μg/ml leupeptin, 2 μg/ml aprotinin, and 25 μg/ml ALLN). After sonication, the extracts were centrifuged at 13,000 rpm for 5 min at 4°C in a Biofuge 13 (Heraeus). The supernatants were further centrifuged at 45,000 rpm for 30 min at 4°C in a tabletop ultracentrifuge (Beckman). The protein concentrations of the resulting supernatants were assayed using the Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL). The supernatants were stored in aliquots at −80°C until use.
Biochemical assay for caspase activity. To detect caspase activity biochemically, a volume of nerve or thymocyte extract containing usually 60 μg of protein was incubated in buffer A and 2 mm additional MgCl2 with 0.1 mm fluorogenic peptide caspase substrate (either zDEVD-AFC, zVEID-AFC, or zYVAD-AFC) at 37°C in a final volume of 165 μl. Aliquots (25 μl) were taken at 0, 15, 30, 45, 60, and 120 min and were added to 12.5 μl of 1% Na acetate·3H2O in 175 mm acetic acid to stop the reaction. The samples were then stored at −20°C until analysis.
The amount of cleaved AFC in each of three aliquots of each sample was measured in a 96-well plate luminescence spectrometer (Perkin-Elmer, Norwalk, CT; excitation at 400 nm and emission at 505 nm). The mean fluorescence value for each time point was converted to micromoles of AFC using a standard curve prepared for each experiment. The specific activity of caspases in an extract was defined as the maximum rate of AFC released (Kmax in micromoles per hour) divided by the amount of protein in the extract (in micrograms), where Kmax was determined from the maximum slope of a plot of micromoles of AFC released versus time.
RESULTS
Lack of activated caspase-3 in transected optic nerve
Caspase-3 and its close relatives are thought to play an important part when many types of neurons, including RGCs, undergo apoptosis. To test whether these caspases are activated during Wallerian degeneration of RGC axons, we cultured transected optic nerves from P7 rats as explants for up to 48 hr. At various times we fixed the nerves and treated frozen sections with the CM1 or Ab206 antibodies, which recognize activated caspase-3 (but not inactive procaspase-3), or with the Ab127 antibodies, which recognize various proteins that have been cleaved by caspase-3 or its close relatives.
As shown in Figure 1a, none of the antibodies stained axons in nerves that had been either fixed immediately after transection or cultured for 14, 19, or 24 hr before fixation. The antibodies did stain the occasional apoptotic glial cell, however, which served as a positive control for the staining procedure (Fig. 1a). Staining with the N52 monoclonal anti-neurofilament antibody showed that after 24 hr in culture most axons in the nerves had degenerated (Fig. 1a). The CM1 antibodies also failed to stain axons in optic nerves that were either cultured as explants for 4, 7, 10, 32, or 48 hr or transected and allowed to degenerate in vivo for 24 or 48 hr (data not shown). Thus procaspase-3 and its close relatives are apparently not activated in optic nerve axons undergoing Wallerian degeneration.
Fig. 1.
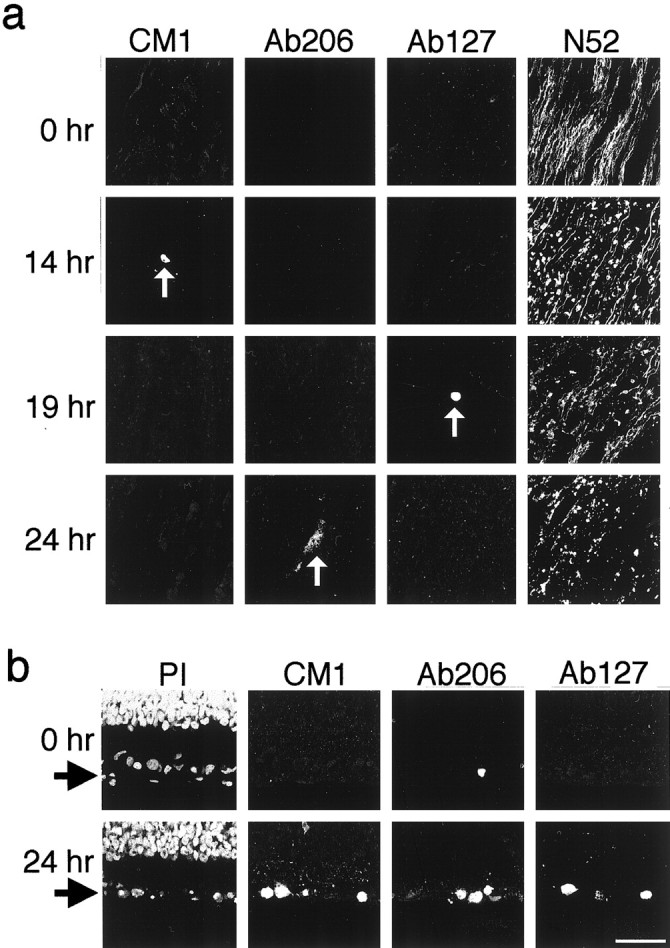
Confocal immunofluorescence micrographs showing that procaspase-3 and its close relatives are not activated in RGC axons undergoing Wallerian degeneration in optic nerve explants (a) but are activated in RGCs undergoing apoptosis in retinal explants (b).a, Longitudinal sections of freshly dissected P7 optic nerve (0 hr) or P7 optic nerve explants cultured for 14, 19, or 24 hr were stained with the N52 anti-neurofilament antibody or with antibodies that specifically recognize either the activated form of caspase-3 (CM1 and Ab206) or proteins that have been cleaved by caspase-3 or its close relatives (Ab127). Note the lack of activated caspase-3 in nerves undergoing Wallerian degeneration, even though neurofilament degradation is apparent. Arrows point to apoptotic glial cells containing activated caspase-3 or its close relatives, which served as positive controls for our staining procedure. b, Cross sections of freshly dissected P7 retinae (0 hr) or P7 retinal explants cultured for 24 hr were stained with PI to visualize nuclei or with the CM1, Ab206, or Ab127 antibodies. Arrows point to the RGC layer. PI and CM1 staining are shown for the same fields. Note the single RGC undergoing naturally occurring apoptosis that was recognized by Ab206 at 0 hr. Scale bar, 50 μm.
Activated caspase-3 in the cell body of axotomized RGCs
As many as 80% of rat RGCs that are axotomized during the first few postnatal weeks undergo apoptosis (Rabacchi et al., 1994). To determine whether procaspase-3 or its close relatives are activated in RGC cell bodies in these conditions, we cultured, fixed, sectioned, and immunostained the retinae corresponding to the optic nerves used in the above experiments. As shown in Figure 1b, after 24 hr in culture many RGCs showed the characteristic nuclear changes of apoptosis, and nearly all of these cells stained with CM1, Ab206, and Ab127 antibodies. In contrast, retinae that were fixed immediately after excision contained relatively few stained RGCs, and all of these cells were apoptotic (Fig. 1b); presumably these cells died in the process of naturally occurring apoptosis. Thus procaspase-3 is activated in the cell body when RGCs undergo either axotomy-induced or naturally occurring apoptosis, but this procaspase and its close relatives apparently are not activated in the degenerating axons of these cells.
Lack of activated caspase-3 in transected sciatic nerve
To determine whether procaspase-3 or its close relatives are activated during Wallerian degeneration in the peripheral nervous system (PNS), we stained explants of transected P7 sciatic nerves with CM1, Ab127, Ab206, and anti-neurofilament antibodies after 14, 19, or 24 hr in culture. Neurofilament staining indicated that the axons had degenerated, yet no axonal CM1, Ab127, or Ab206 staining was seen, although the occasional apoptotic glial cell was stained (Fig.2). The CM1 antibodies also failed to stain axons in sciatic nerves that were cultured as explants for 4, 7, 10, 32, or 48 hr or sciatic nerves of adult C57Bl/6 mice that were transected and allowed to degenerate in vivo for 24 or 48 hr (data not shown). Thus procaspase-3 and its close relatives are apparently not activated in sciatic nerve axons undergoing Wallerian degeneration.
Fig. 2.
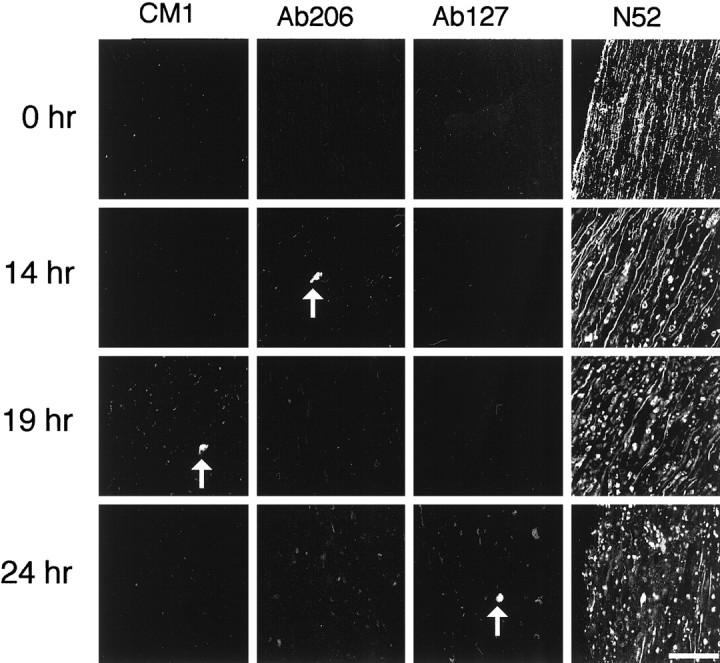
Confocal immunofluorescence micrographs showing that procaspase-3 and its close relatives are not activated during Wallerian degeneration of sciatic nerve. Longitudinal sections of uncut sciatic nerve (0 hr) or sciatic nerve explants cultured for 14, 19, or 24 hr were stained with the N52 anti-neurofilament antibody and with the CM1, Ab206, or Ab127 antibodies. Note the lack of activated caspase-3 in nerves undergoing Wallerian degeneration, even though neurofilaments have been degraded. Arrows point to apoptotic glial cells containing activated caspase-3 or its close relatives. Scale bar, 50 μm.
Lack of caspase activity in extracts of explanted sciatic nerve
To look more generally for procaspase activation during Wallerian degeneration, we used three different fluorogenic peptide caspase substrates to test for caspase activity in cytosolic extracts of explanted sciatic nerves. Because the extracts were prepared from whole sciatic nerve, they would be expected to contain both glial and axonal proteins. When zDEVD-AFC, which is cleaved by caspases 2, 3, and 7 (Thornberry et al., 1997), was used as a substrate to test extracts of sciatic nerve after 4, 10, 14, 19, or 24 hr in explant culture, little caspase activity was detected. The broad-spectrum protein kinase inhibitor STS, especially when used together with the protein synthesis inhibitor CHX, induces caspase-dependent apoptosis in a variety of cell types (Weil et al., 1996). Extracts from P0 rat thymus cells treated with STS and CHX, similar to those we used as a positive control in related caspase activity assays (Weil et al., 1998), exhibited much greater activity (Fig.3). We also treated explanted sciatic nerves with STS and CHX for 4 hr before preparing the extracts, which resulted in an approximately fivefold increase of cleavage activity compared with that of extracts from untreated nerve (Fig. 3). [Although a 10 hr treatment with STS and CHX resulted in greater caspase activation (Fig. 3), in most experiments we used a 4 hr treatment as a way to monitor the sensitivity of the assay.] Immunostaining with the CM1 antibodies of sections of nerves treated with STS and CHX for various times revealed robust procaspase-3 activation in glial cells, making it impossible to determine whether individual axons were also CM1+ (data not shown). Similar results were obtained when zYVAD-AFC (a substrate for caspases 1, 4, and 5) or zVEID-AFC (a substrate for caspases 6, 8, and 9) (Thornberry et al., 1997) was used as a substrate to access caspase activity in nerve extracts (data not shown). Thus little, if any, procaspase activation seems to occur during Wallerian degeneration in sciatic nerve.
Fig. 3.

Caspase activity in cytosolic extracts of sciatic nerve and thymocytes. Extracts were prepared from P0 thymocytes treated with STS and CHX (Thy + STS) or from P7 sciatic nerves that were cultured as explants for various times or that were treated with STS and CHX for 4 hr (4 h + STS) or 10 hr (10 h + STS). The extracts were assayed for their ability to cleave the fluorogenic caspase substrate zDEVD-AFC. There is little caspase activity in extracts from nerves undergoing Wallerian degeneration. Data from a representative experiment are shown. The experiment was repeated three times with similar results, and similar results were obtained when zVEID-AFC or zYVAD-AFC was used as a substrate.
Effect of caspase inhibitors on transected optic nerve and axotomized RGCs
We next tested whether cell-permeable peptide caspase inhibitors can suppress or delay Wallerian degeneration of the optic nerve. Treatment of P7 optic nerve explants with either zVAD-fmk (Fig.4a) or BocD-fmk (data not shown) had no appreciable effect on axotomy-induced neurofilament breakdown after 14, 19, 24, or 48 hr when compared with nerves that were either treated with the chemically similar cathepsin B peptide inhibitor zFA-fmk (Fig. 4a) or left untreated (see Fig.1a). The caspase inhibitors were also ineffective in preventing axolemma breakdown, as assessed by staining with antibodies against either the cell-surface antigen Thy-1.1 or the cytoplasmic enzyme PGP 9.5 (data not shown). The ability of zVAD-fmk to block the binding of the CM1 antibodies to activated caspase-3 (A. Srinivasan, unpublished observations) provided evidence that the inhibitor penetrated throughout the nerves under our experimental conditions; in nerves treated with zVAD-fmk, CM1+ glial cells were not detected (data not shown), whereas they were always present in untreated optic nerves (see Fig. 1a)
Fig. 4.

Confocal immunofluorescence micrographs of sections of P7 optic nerves (a) and retinae (b) showing that caspase inhibitors inhibit apoptosis of axotomized RGCs but not Wallerian degeneration of their axons. Explants were cultured for 24 hr in the presence of the caspase inhibitor zVAD-fmk or the cathepsin B inhibitor zFA-fmk.a, Longitudinal sections of optic nerve were stained with the N52 anti-neurofilament antibody. Note that neither inhibitor prevented the breakdown of neurofilaments. b, Left,Cross sections of retina were stained with propidium iodide. Thearrowheads point to the RGC layers. Note that pyknotic RGCs are apparent in explants treated with zFA-fmk but not in those treated with zVAD-fmk. Right, The bar graph shows the number of pyknotic (pyk.) cells per field in the RGC layer of the retinae (expressed as the mean ± SD of three experiments). Scale bar, 50 μm.
In contrast, treatment of P7 retinal explants with zVAD-fmk greatly inhibited the apoptosis of RGCs that occurred after 24 hr in explant culture; whereas explants treated with zFA-fmk contained many apoptotic RGCs, explants treated with zVAD-fmk contained very few (Fig.4b). It was reported previously that intraocular injection of zVAD-fmk or other caspase inhibitors also rescues axotomized RGCs in vivo (Kermer et al., 1998; Chaudhary et al., 1999). Thus, although caspase inhibitors do not suppress the Wallerian degeneration of a transected RGC axon, they do suppress the axotomy-induced apoptosis of the RGC cell body.
Effect of caspase inhibitors on transected DRG axons in explant culture
To determine whether caspase inhibitors can suppress or delay Wallerian degeneration in the PNS, we tested them on DRG explants. The explants were cultured in NGF and allowed to extend axons for 7–10 d. We then transected the proximal portions of the axons and removed the cell bodies. Treatment with zVAD-fmk did not inhibit axotomy-induced axolemma disintegration or neurofilament breakdown seen 24 hr after the axons were cut, when compared with untreated explants (Fig.5). Similar results were obtained when neurofilaments were stained 10, 12, 19, or 48 hr after transection or when axon degeneration was assessed by staining with antibodies against Thy-1.1 or PGP 9.5 (data not shown). Axons were also repeatedly examined for up to 48 hr after transection by phase-contrast microscopy; zVAD-fmk was ineffective at all times. Because neurofilament cleavage in Wallerian degeneration has been shown to depend on the Ca2+-dependent protease calpain (George et al., 1995), we used the calpain inhibitor ALLN as a positive control; as expected, ALLN prevented the breakdown of some neurofilaments (Fig. 5) but not the axolemma, assessed by either DIC microscopy (Fig. 5) or staining with antibodies against PGP 9.5 or Thy-1.1 (data not shown).
Fig. 5.

Differential interference contrast (DIC) and confocal immunofluorescence micrographs showing the effect of caspase inhibitors on transected DRG axons in culture. P0 DRG explants were cultured for 7d, treated with zVAD-fmk or the calpain inhibitor ALLN for 1 hr, and then axotomized. After 24 hr, the transected axons were fixed and then studied by DICmicroscopy or stained with the N52 anti-neurofilament antibody and then studied with confocal immunofluorescence microscopy. Note that ALLN prevented the breakdown of some neurofilaments but not the axolemma, whereas zVAD-fmk had no effect on either. Different fields are shown for DIC images (scale bar, 33 μm) and immunofluorescence images (scale bar, 50 μm).
Activated caspase-3 in DRG neurons deprived of NGF
When dissociated DRG neurons that had been cultured in the presence of NGF for 7 d were deprived of NGF and treated with anti-NGF antibodies for 1–2 d, many of the cells underwent apoptosis. The apoptotic neuronal cell bodies and many of their axons stained brightly with CM1 antibodies (Fig. 6), suggesting that procaspase-3 is activated throughout the cell when the neuron undergoes NGF deprivation-induced apoptosis.
Fig. 6.

Confocal immunofluorescence micrograph of activated caspase-3 in apoptotic DRG neurons deprived of NGF. Dissociated P0 DRG neurons were cultured for 7 d in medium containing NGF and were then deprived of NGF and treated with anti-NGF antibodies for 24 hr. The cells were then fixed and stained with CM1 antibodies. Note that activated caspase-3 is present in the cell body and axons of the two neurons. The CM1 staining extended throughout the entire length of the axons (∼1 mm; data not shown). Scale bar, 50 μm.
To determine whether procaspase-3 is activated locally in neurites when they degenerate in response to local deprivation of NGF, we cultured dissociated DRG neurons in three-chambered compartmented culture dishes (Campenot, 1982). Dissociated neurons were added to the central compartment, and after 7 d they had extended axons into the two distal compartments. We then replaced the medium in the right-hand compartments with medium lacking NGF and containing anti-NGF antibodies. We repeatedly monitored the breakdown of the axolemma (see below) by phase-contrast microscopy. After 4d, the extent of the breakdown was similar to that of axons of dissociated DRG neurons that had been deprived of NGF for 24 hr, which stained brightly with CM1. Thus, we fixed the neurons growing in compartmented culture dishes after 4 d and stained them with CM1 antibodies. CM1 staining was not detected in any of the three compartments, indicating that procaspase-3 had not been activated (data not shown). Thus, procaspase-3 is activated throughout the neuron when it undergoes apoptosis in response to global NGF deprivation, but it is not activated in axons that degenerate selectively in response to local NGF deprivation.
Because zVAD-fmk inhibited the apoptosis of dissociated DRG neurons deprived of NGF for 2 d (data not shown), we sought to determine whether zVAD-fmk could suppress or delay localized axon degeneration induced by local NGF deprivation. We cultured DRG neurons in three-chambered culture dishes as described above and after 7 d replaced the medium in some of the right-hand compartments with medium lacking NGF and containing anti-NGF antibodies and either zVAD-fmk or zFA-fmk. After 4 d, axons cultured in the presence of zVAD-fmk disintegrated extensively (Fig. 7), as did untreated axons and axons treated with zFA-fmk (data not shown). Repeated monitoring of distal axons by phase-contrast microscopy before fixation indicated that zVAD-fmk was ineffective at all times. Thus, localized axon degeneration attributable to local NGF deprivation also seems to be caspase independent.
Fig. 7.

DIC and immunofluorescence micrographs showing the effect of caspase inhibitors on axons locally deprived of NGF. P0 DRG neurons were dissociated and plated in the central compartment of a three-chambered culture dish, with NGF in all of the compartments. After 7 d, the neurons had extended axons into the two distal compartments, and the medium in some of the right-hand distal chamber was replaced with medium that lacked NGF and contained both anti-NGF antibodies and zVAD-fmk. Four days later, the neurons were fixed and then studied by DIC microscopy or stained with the N52 anti-neurofilament antibody and then studied with confocal immunofluorescence microscopy. Many of the axons in compartments deprived of NGF and treated with zVAD-fmk (shown) degenerated; axons in compartments deprived of NGF without zVAD-fmk (data not shown) showed comparable degeneration. Different fields are shown for DIC and immunofluorescence images. Scale bar, 100 μm.
DISCUSSION
Because selective axon degeneration may involve an active and regulated self-destruction program, rather than a passive wasting away, we have investigated whether it depends on caspases, which mediate apoptosis. There had been few attempts to determine whether caspases are involved in selective axon degeneration, but the published reports left the issue unresolved (see below for details). Several reports, for example, suggested that caspases may be involved; two described a caspase-dependent apoptosis-like process in isolated synaptic terminals (Mattson et al., 1998a,b), and another concluded that caspases are required for some forms of axon degeneration (Ivins et al., 1998). Furthermore, both Wallerian degeneration and neuronal apoptosis are delayed by elevated intracellular cAMP or moderately increased intracellular Ca2+ (Franklin and Johnson, 1994; Buckmaster et al., 1995). The inability of axons to synthesize RNA or protein does not exclude a role for caspases, because these enzymes are expressed constitutively (Weil et al., 1996); indeed apoptosis can occur in the absence of RNA or protein synthesis (for review, see Martin, 1993) or even of a nucleus (Jacobson et al., 1994), although the activation of caspase-dependent apoptosis often requires transcription (Martin, 1993). In contrast, overexpression in mouse neurons of the human Bcl-2 protein, a well known inhibitor of many forms of caspase-dependent apoptosis, did not inhibit Wallerian degeneration (Dubois-Dauphin et al., 1994; Burne et al., 1996) or axon degeneration in a mouse model of neurodegenerative disease (Sagot et al., 1995).
To address directly whether selective axon degeneration involves caspases, we focused our attention on two simple models of selective axon degeneration: Wallerian degeneration and localized axon degeneration induced by local deprivation of neurotrophin. We asked whether caspases are activated during either of these forms of degeneration and whether either form is prevented or retarded by caspase inhibitors.
Wallerian degeneration seems to be molecularly distinct from caspase-dependent apoptosis in the same neurons
We find that procaspase-3 and its close relatives are not activated during Wallerian degeneration, even when procaspase-3 is activated in the axotomized cell body of the same neurons. Moreover, we do not detect caspase activity biochemically in axons undergoing Wallerian degeneration, and caspase inhibitors do not prevent or retard Wallerian degeneration, although they inhibit the apoptosis that occurs in the axotomized cell body of the same neurons. Thus the molecular mechanisms that mediate Wallerian degeneration and caspase-dependent apoptosis are apparently distinct.
Axon degeneration induced by local deprivation of neurotrophin seems to be molecularly distinct from caspase-dependent apoptosis in the same neurons
We have also studied localized axon degeneration induced by local deprivation of neurotrophin in a compartmented cell-culture system. Although procaspase-3 is activated throughout neurons that undergo apoptosis in response to global deprivation of neurotrophin, it is not activated in axons that degenerate locally in response to local neurotrophin deprivation. As with Wallerian degeneration, caspase inhibitors neither inhibit nor retard this form of axon degeneration.
Experiments on neurons from Wldsmice suggest that a common mechanism may underlie the two models of selective axon degeneration that we have studied.Wlds neurons also exhibit delayed axon degeneration in response to neurotrophin deprivation; when cultured superior cervical ganglia neurons fromWlds mice are deprived of NGF, the apoptosis of the neuronal cell body occurs normally, whereas the neurites remain viable (Deckwerth and Johnson, 1994).
Neurons seem to have distinct programs for selective axon degeneration and apoptosis
It is difficult to exclude caspase involvement in axon degeneration with absolute certainty, especially because there may be rodent caspases still to be identified. Nonetheless, our results indicate that the forms of axon degeneration we have studied are at least molecularly distinct from caspase-dependent apoptosis in the same neurons. Other evidence supports this conclusion. As mentioned above, for example, the Wlds mutation protects axons from both forms of degeneration that we have studied, whereas it does not protect neuronal cell bodies from undergoing apoptosis in response to neurotrophin deprivation (Deckwerth and Johnson, 1994), although it may partially protect axotomized motor neurons (Lapper et al., 1994) and RGCs (Perry et al., 1991) from undergoing apoptosis.
In addition, the Bcl-2 protein can inhibit caspase-dependent apoptosis in neurons (and many other cell types) (for review, see Adams and Cory, 1998), but it seems not to protect axons from degeneration. In transgenic mice expressing the human Bcl-2 protein in neurons, for example, RGC cell bodies are protected from axotomy-induced apoptosis, although their severed axons still rapidly undergo Wallerian degeneration (Dubois-Dauphin et al., 1994; Burne et al., 1996). Similarly, in pmn mice, which have a genetic defect that causes motor neurons to degenerate and the mice to die before 6 weeks of age, overexpression of a human bcl-2 transgene prevents the degeneration of the mutant motor neuron cell bodies but not axons (Sagot et al., 1995). Neither the course of the disease nor the life span of the mice is affected by the bcl-2 transgene, emphasizing the clinical importance of selective axon degeneration.
The neuronal cell body and axon also seem to respond differently to high concentrations of STS. Whereas STS induces caspase-dependent apoptosis in a wide variety of cell types, including neurons (Jacobson et al., 1996; Weil et al., 1996), it does not induce degeneration when locally applied to axons in vitro (Campenot et al., 1991; J. T. Finn, unpublished observations), suggesting that axons lack components of either the apoptosis program or the STS-sensitive apoptosis-activating pathway. Although we do not detect activated caspases in axons treated locally with STS or in axons degenerating in response to axotomy or local neurotrophin deprivation, we do find activated caspase-3 in degenerating axons associated with apoptotic neurons that have been globally treated with STS (J. T. Finn, unpublished observation) or deprived of neurotrophin; moreover, activated caspase-3 has been detected in the processes of neurons undergoing developmental apoptosis (Srinivasan et al., 1998). Thus, caspases may play a part in dismantling the axon when a neuron undergoes apoptosis. In this case a proteolytic cascade of caspase activation may spread from the cell body down the length of the axon.
Our results are perhaps surprising in view of recent reports by Mattson et al. (1998a,b). They reported that treatment of synaptosomes with STS, Fe2+, or amyloid β-peptide caused the synaptosomes to undergo a process that had the features of apoptosis, including caspase activation, exposure of phosphatidylserine (PS), and mitochondrial membrane depolarization. The mitochondrial changes were inhibited by zVAD-fmk, suggesting that axon terminals can undergo a caspase-dependent apoptosis-like process, which would seem to distinguish them from the parent axon.
A recent report by Ivins et al. (1998) does not fit easily with our observations. They reported that local treatment of axons with amyloid β-peptide caused the axons to degenerate with associated caspase activation and PS exposure. The PS exposure could be inhibited by zVAD-fmk, suggesting that axonal caspases are involved in this form of axon degeneration. In contrast to our findings and those of Campenot et al. (1991), Ivins et al. (1998) also found that local treatment with STS caused axon degeneration. Although Ivins et al. (1998) did not address this difference between their results and those of Campenot et al. (1991), it is possible that the discrepancy reflects the type of compartmented chamber used; whereas we and Campenot et al. (1991) used specially constructed, thick-walled Teflon chambers purchased from Tyler Research Instruments, Ivins et al. (1998) made their chambers from a hemisected Teflon ring, with only a glass coverslip serving as the barrier between the neurites and the cell bodies. Also, Ivins et al. (1998) did not exclude the possibility that the axon degeneration they observed was secondary to effects on the neuronal cell bodies.
Why might neurons have a caspase-independent axonal self-destruction program?
One advantage of a caspase-independent axon degeneration program is that it would enable a neuron to confine the destruction process to the axon, without endangering the life of the cell, which might be difficult to achieve with a self-destruction program that depended on a caspase cascade. Such spatial control would be especially useful for eliminating either unwanted axon branches during development or injured axons in the adult.
The challenge now is to determine the molecular nature of the axon degeneration program and how it is controlled. Because of the clinical importance of selective axon degeneration in a variety of disorders, including multiple sclerosis (Scolding and Franklin, 1998) and acquired immune deficiency syndrome (Bradley et al., 1998), it is surprising that its mechanism(s) has received so little attention. TheWlds mouse should provide a powerful approach to the problem, because the mutation seems to affect axon degeneration specifically. It is possible that the mutation affects the regulation of the putative axon degeneration program, rather than the program itself, because the mutant axons are capable of rapid degeneration under some circumstances (Glass et al., 1994). TheWlds phenotype is caused by an autosomal-dominant mutation (Perry et al., 1990a) that involves an 85 kb tandem triplication (Coleman et al., 1998) in the distal region of chromosome 4 (Lyon et al., 1993). The nature of the specific gene(s) involved or how the mutation delays Wallerian (Lunn et al., 1989) and neurotrophin deprivation-induced axon degeneration (Deckwerth and Johnson, 1994) remains unknown.
Footnotes
J.T.F. was supported by a Hitchings–Elion Fellowship from the Burroughs-Wellcome Fund. M.W. was supported by a project grant from Action Research for the Crippled Child. M.C.R. and this work were supported by a Medical Research Council Program Grant. We thank Pierre-Alain Fernandez, Toru Kondo, Anne Mudge, and V. Hugh Perry for comments on this manuscript, Julia Burne and Robert Campenot for discussion and preliminary experiments, Tom Deckwerth for discussion, Anne Mudge for help with the DRG and sciatic nerve preparations, and Michele Binder for help with preparing this manuscript.
Correspondence should be addressed to Dr. John T. Finn, Center for International Security and Cooperation, Encina Hall, Stanford University, Stanford, CA 94305-6165. E-mail: jfinn@stanford.edu.
Dr. Weil's present address: Department of Cell Research and Immunology, Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israel.
REFERENCES
- 1.Adams JM, Cory S. The bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KC. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley WG, Shapshak P, Delgado S, Nagano I, Stewart R, Rocha B. Morphometric analysis of the peripheral neuropathy of AIDS. Muscle Nerve. 1998;21:1188–1195. doi: 10.1002/(sici)1097-4598(199809)21:9<1188::aid-mus10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Buckmaster EA, Perry VH, Brown MC. The rate of Wallerian degeneration in cultured neurons from wild type and C57Bl/Wlds mice depends on time in culture and may be extended in the presence of elevated K+ levels. Eur J Neurosci. 1995;7:1596–1602. doi: 10.1111/j.1460-9568.1995.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 6.Burne JF, Staple JK, Raff MC. Glial cells are increased proportionally in transgenic optic nerves with increased numbers of axons. J Neurosci. 1996;16:2064–2073. doi: 10.1523/JNEUROSCI.16-06-02064.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- 8.Campenot RB. Construction and use of compartmented cultures. In: Federoff S, Richardson A, editors. Protocols for neural cell culture, Second Edition. Humana; Totowa, NJ: 1997. pp. 107–116. [Google Scholar]
- 9.Campenot RB, Walji AH, Draker DD. Effects of sphingosine, staurosporine, and phorbol ester on neurites of rat sympathetic neurons growing in compartmented cultures. J Neurosci. 1991;11:1126–1139. doi: 10.1523/JNEUROSCI.11-04-01126.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhary P, Ahmed F, Quebada P, Sharma SC. Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Brain Res Mol Brain Res. 1999;67:36–45. doi: 10.1016/s0169-328x(99)00032-7. [DOI] [PubMed] [Google Scholar]
- 11.Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci USA. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowan WM, Fawcett JW, O'Leary DD, Stanfield BB. Regressive events in neurogenesis. Science. 1984;225:1258–1265. doi: 10.1126/science.6474175. [DOI] [PubMed] [Google Scholar]
- 13.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 14.Deckwerth TL, Johnson EM. Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis). Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- 15.Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou JC. Neonatal motoneurons overexpressing the bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci USA. 1994;91:3309–3313. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franklin JL, Johnson EM. Block of neuronal apoptosis by a sustained increase of steady-state free Ca2+ concentration. Philos Trans R Soc Lond B Biol Sci. 1994;345:251–256. doi: 10.1098/rstb.1994.0102. [DOI] [PubMed] [Google Scholar]
- 17.Friedlander RM, Yuan J. ICE, neuronal apoptosis and neurodegeneration. Cell Death Differ. 1998;5:823–831. doi: 10.1038/sj.cdd.4400433. [DOI] [PubMed] [Google Scholar]
- 18.George ER, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57B1/01a mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- 20.Glass JD, Schryer BL, Griffin JW. Calcium-mediated degeneration of the axonal cytoskeleton in the Ola mouse. J Neurochem. 1994;62:2472–2475. doi: 10.1046/j.1471-4159.1994.62062472.x. [DOI] [PubMed] [Google Scholar]
- 21.Griffin JW, George EB, Chaudhry V. Wallerian degeneration in peripheral nerve disease. Baillieres Clin Neurol. 1996;5:65–75. [PubMed] [Google Scholar]
- 22.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci. 1999;19:3043–3049. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivins KJ, Bui ETN, Cotman CW. β-Amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis. 1998;5:365–378. doi: 10.1006/nbdi.1998.0228. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson MD, Burne JF, Raff MC. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobson MD, Weil M, Raff MC. Role of Ced-3/ICE-family proteases in staurosporine-induced programmed cell death. J Cell Biol. 1996;133:1041–1051. doi: 10.1083/jcb.133.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kermer P, Klocker N, Labes M, Bahr M. Inhibition of CPP32-like proteases rescues axotomized retinal ganglion cells from secondary cell death in vivo. J Neurosci. 1998;18:4656–4662. doi: 10.1523/JNEUROSCI.18-12-04656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 28.Lapper SR, Brown MC, Perry VH. Motor neuron death induced by axotomy in neonatal mice occurs more slowly in a mutant strain in which Wallerian degeneration is very slow. Eur J Neurosci. 1994;6:473–477. doi: 10.1111/j.1460-9568.1994.tb00289.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 30.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 31.Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH. A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proc Natl Acad Sci USA. 1993;90:9717–9720. doi: 10.1073/pnas.90.20.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin SJ. Protein or RNA synthesis inhibition induces apoptosis of mature human CD4+ T cell blasts. Immunol Lett. 1993;35:125–134. doi: 10.1016/0165-2478(93)90080-l. [DOI] [PubMed] [Google Scholar]
- 33.Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998a;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- 34.Mattson MP, Partin J, Begley JG. Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998b;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 35.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 37.O'Leary DD, Koester KS. Development of projection neuron types, axon pathways, and patterned connections of the mammalian cortex. Neuron. 1993;10:991–1006. doi: 10.1016/0896-6273(93)90049-w. [DOI] [PubMed] [Google Scholar]
- 38.Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur J Neurosci. 1990a;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 39.Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that very slow Wallerian degeneration in C57Bl/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci. 1990b;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 40.Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57Bl/Ola mice. Eur J Neurosci. 1991;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 41.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 42.Rabacchi SA, Bonfanti L, Liu XH, Maffei L. Apoptotic cell death induced by optic nerve lesion in the neonatal rat. J Neurosci. 1994;14:5292–5301. doi: 10.1523/JNEUROSCI.14-09-05292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scolding N, Franklin R. Axon loss in multiple sclerosis. Lancet. 1998;352:340–341. doi: 10.1016/S0140-6736(05)60463-1. [DOI] [PubMed] [Google Scholar]
- 45.Shaw G, Osborn M, Weber K. Reactivity of a panel of neurofilament antibodies on phosphorylated and dephosphorylated neurofilaments. Eur J Cell Biol. 1986;42:1–9. [PubMed] [Google Scholar]
- 46.Siman R, Bozyczko-Coyne D, Bhat R. Immunolocalization of caspase proteolysis: evidence for widespread caspase-mediated apoptosis of neurons and glia in the postnatal rat brain. Neuroscience. 1999;92:1425–1442. doi: 10.1016/s0306-4522(99)00034-2. [DOI] [PubMed] [Google Scholar]
- 47.Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 48.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 49.Waller A. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alternatives produced thereby in the structure of their primitive fibers. Philos Trans R Soc Lond B Biol Sci. 1850;140:423. [Google Scholar]
- 50.Weil M, Jacobson MD, Coles HSR, Davies TJ, Gardener RL, Raff KD, Raff MC. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weil M, Jacobson MD, Raff MC. Are caspases involved in the death of cells with a transcriptionally inactive nucleus? Sperm and chicken erythrocytes. J Cell Sci. 1998;111:2707–2715. doi: 10.1242/jcs.111.18.2707. [DOI] [PubMed] [Google Scholar]
- 52.Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, Senaldi G, Howard T, Lowe SW, Mak TW. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]