

Abstract
The adhesive cell surface molecule P0 is the most abundant glycoprotein in peripheral nerve myelin and fulfills pivotal functions during myelin formation and maintenance. Mutations in the corresponding gene cause hereditary demyelinating neuropathies. In mice heterozygously deficient in P0(P0+/− mice), an established animal model for a subtype of hereditary neuropathies, T-lymphocytes are present in the demyelinating nerves. To monitor the possible involvement of the immune system in myelin pathology, we cross-bred P0+/− mice with null mutants for the recombination activating gene 1 (RAG-1) or with mice deficient in the T-cell receptor α-subunit. We found that in P0+/− mice myelin degeneration and impairment of nerve conduction properties is less severe when the immune system is deficient. Moreover, isolated T-lymphocytes from P0+/− mice show enhanced reactivity to myelin components of the peripheral nerve, such as P0, P2, and myelin basic protein. We hypothesize that autoreactive immune cells can significantly foster the demyelinating phenotype of mice with a primarily genetically based peripheral neuropathy.
Keywords: Charcot-Marie-Tooth disease, myelin degeneration, Schwann cell, P0, myelin protein zero, immune system, immune deficiency, T-lymphocytes, macrophages, electron microscopy, electrophysiology
Myelin protein zero (MPZ or P0), the most abundant glycoprotein of the myelin sheath of peripheral nerves, is a transmembrane adhesion molecule of the Ig superfamily that fulfills multiple functions during myelin development and maintenance (for review, see Martini and Schachner, 1997). Its pivotal role in the peripheral nervous system is reflected by the fact that mutations in the corresponding gene cause inherited neuropathies, such as Charcot-Marie-Tooth (CMT) disorder, type 1B, the Déjérine-Sottas syndrome, and congenital hypomyelination (for review, see De Jonghe et al., 1997; Martini et al., 1998; Nelis et al., 1999). These disorders are typically associated with muscular weakness and atrophy, sensory dysfunction, and skeletal deformities. Biopsies from CMT1B patients revealed that, dependent on the mutation in the P0 gene, divergent pathological hallmarks can be found. Such hallmarks comprise either formation of myelin tomacula, i.e., focally thickened myelin sheaths of reduced stability or, alternatively, myelin decompaction and demyelination (Thomas et al., 1994; Gabreëls-Festen et al., 1996; Tachi et al., 1997). Recently, we have shown that heterozygous P0 null mutant mice (P0+/−) are appropriate models for mild CMT1B forms with an initially normal myelin formation followed by myelin decompaction and demyelination starting at 4 months of life (Martini et al., 1995a; for review, see Martini, 1997). Although it is well accepted that various heterozygous mutations or the reduction in gene dosage of P0 lead to demyelination (for review, see Martini et al., 1998), the molecular and cellular mechanisms that lead to myelin degeneration are not yet understood. Based on the observation that nerves of patients suffering from inherited neuropathies (Schmidt et al., 1996) or nerves of P0+/− mice contain endoneurial T-lymphocytes (Schmid et al., 1996; Shy et al., 1997), we tested the hypothesis that the immune system might be involved in the development of the demyelinating phenotype. We, therefore, cross-bred P0+/− mice with homozygous null mutants for the recombination activating gene 1 (RAG-1) or with mice deficient in the T-cell receptor α-subunit (TCRα), i.e., with mutants that are deficient in mature T- and B-lymphocytes or only T-lymphocytes, respectively. We found that, in the absence of an intact immune system, myelin degeneration in P0+/− mice is significantly reduced in comparison to P0+/− mice with normal immune cells. Moreover, isolated T-lymphocytes from P0+/− mice show enhanced reactivity to myelin components of the peripheral nerve, such as P0, P2, and myelin basic protein (MBP). We conclude that the immune system may be functionally involved in the demyelinating phenotype of mice with a primarily genetically mediated peripheral neuropathy.
MATERIALS AND METHODS
Animals and determination of genotypes. Mice deficient in the gene for protein zero (P0−/−; Giese et al., 1992) or the recombination activating gene 1 (RAG-1−/−; Mombaerts et al., 1992) were generated as described previously and backcrossed to the C57BL/6 strain for six to eight generations. Double mutants were generated by cross-breeding heterozygous P0-mutants (P0+/− mice) with RAG-1−/− mice under specific pathogen-free conditions (Institute of Laboratory Animal Science, University of Zürich) in analogy to the breeding protocol described previously (Martini et al., 1995b; Carenini et al., 1997). Mice deficient for the T-cell receptor α subunit (TCRα−/−; Philpott et al., 1992) were bred on a mixed background (129/Ola/Hsd, BALB/c, C57Bl/6) and cross-bred with P0+/−mice as described above. Genotypes of P0-mutants were determined by conventional PCR using oligonucleotides 5′-TCAGTTCCTTGTCCCCCGCTCTC-3′, 5′-GGCTGCAGGGTCGCTCGGTGTTC-3′, and 5′-ACTTGTCTCTTCTGGGTAATCAA-3′ leading to 334 or 500 bp products for the P0 null mutation or wild-type allele, respectively. The phenotype of the RAG-1 or the TCRα null mutation was determined by fluorescence activated cell sorting (FACS) analysis of peripheral blood cells stained with phycoerythrin (PE)-coupled anti-CD4 and fluorescein-isothiocyanate-coupled anti-CD8 antibodies (PharMingen, Hamburg, Germany) using a FACScan instrument (Becton Dickinson, Heidelberg, Germany). To rule out the possibility that the effects observed were a consequence of differences in the genetic background, all pathological alterations were investigated in littermates. For all pathological studies, the femoral quadriceps nerve was chosen that contains ∼500 myelinated axons in wild-type mice (Lindberg et al., 1999), thus being an ideal paradigm for quantitative analyses. All experiments described here on animals were approved by the Veterinary Administration of Zürich, Switzerland and by the Regierung von Unterfranken (Würzburg, Germany).
Immunohistochemistry. Fourteen-micrometer-thick serial sections from fresh frozen femoral nerve were immunostained as previously described (Guénard et al., 1999). For detection of T-lymphocytes, anti-mouse CD8 (1:50 of supernatants of hybridoma cell lines TS169; Cobbold et al., 1984) were used. For detection of macrophages, anti-mouse F4/80 (1:500; Serotec, Oxford, UK) was used. Sections of spleen of wild-type mice were used as positive controls, and for negative control the primary antibody was omitted. Positive profiles were counted on cryosections using a Zeiss Axiophot microscope, and the mean number of positive profiles per mm2 section was calculated. Statistical analysis of the number of immune cells in cryosections was performed by a two-way ANOVA test.
Electrophysiology. Nerve conduction properties of sciatic nerves from 13-month-old myelin mutants homozygously deficient in RAG-1 (P0+/−/RAG-1−/−) and myelin mutants heterozygously deficient in RAG-1 (P0+/−/RAG-1+/−) as well as from myelin wild-type mice either homozygously or heterozygously deficient in RAG-1 (P0+/+/RAG-1−/−, P0+/+/RAG-1+/−) were determined by established electrophysiological methods as previously described in detail (Zielasek et al., 1996). In brief, anesthetized mice (Hypnorm, Janssen, Belgium) received subcutaneous stimulating electrodes at the sciatic notch (proximal stimulation) and above the ankle (distal stimulation). Recording electrodes were placed subcutaneously close to the hallux and between digits 2 and 3. Studies were performed with a current 50% higher than that needed to elicit maximal stimulation. In all experiments, the investigator (J.Z.) was not aware of the genotype of the mice. For motor nerve conduction studies, the following parameters were measured: proximal and distal M-response latencies, proximal and distal F-wave latencies, and amplitudes of compound muscle action potentials. Nerve conduction velocities were calculated from the latency measurements and the distance of the stimulation sites. In analogy to the different P0/RAG-1 mutants, nerve conduction properties from 13-month-old P0+/−/TCRα−/−, P0+/−/TCRα+/−, P0+/+/TCRα−/−and P0+/+/TCRα+/−mice were determined. Statistical analysis was performed using a one-sided t test.
Tissue preservation for electron microscopy. Femoral nerves of mice were processed for light and electron microscopy as previously described (Martini et al., 1995b; Lindberg et al., 1999). Briefly, mice were transcardially perfused with 0.1 m cacodylate buffer containing 4% paraformaldehyde and 2% glutaraldehyde, and nerves were post-fixed in the same fixative overnight. After osmification and dehydration, nerves were embedded in Spurr's medium.
Morphometry. For light microscopy, semithin sections (0.5-μm-thick) were stained with alkaline methylene blue. For electron microscopy, ultrathin sections (80 nm) were contrasted with lead citrate. Quantification of total nuclei and myelin competent axons was performed on cross sections of the quadriceps branch of the femoral nerve. For this purpose, all nuclei were counted on semithin sections at a final magnification of 600×. The number of myelin competent axons (>2 μm in diameter) that were not or only thinly myelinated (less than five turns) was determined by electron microscopy. Analysis of morphometric parameters such as the g-ratio (myelin thickness; Friede, 1972), the diameter of axons (Friede, 1972), and the endoneurial space was done by electron microscopy on randomly selected parts of ultrathin cross sections of the quadriceps branch of the femoral nerve. The endoneurial space was determined as the fraction of total nerve area not occupied by fibers. Electron micrographs of two randomly selected parts per nerve cross section were digitized with a scanner (HP 4C). Morphometric parameters were evaluated at a final magnification of 8000× using the Image Tool Application, version 1.28 program (University of Texas UTHSCSA). Statistical significance of differences between mean values of P0+/−/RAG1−/−versus P0+/−/RAG1+/−mice was determined by double-sided Student's t test using Microsoft Excel software.
Culture of splenocytes and proliferation assay. Splenocytes from four myelin wild-type (P0+/+) and four P0+/− mice at the age of 8 months were seeded into 96-well microtiter plates at a density of 1.5 × 106/ml (Nunc, Wiesbaden, Germany). Splenocytes were exposed to recombinant human (rh) P0, rhP2 (Weishaupt et al., 1995), rat MBP (10 μg/ml each), or concanavalin A (Con A; 2.5 μg/ml), as described (Pette et al., 1990; Stienekemeier et al., 1999) except that we used 1% autologous serum and added 5 × 10−5m 2-mercaptoethanol. After 3 d, murine interleukin (IL)-2 (2.5 ng/ml; R & D Systems, Wiesbaden, Germany) was added. On day 8, the nonadherent cells from each plate were split into two new microtiter plates. Cells were reactivated on day 14 by addition of irradiated, syngeneic splenocytes to both subcultures, whereas the respective antigens were added to only one as described for the split well technique (Pette et al., 1990). After 48 hr, cells were pulsed with tritiated thymidine and harvested 16 hr later (Stienekemeier et al., 1999). Corresponding cultures were compared for specific proliferation. Wells showing a thymidine incorporation of >1000 counts per minute and a stimulation index of >3 in comparison to control wells without antigen were considered positive. For each antigen, at least 96 primary wells were screened. Statistical analysis was performed with the Mann–Whitney Utest using the Prism computer program (Graph Pad, San Diego, CA).p values < 0.05 were considered statistically significant.
RESULTS
Quantification of immune cells in peripheral nerves
In an attempt to confirm previous observations of the presence of immune system derived cells within the nerves of adult P0+/− mice (Schmid et al., 1996), markers for lymphocytes (CD8; Brideau et al., 1980) or for mouse macrophages (F4/80; Austin and Gordon, 1981) were used. At the age of 4 and 6 months, three P0+/+ and three P0+/− mice were considered. At older ages (8 and 21 months), two mice of each genotype were included, with the exception of three P0+/− mice investigated at 21 months. In cross sections of femoral nerves of 4- to 21-month-old mice, CD8-positive cells were always rounded, whereas F4/80-positive cells were usually ramified or showed at least a few slender processes. During the analysis, the investigator (C.D.S.) was not aware of the genotype of the nerve sections. In general, CD8-positive cells as well as F4/80-positive cells were predominantly detectable in the endoneurium of nerves. When comparing nerves from P0+/− versus P0+/+ mice, there was no quantitative difference in the number of CD8-positive cells at 4 months of age (Fig. 1, top left diagram), a stage where the very first pathological changes are detectable in P0+/− mice (Martini et al., 1995a). However, at 6 months and older, the number of CD8-positive profiles was always higher in nerves from P0+/− versus P0+/+ mice, although there was considerable heterogeneity. This heterogeneity was possibly caused by the previously reported nonuniform distribution of lymphocytes along the course of the nerve (Shy et al., 1997; Fig. 1,top left diagram). F4/80-positive endoneurial profiles outnumbered CD8-positive cells by a factor of ∼20. In contrast to CD8-positive profiles, the numbers of F4/80-positive cells were already elevated in 4-month-old P0+/− mice when compared to age-matched P0+/+ mice (Fig. 1, top right diagram).
Fig. 1.
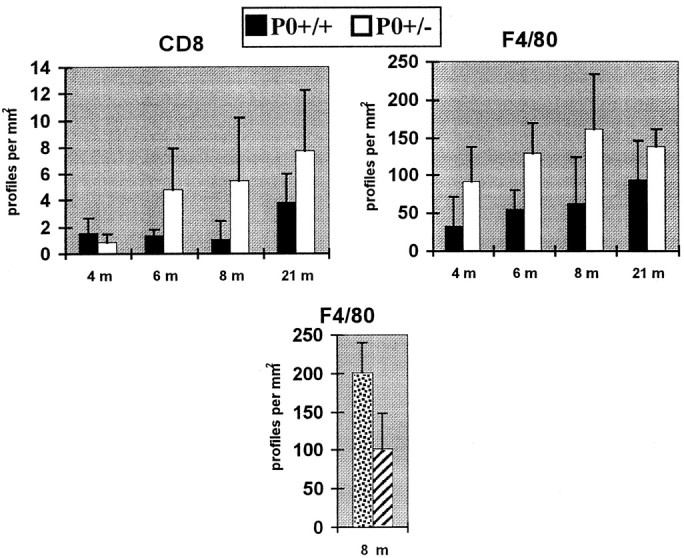
Quantification of CD8- and F4/80-positive cells in cryosections of quadriceps nerves from P0+/+ (top diagrams, filled bars) and P0+/− mice (top diagrams, open bars) and quantification of F4/80-positive cells in quadriceps nerves from P0+/−/TCRα+/−(bottom diagram, stippled bars) and P0+/−/TCRα−/− mice (bottom diagram, hatched bars). Top diagrams, Note elevated levels of immune-derived cells in P0+/− mice. Statistical significance was achieved for F4/80-positive cells, but not for CD8-positive cells, which might be related to the heterogeneous distribution of the lymphocytes along the nerve. Error bars indicate SD. Bottom diagram, Note reduced levels of F4/80-positive cells in quadriceps nerves of P0+/− /TCRα−/−versus P0+/−/TCRα+/−mice at 8 months of age. Error bars indicate SD. p< 0.05 (Student's t test).
Statistical analysis comparing the numbers of immune cells in P0+/+ versus P0+/− mice by a two-way ANOVA test considering all time points investigated resulted inp values of 0.0035 and 0.064 for F4/80- and CD8-positive cells, respectively. Thus, the difference of F4/80-positive cells in P0+/+ versus P0+/− mice was statistically significant, whereas the difference of the CD8-positive cells did not reach statistical significance, which might be related to the considerable heterogeneity in the distribution of the cells (see above).
Generation of double mutants
By cross-breeding P0+/− mice with RAG-deficient (RAG-1−/−) mice and intercrossing the individuals of the F1 generation, we obtained an F2 generation with myelin mutants homozygously deficient in RAG-1 (P0+/−/RAG-1−/−) and myelin mutants heterozygously deficient in RAG-1 (P0+/−/RAG-1+/−). In addition, P0+/+ mice either homozygously or heterozygously deficient in RAG-1 (P0+/+/RAG-1−/−, P0+/+/RAG-1+/−) were obtained. RAG−/− mice are reported to be deficient for mature T- and B-lymphocytes (Mombaerts et al., 1992). Based on FACS and in agreement with previous reports (Mombaerts et al., 1992), CD4- and CD8-single positive T-lymphocytes in peripheral blood were not detectable in RAG-1−/−genotypes. In RAG-1+/− mice, the number of CD4- and CD8-positive T-lymphocytes was indistinguishable from values from RAG-1+/+ genotypes reflecting normal immune cell development (Mombaerts et al., 1992). A corresponding pattern of cross-breeding of P0+/− mice was performed with mice deficient in the TCRα-subunit (TCRα−/− mice) that lack mature TCRαβ-lymphocytes, but contain B-lymphocytes and small populations of TCRβ- and TCRγδ-T-lymphocytes (Philpott et al., 1992; Neuhaus et al., 1996; see Materials and Methods).
Deficiency in the RAG-1 gene leads to milder pathological changes in P0+/− mice
We first investigated semithin sections of femoral quadriceps nerves of P0+/+/RAG-1+/−and P0+/+/RAG-1−/−mice at the age of 13 months. We found that, independent of the RAG-1 genotype, these nerves were of normal phenotype with thick myelin, overall compact appearance with small endoneurial spaces between the nerve fibers, and absence of features indicative of demyelination (Table 1, Fig.2a,b).
Table 1.
Morphometric analysis of femoral quadriceps nerves from 13-month-old P0/RAG-1 mutants
| Genotype | P0+/+/RAG-1+/− (n= 2) | P0+/+/RAG-1−/− (n= 3) | P0+/−/RAG-1+/− (n= 2) | P0+/−/RAG-1−/− (n= 3) |
|---|---|---|---|---|
| Number of nuclei* | 10.5 ± 6.4 | 13.7 ± 3.2 | 68.0 ± 2.8 | 36.3 ± 13.5 |
| Endoneurial space* | 22.5 ± 0.9 | 26.4 ± 3.9 | 64.1 ± 1.4 | 37.8 ± 5.6 |
| g-ratio* | 0.74 ± 0.003 | 0.73 ± 0.012 | 0.81 ± 0.004 | 0.71 ± 0.020 |
| Axon diameter** | 7.4 ± 0.2 | 6.7 ± 0.2 | 5.6 ± 0.2 | 5.1 ± 0.7 |
*Values from P0+/−/RAG-1+/−mice are significantly different from values from P0+/−/RAG-1−/− littermates (p < 0.05).
**Only axons with compacted myelin were measured. Mean values ± SDs are indicated. Values in brackets are numbers of animals investigated.
Fig. 2.
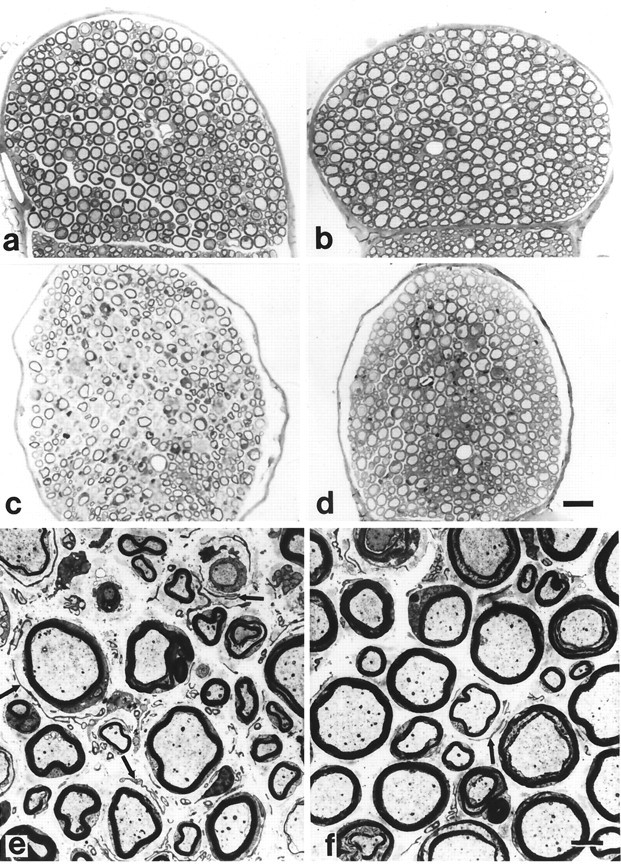
Representative semithin cross sections of femoral quadriceps nerves from P0+/+/RAG-1+/−(a), P0+/+/RAG-1−/−(b), P0+/−/RAG-1+/−(c), and P0+/−/RAG-1−/−mice (d) and ultrathin cross sections of femoral quadriceps nerves from P0+/−/RAG-1+/−(e) and P0+/−/RAG-1−/−mice (f) at the age of 13 months. In P0+/−/RAG-1+/− mice (c, e), numerous thinly myelinated and demyelinated axons are visible as well as extended endoneurial spaces with supernumerary Schwann cells (e, f, arrows). In P0+/−/RAG-1−/−mice (d, f), these pathological characteristics are less pronounced. Note normal appearance of axon-Schwann cell units in P0+/+/RAG-1+/−(a) and P0+/+/RAG-1−/− mice (b). Scale bars: a–d, 20 μm (ind); e, f, 5 μm (inf).
Next, we compared quadriceps femoral nerves from 13-month-old P0+/−/RAG-1+/−mice with nerves from P0+/−/RAG-1−/−littermates. In semithin sections, all mice of P0+/− genotype showed pathological alterations indicative of compromised myelin maintenance (Fig. 2c,d). However, two principle categories of pathological severity could be distinguished by an investigator who was not aware of the genotype (R.M.). In all cases, the more severely affected nerves were from P0+/−/RAG-1+/−mice, whereas the nerves with less pronounced pathological alterations were derived from immune-deficient P0+/−/RAG-1−/−mice (Fig. 2c,d). The nerves of the latter mutants were characterized by thicker myelin sheaths and significantly lower numbers of cell nuclei (Table 1) that mostly belonged to typical Schwann cell onion bulbs. Such supernumerary Schwann cells are a well established indicator of demyelination in mice and humans (for review, see Martini, 1997) so that their reduced numbers in the P0+/−/RAG-1−/−mutants reflect a lower degree of myelin degeneration. As a consequence of the lower number of supernumerary Schwann cells, the nerves of the P0+/−/RAG-1−/−mice appeared more compact in that the myelinated fibers were separated by smaller endoneurial spaces. The number of myelin-competent, i.e., thick caliber axons was similar in both P0+/−/RAG-1+/−and P0+/−/RAG-1−/−mutants (data not shown).
Next, we quantified the pathological alterations in the different mutants by electron microscopy. As exemplified in Figure 2,e and f, and in agreement with the light-microscopic findings, pathological features were less severe in P0+/−/RAG-1−/−versus P0+/−/RAG-1+/−mice (Table 1). Particularly striking were the thicker myelin sheaths in the P0+/−/RAG-1−/−mice, as indicated by their significantly lower g-ratio values (see Materials and Methods) when compared to the P0+/−/RAG-1+/−littermates. Supporting the impression by light microscopy, the endoneurial space was significantly less expanded in P0+/−/RAG-1−/−mice in comparison to the P0+/−/RAG-1+/−littermates when quantified on electron micrographs (Table 1).
We also investigated the number of F4/80-positive macrophages in P0+/−/RAG-1−/−versus P0+/−/RAG-1+/−mice. Most interestingly, macrophages were reduced in numbers in one P0+/−/RAG-1−/−mouse (140 per mm2) versus a P0+/−/RAG-1+/−littermate (346 per mm2) at 13 months of age. Although this observation refers to only one individual of each genotype, we consider the reduced number of macrophages in the P0+/−/RAG-1−/−mouse as relevant, because analogous investigations in other immune-deficient mice (P0+/−/TCRα+/−and P0+/−/TCRα−/−mice) with higher numbers of individuals investigated (n = 3) revealed similar observations (see below; Fig.1, bottom diagram).
Deficiency in the TCRα gene leads to milder pathological changes in P0+/− mice
We also compared quadriceps nerves from 8-month-old P0+/−/TCRα+/−(n = 4) versus P0+/−/TCRα−/−(n = 3) mice both at the light- and electron-microscopic level. Similar to the findings in P0+/−/RAG-1+/−and P0+/−/RAG-1−/−mice, the pathological alterations in the P0+/−/TCRα−/−mice were less pronounced than in the P0+/−/TCRα+/−littermates. For instance, in P0+/−/TCRα−/−mice, the number of Schwann cell nuclei was significantly reduced when compared to values from P0+/−/TCRα+/−littermates (26.7 ± 11.2 vs 53.8 ± 13.3; p< 0.05). Furthermore, the endoneurial space was less expanded in P0+/−/TCRα−/−mice in comparison to the P0+/−/TCRα+/−littermates (42.1 ± 6.3% vs 65.0 ± 11.3% of total nerve area; p < 0.05). Also, myelin was thicker in P0+/−/TCRα−/−mice than in P0+/−/TCRα+/−mice, although the differences between g-ratios were not statistically significant (0.75 ± 0.027 vs 0.78 ± 0.015). We also investigated the number of F4/80-positive macrophages in quadriceps nerves of P0+/−/TCRα−/−mice in comparison to the P0+/−/TCRα+/−littermates at 8 months of age. Similar to the findings in P0+/−/RAG-1−/−versus P0+/−/RAG-1+/−mice, numbers of macrophages were reduced in the immune-deficient myelin mutants (Fig. 1, bottom diagram).
Deficiency in the RAG-1 gene leads to less impaired conduction properties in P0+/− mice
To test whether the immune system has an impact on conduction properties of peripheral nerves of P0+/− mutants, mice were subjected to an electrophysiological investigation by an examiner not aware of the genotypes.
In sciatic nerves of 13-month-old P0+/+/RAG-1+/−and P0+/+ /RAG-1−/−mice, conduction properties were comparable to those previously described for P0+/+ mice (Zielasek et al., 1996; Fig. 3.). In both P0+/−/RAG-1+/−and P0+/−/RAG-1−/−mice, conduction properties were impaired when compared with littermates of P0+/+genotype. However, when comparing P0+/−/RAG-1+/−and P0+/−/RAG-1−/−mice, the latter mutants showed better conduction properties. This was reflected by significantly shorter latencies of M-responses and of F-waves after distal or proximal stimulations (Fig. 3). Amplitudes of compound muscle action potentials were not significantly different in P0+/−/RAG-1+/−versus P0+/−/RAG-1−/−mutants (13.9 ± 4.1 mV vs 11.8 ± 3.7 mV after distal stimulations; n = 4 for each genotype).
Fig. 3.

Schematic representation of parameters of conduction properties of sciatic nerves of P0+/+/RAG-1+/−, P0+/+/RAG-1−/−, P0+/−/RAG-1+/−, and P0+/−/RAG-1−/−mice (asterisks). Note that in P0+/−/RAG-1−/−mice, nerve conduction properties are significantly improved when compared to values from P0+/−/RAG-1+/−mice. Each black dot represents the value of one sciatic nerve from one single mouse investigated; short horizontal bars represent mean values ± SDs.
Deficiency in the TCRα gene leads to less impaired nerve conduction properties in P0+/− mice
In addition, we investigated conduction properties in P0+/+/TCRα+/−, P0+/+/TCRα−/−, P0+/−/TCRα+/−, and P0+/−/TCRα−/−mice at 13 months of age. Similar to P0/RAG-1-mice, conduction properties in P0+/−/TCRα−/−mice were less affected than in P0+/−/TCRα+/−littermates. Motor nerve conduction velocities were significantly higher in the P0+/−/TCRα−/−mice (37 ± 7 m/sec vs 26 ± 4 m/sec; p < 0.05; n = 3 for each genotype). Other parameters, such as latencies of M-responses and of F-waves tended to show less impaired nerve conduction without reaching statistical significance, probably because of the low number of mice available (data not shown).
Increased frequency of myelin-reactive splenocytes in P0+/− mice
Based on the observation that the immune system appears to be involved in demyelination in P0+/− mice, we tested the hypothesis that in P0+/− mice an increased proportion of T-lymphocytes might recognize components of peripheral nerve myelin as autoantigens. We used the split-well technique (Pette et al., 1994) and compared the specific proliferative responses of isolated splenocytes from 8-month-old P0+/+ and P0+/− mice, i.e., at an age of advanced myelin degeneration in P0+/− mice. Interestingly, the frequencies of cells proliferating in response to different myelin components were significantly higher in P0+/− versus P0+/+ mice (Fig.4). By contrast, stimulation with concanavalin A caused comparable proliferative responses in both P0+/+ and P0+/− mice (Fig. 4).
Fig. 4.

Schematic representation of the proliferative response of splenocytes from 8-month-old P0+/+ (filled bars) and P0+/− mice (open bars) to myelin components and to concanavalin A. Stimulation of isolated splenocytes from P0+/+ or P0+/− mice by different myelin antigens was performed by split-well technique. Each columnrepresents the mean of the percentage of positive wells of four mice ± SEM. All myelin components tested elicited a higher proliferative response in P0+/− versus P0+/+ mice, and statistical significance (p < 0.05) was achieved when the proliferative responses against the three myelin components were summed up. Stimulation with concanavalin A (Con A) was taken as positive control for the proliferative capacity of splenocytes from P0+/+ and P0+/− mice.
DISCUSSION
In the present study, we demonstrate that in a mouse model for hereditary neuropathies, demyelination is less severe when the immune system is impaired. Removal of mature T-lymphocytes and amelioration of the pathological phenotype was achieved by two different genetic immune defects (RAG-1−/− and TCRα−/−) thus supporting a role of the immune system in the demyelinating phenotype of P0+/− mice. Another and independent argument in favor of this view is the observation that in splenocyte preparations from P0+/− mice the frequencies of leukocytes proliferating in response to myelin components was higher than in preparations from P0+/+ mice. Although we did not investigate the cytokine release of these myelin-specific T-lymphocytes, we suggest that the elevated frequency of proliferating T-lymphocytes in P0+/−mice reflects the involvement of the immune system in the myelin pathology of P0+/− mice. The highest frequency of proliferating cells was observed when P0 was offered to leukocytes from P0+/− mice. Why P0 appears to be a main immunogenic component in P0+/− mice is presently not known. In experimental autoimmune neuritis (EAN) in rat, P2 is a much stronger neuritogen than P0 (Linington et al., 1992), but there might be differences among species. On the other hand, even in heterozygous mutants, P0 is still the most abundant peripheral myelin protein (Giese et al., 1992) which could explain a substantial role of P0 as an immunogenic component. This does not preclude the possibility that other myelin components that have not been tested could elicit even stronger immune responses.
The mechanism leading to an involvement of the immune system during demyelination in P0+/−mice is presently not known. It is possible that the reduction of P0 may result in an intrinsic instability of myelin leading to an initial attraction of macrophages by Schwann cell-derived cytokines followed by a macrophage-mediated attraction of T-lymphocytes. An argument in favor of this model is our observation that the number of macrophages is increased already at 4 months of age in P0+/− mice, whereas the number of T cells is not yet elevated at this age. The activation of autoimmune T-lymphocytes by antigen-presenting macrophages could eventually result in a local cascade of cellular and humoral immune reactions leading to demyelination (Gold et al., 1999) and/or to a continued attraction and activation of macrophages by T-lymphocyte-derived cytokines. The reduced numbers of macrophages in the less impaired nerves of immune-deficient myelin mutants could indicate a devastating role of macrophages during genetically induced demyelination, as is the case in primarily immune-mediated disorders of the nervous system, such as EAE and EAN (Huitinga et al., 1990; Jung et al., 1993). However, it is still possible that the lower number of macrophages is rather the consequence of than the cause for a less severe pathological status of the immune deficient myelin mutants. Thus, further studies are needed to characterize the roles of macrophages in genetically mediated demyelination.
In contrast to acute and primarily immune-mediated disorders, such as EAN, we observed only few T-lymphocytes within the nerve. As mentioned above, activated T-lymphocytes can lead to a significant damage by direct cytotoxicity or by T-cell-derived signals attracting other immune cells, such as macrophages. In addition, chronic instability of myelin induced by a gene defect may favor the perpetuation of immune reactions to self by repeated exposure of antigen or antigens toward patrolling T-lymphocytes. Furthermore, Schwann cells of P0+/− mice with an intrinsically reduced stability of myelin might react very sensitive to a mild inflammatory attack carried by low numbers of cells. An alternative interpretation could be that the relatively few lymphocytes in the diseased nerve are not the only culprits leading to an aggravation of the pathology. It is possible that mice deficient in the immune system may generally express lower levels of gliotoxic cytokines than mice with a wild-type immune system and that the intrinsically instable Schwann cells of P0+/− mice react particularly sensitive on such cytokines.
A relevant question concerns the clinical implications of our findings. Most forms of P0-related hereditary neuropathies are caused by heterozygous mutations that only seldom result in a pure reduction of gene dosis (Scherer and Chance, 1995; Warner et al., 1996). Rather, toxic gain-of-function and dominant-negative effects might be the leading detrimental mechanisms in most of the P0-mutation-mediated disorders (Warner et al., 1996; Wong and Filbin, 1996; Scherer, 1997; Zhang and Filbin, 1998). This, however, does not argue against the possibility that in patients suffering from such detrimental processes immune-mediated mechanisms are involved as a secondary cause as we have shown for the P0+/− mice. Rather, it is plausible to assume that the involvement of the immune system as an aggravating component is not confined to hereditary neuropathies caused by P0-mutations, but might be widespread among the different types of hereditary neuropathies. Interestingly, in a variety of CMT patients of unknown genotype, elevated levels of activated T-lymphocytes have been detected in the peripheral blood (Williams et al., 1987, 1992). Furthermore, some patients are reported to develop a sudden worsening of their clinical neuropathy and show inflammatory infiltrates in nerve biopsies (Malandrini et al., 1999) and clinically respond to corticosteroids (Dyck et al., 1993). Taking into consideration that immune reactions can be superimposed on the clinical and pathological phenotype of CMT, it might be worthwhile to investigate whether among patients with chronic inflammatory demyelinating polyradiculopathy some might carry CMT mutations that would lead to a more pronounced immune reaction. In analogy, it is intriguing to speculate that mutations in myelin and other genes of the CNS could elicit similar immune reactions that could manifest themselves in multiple sclerosis and modulate the disease course.
Our finding that in P0+/− mice, the immune system contributes to demyelination might offer the possibility to understand the pathological mechanisms of demyelination in at least some forms of inherited neuropathies. It might, therefore, be worthwhile to reconsider treatment strategies with appropriate immunomodulators or suppressors to ameliorate the immune-mediated pathological components of the still untreatable monogenic disorders. In addition, the knowledge about the participation of the immune system in primarily genetically based neuropathies might extend our understanding of the pathomechanisms occurring in neurodegenerative disorders in general.
Footnotes
This study was supported by the Swiss National Science Foundation (NF 31-45890.95 to R.M.), Gemeinnützige Hertie-Stiftung (GHS 2/3378/96 to R.M. and M. Sch.), the Deutsche Forschungsgemeinschaft (Priority Program “Microglia,” MA1053/3-1, to R.M.), Research Funds of the University of Würzburg European Union (Clinical, genetic and functional analysis of peripheral neuropathies), and by the German Ministry for Education and Research. We are grateful to Professor Rolf Zinkernagel (University of Zürich) for valuable advice and discussions, Heinrich Blazyca and Kathrin Rohner for excellent technical assistance, and Dr. Stefano Carenini for discussions. We are indebted to Dr. Imme Haubitz (Department of Mathematics and Statistics, University of Würzburg) for performing the statistical analysis of the quantification of immune cells in peripheral nerves and of the splenocyte proliferation assay. We also thank Dr. M. Koltzenburg for the possibility to use electronic equipment.
Correspondence should be addressed to Rudolf Martini, Department of Neurology, Section of Developmental Neurobiology, University of Würzburg, D-97080 Würzburg, Germany. E-mail:neuk176@rzkli.uni-wuerzburg.de.
REFERENCES
- 1.Austin JM, Gordon S. A monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;10:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 2.Brideau RJ, Carter PB, McMaster WR, Mason DW, Williams AF. Two subsets of rat T lymphocytes defined with monoclonal antibodies. Eur J Immunol. 1980;10:609–615. doi: 10.1002/eji.1830100807. [DOI] [PubMed] [Google Scholar]
- 3.Carenini S, Montag D, Cremer H, Schachner M, Martini R. Absence of the myelin-associated glycoprotein (MAG) and the neural cell adhesion molecule (N-CAM) interferes with the maintenance, but not with the formation of peripheral myelin. Cell Tissue Res. 1997;287:3–9. doi: 10.1007/s004410050727. [DOI] [PubMed] [Google Scholar]
- 4.Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 1984;312:548–551. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 5.De Jonghe P, Timmerman V, Nelis E, Martin JJ, Van Broeckhoven C. Charcot-Marie-Tooth disease and related peripheral neuropathies. J Periph Nerv Syst. 1997;2:370–387. [PubMed] [Google Scholar]
- 6.Dyck PJ, Chance P, Lebo R, Carney JA. Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral Neuropathy. WB Saunders; Philadelphia: 1993. pp. 1094–1136. [Google Scholar]
- 7.Friede RL (1972) Control of myelin formation by axon caliber (with a model of the control mechanism). J Comp Neurol 233–252. [DOI] [PubMed]
- 8.Gabreëls-Festen AA, Hoogendijk JE, Meijerink PH, Gabreëls FJ, Bolhuis PA, van Beersum S, Kulkens T, Nelis E, Jennekens FG, de Visser M, van Engelen BG, Van Broeckhoven C, Mariman EC. Two divergent types of nerve pathology in patients with different P0 mutations in Charcot-Marie-Tooth disease. Neurol. 1996;47:761–765. doi: 10.1212/wnl.47.3.761. [DOI] [PubMed] [Google Scholar]
- 9.Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- 10.Gold R, Archelos JJ, Hartung H-P. Mechanisms of immune regulation in the peripheral nervous system. Brain Pathol. 1999;9:343–360. doi: 10.1111/j.1750-3639.1999.tb00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guénard V, Schweitzer B, Flechsig E, Hemmi S, Martini R, Suter U, Schachner M. Effective gene transfer of lacZ and P0 into Schwann cells of P0-deficient mice. Glia. 1999;25:165–178. [PubMed] [Google Scholar]
- 12.Huitinga I, v Rooijen N, Groot CJA, Uitdehaag BMJ, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990;172:1025–1033. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung S, Huitinga I, Schmidt B, Zielasek J, Dijkstra CD, Toyka KV, Hartung HP. Selective elimination of macrophages by dichlormethylene diphosphonate-containing liposomes suppresses experimental autoimmune neuritis. J Neurol Sci. 1993;119:195–202. doi: 10.1016/0022-510x(93)90134-k. [DOI] [PubMed] [Google Scholar]
- 14.Lindberg RLP, Martini R, Baumgartner M, Erne B, Borg J, Zielasek J, Ricker K, Steck A, Toyka KV, Meyer UA. Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest. 1999;103:1127–1134. doi: 10.1172/JCI5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linington C, Lassmann H, Ozawa K, Kosin S, Mongan L. Cell adhesion molecules of the immunoglobulin supergene family as tissue-specific autoantigens: induction of experimental allergic neuritis (EAN) by PO protein-specific T cell lines. Eur J Immunol. 1992;22:1813–1817. doi: 10.1002/eji.1830220721. [DOI] [PubMed] [Google Scholar]
- 16.Malandrini A, Villanova M, Dotti MT, Frederico A. Acute inflammatory neuropathy in Charcot-Marie-Tooth disease. Neurology. 1999;52:859–861. doi: 10.1212/wnl.52.4.859. [DOI] [PubMed] [Google Scholar]
- 17.Martini R. Animal models for inherited peripheral neuropathies. J Anat. 1997;191:321–336. doi: 10.1046/j.1469-7580.1997.19130321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martini R, Schachner M. Molecular bases of myelin formation as revealed by investigations on mice deficient in glial cell surface molecules. Glia. 1997;19:298–310. [PubMed] [Google Scholar]
- 19.Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat Genet. 1995a;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- 20.Martini R, Mohajeri MH, Kasper S, Giese KP, Schachner M. Mice doubly deficient in the genes for P0 and myelin basic protein show that both proteins contribute to the formation of the major dense line in peripheral nerve myelin. J Neurosci. 1995b;15:4488–4495. doi: 10.1523/JNEUROSCI.15-06-04488.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martini R, Zielasek J, Toyka KV. Inherited demyelinating neuropathies: from gene to disease. Curr Opin Neurol. 1998;11:545–556. doi: 10.1097/00019052-199810000-00018. [DOI] [PubMed] [Google Scholar]
- 22.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 23.Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Genet. 1999;13:11–28. doi: 10.1002/(SICI)1098-1004(1999)13:1<11::AID-HUMU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 24.Neuhaus O, Emoto M, Kaufmann SH. Constitutive biological activity of thymus-independent TCR-alpha-beta+ intestinal intraepithelial lymphocytes in TCR-alpha−/− gene disruption mice. Immunol Lett. 1996;54:53–57. doi: 10.1016/s0165-2478(96)02647-8. [DOI] [PubMed] [Google Scholar]
- 25.Pette M, Fujita K, Wilkinson D, Altmann DM, Trowsdale J, Giegerich G, Hinkkanen A, Epplen JT, Kappos L, Wekerle H. Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc Natl Acad Sci USA. 1990;87:7968–7972. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pette M, Linington C, Gengaroli C, Grosse-Wilde H, Toyka KV, Hartung HP. T lymphocyte recognition sites on peripheral nerve myelin P0 protein. J Neuroimmunol. 1994;54:29–34. doi: 10.1016/0165-5728(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 27.Philpott KL, Viney JL, Kay G, Rastan S, Gardiner EM, Chae S, Hayday AC, Owen MJ. Lymphoid development in mice congenitally lacking T cell receptor alpha beta-expressing cells. Science. 1992;256:1448–1452. doi: 10.1126/science.1604321. [DOI] [PubMed] [Google Scholar]
- 28.Scherer SS. Molecular genetics of demyelination: new wrinkles on an old membrane. Neuron. 1997;18:13–16. doi: 10.1016/s0896-6273(01)80042-8. [DOI] [PubMed] [Google Scholar]
- 29.Scherer SS, Chance PF. Myelin genes: getting the dosage right. Nat Genet. 1995;11:226–228. doi: 10.1038/ng1195-226. [DOI] [PubMed] [Google Scholar]
- 30.Schmid CD, Schnell L, Schachner M, Martini R. Peripheral nerves of a mouse model for Charcot-Marie-Tooth neuropathy 1B show infiltration of macrophages and CD4- and CD8-positive lymphocytes. Soc Neurosci Abstr. 1996;22:835.819. [Google Scholar]
- 31.Schmidt B, Toyka KV, Kiefer R, Full J, Hartung HP, Pollard J. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barre syndrome and chronic inflammatory demyelinating neuropathy. Muscle Nerve. 1996;19:474–487. doi: 10.1002/(SICI)1097-4598(199604)19:4<474::AID-MUS8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.Shy ME, Arroyo E, Sladky J, Menichella D, Jiang H, Xu W, Kamholz J, Scherer SS. Heterozygous P0 knockout mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP). J Neuropathol Exp Neurol. 1997;56:811–821. [PubMed] [Google Scholar]
- 33.Stienekemeier M, Herrmann T, Kruse N, Weishaupt A, Weilbach FX, Giegerich G, Theofilopoulos A, Jung S, Gold R. Heterogeneity of T-cell receptor usage in experimental autoimmune neuritis in the Lewis rat. Brain. 1999;122:523–535. doi: 10.1093/brain/122.3.523. [DOI] [PubMed] [Google Scholar]
- 34.Tachi N, Kozuka N, Ohya K, Chiba S, Sasaki K. Tomaculous neuropathy in Charcot-Marie-Tooth disease with myelin protein zero gene mutation. J Neurol Sci. 1997;153:106–109. doi: 10.1016/s0022-510x(97)00202-5. [DOI] [PubMed] [Google Scholar]
- 35.Thomas FP, Lebo RV, Rosoklija G, Ding XS, Lovelace RE, Latov N, Hays AP. Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome. Acta Neuropathol (Berl) 1994;87:91–97. doi: 10.1007/BF00386259. [DOI] [PubMed] [Google Scholar]
- 36.Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodny EH, Karpati G, Carpenter S, Watters GV, Wheeler C, Witt D, Bodell A, Nelis E, Van Broeckhoven C, Lupski JR. Clinical phenotypes of different MPZ (P0) mutations may include Charcot Marie Tooth type 1B, Dejerine Sottas, and congenital hypomyelination. Neuron. 1996;17:451–460. doi: 10.1016/s0896-6273(00)80177-4. [DOI] [PubMed] [Google Scholar]
- 37.Weishaupt A, Giegerich G, Jung S, Gold R, Enders U, Pette M, Hayasaka K, Hartung HP, Toyka KV. T cell antigenic and neuritogenic activity of recombinant human peripheral myelin P2 protein. J Neuroimmunol. 1995;63:149–156. doi: 10.1016/0165-5728(95)00139-5. [DOI] [PubMed] [Google Scholar]
- 38.Williams LL, Shannon BT, O'Dougherty M, Wright FS. Activated T cells in type I Charcot-Marie-Tooth disease: evidence for immunologic heterogeneity. J Neuroimmunol. 1987;16:317–330. doi: 10.1016/0165-5728(87)90108-1. [DOI] [PubMed] [Google Scholar]
- 39.Williams LL, Kissel JT, Shannon BT, Wright FS, Mendell JR. Expression of Schwann cell and peripheral T-cell activation epitopes in hereditary motor and sensory neuropathy. J Neuroimmunol. 1992;36:147–155. doi: 10.1016/0165-5728(92)90046-n. [DOI] [PubMed] [Google Scholar]
- 40.Wong MH, Filbin MT. Dominant-negative effect on adhesion by myelin P0 protein truncated in its cytoplasmic domain. J Cell Biol. 1996;134:1531–1541. doi: 10.1083/jcb.134.6.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K, Filbin MT. Myelin Po protein mutated at Cys21 has a dominant-negative effect on adhesion of wild type Po. J Neurosci Res. 1998;53:1–6. doi: 10.1002/(SICI)1097-4547(19980701)53:1<1::AID-JNR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 42.Zielasek J, Martini R, Toyka KV. Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve. 1996;19:946–952. doi: 10.1002/(SICI)1097-4598(199608)19:8<946::AID-MUS2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]