

Abstract
Alzheimer's disease (AD) is characterized by the extracellular deposition of β-amyloid fibrils within the brain and the subsequent association and phenotypic activation of microglial cells associated with the amyloid plaque. The activated microglia mount a complex local proinflammatory response with the secretion of a diverse range of inflammatory products. Nonsteroidal anti-inflammatory drugs (NSAIDs) are efficacious in reducing the incidence and risk of AD and significantly delaying disease progression. A recently appreciated target of NSAIDs is the ligand-activated nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ). PPARγ is a DNA-binding transcription factor whose transcriptional regulatory actions are activated after agonist binding. We report that NSAIDs, drugs of the thiazolidinedione class, and the natural ligand prostaglandin J2 act as agonists for PPARγ and inhibit the β-amyloid-stimulated secretion of proinflammatory products by microglia and monocytes responsible for neurotoxicity and astrocyte activation. The activation of PPARγ also arrested the differentiation of monocytes into activated macrophages. PPARγ agonists were shown to inhibit the β-amyloid-stimulated expression of the cytokine genes interleukin-6 and tumor necrosis factor α. Furthermore, PPARγ agonists inhibited the expression of cyclooxygenase-2. These data provide direct evidence that PPARγ plays a critical role in regulating the inflammatory responses of microglia and monocytes to β-amyloid. We argue that the efficacy of NSAIDs in the treatment of AD may be a consequence of their actions on PPARγ rather than on their canonical targets the cyclooxygenases. Importantly, the efficacy of these agents in inhibiting a broad range of inflammatory responses suggests PPARγ agonists may provide a novel therapeutic approach to AD.
Keywords: Alzheimer's disease, β-amyloid, microglia, THP-1 monocytes, signal transduction, tyrosine kinase, inflammation, neurotoxicity, PPARγ, cyclooxygenase, TNFα, IL-6, NSAIDs, cytokines, COX-2
Alzheimer's disease (AD) is characterized by progressive cognitive impairment that is a consequence of extensive neuronal loss (Berg et al., 1993; Braak and Braak, 1997,1998; Braak et al., 1998). The principal pathological feature of the disease is the extracellular deposition of fibrillar amyloid and its compaction into senile plaques. The senile plaque is the focus of a complex cellular reaction involving the activation of both microglia and astrocytes adjacent to the amyloid plaque (McGeer and McGeer, 1998,1999; Kalaria, 1999). Microglia are the most abundant and prominent cellular component associated with the plaque. The plaque-associated microglia exhibit a “reactive” or “activated” phenotype and possess a ramified morphology whose processes envelop and invest the plaque. Microglia are the principal immune cell in the brain originating from mesodermally derived macrophages that become permanently resident in the brain during development (Streit and Kincaid-Colton, 1995). Like macrophages, microglia respond to various stimuli by acquisition of a reactive phenotype as evidenced by the elevated expression of a number of cell surface molecules, including major histocompatibility complex class II antigens, CD45, complement receptors CR3 (MAC-1) and CR4, immunoglobulin receptors FcγRI and FcγRII, and intercellular adhesion molecule-1 (McGeer et al., 1993; McGeer and McGeer, 1995;Kalaria, 1999). Activated microglia, like activated macrophages, secrete a diverse range of acute-phase proteins including α-anti-chymotrypsin, α-anti-trypsin, serum amyloid P, C-reactive protein, and complement components, among others (McGeer and Rogers, 1992). Importantly, activation of microglia results in the synthesis and secretion of the proinflammatory cytokines interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNFα) and the chemokine macrophage chemotactic protein-1 (McGeer and McGeer, 1995).
The appearance of inflammatory products in the AD brain has been interpreted as evidence of a chronic inflammatory component in the disease process and led to studies exploring the effect of anti-inflammatory drug treatment on the incidence of AD-related dementia (McGeer and Rogers, 1992; Aisen, 1997; McGeer and McGeer, 1997). In patient populations treated for extended periods with nonsteroidal anti-inflammatory drugs (NSAIDs), most notably rheumatoid arthritics and lepers, the risk of AD was dramatically reduced. A retrospective analysis of 17 independent epidemiological studies revealed a significant protective effect of these drugs (McGeer and McGeer, 1996). More recently, a longitudinal study of ∼1700 patients demonstrated that NSAID treatment reduced the risk of AD by ∼60% (Stewart et al., 1997). Results from other smaller studies demonstrated that NSAID treatment attenuated the loss of cognitive abilities and disease progression in AD patients (Rogers et al., 1993; Rich et al., 1995) and greatly reduced the number of plaque-associated reactive microglia (Mackenzie and Munoz, 1998). However, a major impediment to use of this class of drugs for treatment of AD is a number of serious side effects associated with the chronic use of NSAIDs.
The effects of NSAIDs are well described (Bjorkman, 1998; Bolten, 1998;Dubois et al., 1998; McCormack, 1998; Pairet and Van Ryn, 1998; Vane and Botting, 1998). The principal targets of NSAID action are thought to be the cyclooxygenases, the rate-limiting enzymes responsible for the conversion of arachadonic acid into inflammatory mediators, including prostaglandin E2 (PGE2). However, our understanding of the mechanism of drug action has been fundamentally changed recently with the discovery of new molecules that mediate their biological effects. Initially, NSAIDs were thought to act via inhibition of the ubiquitously and constitutively expressed cyclooxygenase-1 (COX-1); however, the discovery of a second cyclooxygenase inhibitable by NSAIDs, COX-2, forced a reevaluation of this view. COX-2 is an immediate early gene whose expression is rapidly induced in some cell types after stimulation and acts acutely to generate PGE2 and related molecules (Kaufmann et al., 1997). Paradoxically, therapeutic benefits of NSAIDs are typically observed at doses much greater than those required to inhibit the cyclooxygenases, suggesting that there are other targets of NSAID actions (Brooks and Day, 1991; Meade et al., 1993; Lehmann et al., 1997; Jiang et al., 1998).
It has been appreciated recently that NSAIDs can act to regulate gene expression directly via their interaction with a class of nuclear receptor superfamily members, termed peroxisome proliferator-activated receptors (PPAR) (Lehmann et al., 1997). The PPARs are lipid-activated DNA-binding proteins structurally related to the steroid and retinoic acid receptor families (Lemberger et al., 1996; Kliewer et al., 1999). PPARs are associated with sequence-specific promoter elements and transcriptionally regulate gene expression after ligand binding (Ricote et al., 1998). There are three PPAR isoforms (PPAR α, γ, and δ) that are differentially expressed. The natural ligands for this receptor family are fatty acids and lipid metabolites, with each PPAR family member displaying a distinct pattern of ligand specificity.
The PPAR family has well described roles in adipocytes and serves to regulate the expression of enzymes of lipid metabolism in these cells (Lemberger et al., 1996; Spiegelman, 1998). An appreciation of the function of these receptors has been substantially enlarged by the recent finding that the PPARγ isoform is expressed in monocytes and macrophages in which its principal action is to suppress the expression of the proinflammatory cytokines IL-1β, TNFα, and IL-6 and other proinflammatory products (Jiang et al., 1998; Ricote et al., 1998). Importantly, activation of PPARγ acts to negatively regulate macrophage activation and cytokine expression by antagonizing the activity of the transcription factors NFkB, AP-1, and STAT proteins (Lemberger et al., 1996; Ricote et al., 1998). The natural ligand for PPARγ is the J class prostaglandin PGJ2 (15d-PGJ2) and its immediate metabolites (Forman et al., 1995; Kliewer et al., 1995; Yu et al., 1995). The J class of prostaglandins diffuses across the membrane and directly binds to PPARγ in a manner directly analogous to the mechanism of action of steroid hormones. Importantly, the PPARγ isoform can also be activated by indomethacin and other NSAIDs (Lehmann et al., 1997), the antidiabetic drugs of the thiazolidinedione class (Lehmann et al., 1995), as well as the naturally occurring fatty acid docosahexaenoic acid (DHA) (Yu et al., 1995).
We report that PPARγ agonists act broadly to inhibit the production of proinflammatory and neurotoxic products elaborated by β-amyloid (Aβ)-stimulated microglial cells and monocytes. We argue that the efficacy of NSAIDs in the treatment of AD and perhaps other inflammatory diseases is likely to be attributable primarily to their actions on PPARγ rather than on their established targets—the cyclooxygenases. The capacity of PPARγ agonists to suppress the expression of proinflammatory cytokines and the action of NSAIDs at this receptor suggests that agents that selectively activate PPARγ in microglia may be of value in the treatment of AD or other inflammatory diseases.
MATERIALS AND METHODS
Materials. The anti-COX-2 and anti-MAC-1 antibodies were obtained from Transduction Laboratories (Lexington, KY) and Boehringer Mannheim (Indianapolis, IN), respectively. Anti-glial fibrillary acidic protein (anti-GFAP) antibody was from Accurate Chemicals (Westbury, NY). Anti-MAP2 antibody was purchased from Sigma (St. Louis, MO). Anti-extracellular signal-regulated kinase 2 (anti-ERK2) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phosphotyrosine antibody 4G10 was from Upstate Biotechnology (Lake Placid, NY). Goat anti-mouse F(ab)2 was obtained from Cappel (West Chester, PA). Anti-5-bromo-2′-deoxyuridine (anti-BrdU) antibody was from Harlan Bioproducts for Science (Indianapolis, IN). Affinity-purified horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from Boehringer Mannheim. Peptides corresponding to amino acids 25–35 and 1–40 of human β-amyloid were purchased from Bachem (Philadelphia, PA). Scrambled β-amyloid 25–35 peptide was synthesized at Gliatech (Cleveland, OH). β-Amyloid peptides were resuspended in sterile dH20. Fibrillar β-amyloid 1–40 and 25–35 peptides were prepared by reconstitution of the lyophilized peptides in sterile distilled water, followed by incubation for 1 week at 37°C. Phorbol 12-myristate 13-acetate (TPA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Concanavalin A (Con A) were purchased from Sigma. BrdU was purchased from Boehringer Mannheim. Ciglitazone was obtained from Biomol(Plymouth Meeting, PA). DHA and prostaglandin J2 (15d-PGJ2) were obtained from Calbiochem (San Diego, CA). Troglitazone was a gift from Dr. Charles Burant (Parke-Davis, Ann Arbor, MI).
Tissue culture. THP-1 cells are a monocytic cell line derived from peripheral blood of a human with acute monocytic leukemia and were purchased from the American Type Culture Collection (Rockville, MD). THP-1 cells were grown in RPMI 1640 (Whittaker Bioproducts, Walkersville, MD) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 5 × 10−5m 2-mercaptoethanol, 5 mm HEPES, and 2 μg/ml gentamycin in 5% CO2. Microglial and astrocyte cultures were derived from postnatal day 1 to 2 mouse brain (C57Bl/6J) as described previously (McDonald et al., 1997). Astrocytes were recovered after harvesting of microglia and were serially passaged to enrich for astrocytes. For experiments, astrocytes were plated into Neurobasal media with B27 supplements (∼70% confluency) for stimulation with conditioned media, drugs, and ligands. Neurons were cultured from cortices of embryonic day 16 (E16) mice (C57Bl/6J). Meninges-free cortices were isolated and digested in 0.25% trypsin and 1 mm EDTA for 15 min at 37°C. The trypsin was inactivated with DMEM containing 20% heat-inactivated FCS. Cortices were transferred to Neurobasal media with B27 supplements, triturated, and plated onto poly-l-lysine (0.05 mg/ml)-coated tissue culture wells. Neurons were grown in Neurobasal media (4.0 × 104/24-well tissue culture plate) with B27 supplements for 5 d in vitro before use.
Cell stimulation. THP-1 cells and microglia were stimulated by first removing their respective media and replacing the media with serum-free RPMI for suspension stimulation or by plating the cells onto Aβ peptides bound to the surface of the dish (48 pmol/mm2). Bound fibrillar peptides were prepared as described previously (Lagenaur and Lemmon, 1987). Briefly, tissue culture wells were coated with nitrocellulose, and peptides were added to the coated wells and allowed to dry, immobilizing the peptides. THP-1 cells and microglia (1.8 × 104 cells) were added to wells containing the bound peptides in 48-well tissue culture dishes in 0.5 ml of Neurobasal media for 48 hr in the presence or absence of the indicated drugs. The conditioned media were clarified by centrifugation and added to neuronal and astrocytic cultures for 72 hr. Microglia were fixed and immunostained for MAC-1 expression. Neurons were fixed, stained, and counted using mouse anti-MAP2 antibody. BrdU was added (final concentration, 10 μm) to astrocyte cultures during stimulation with conditioned media, drugs, or ligands. Astrocytes were fixed and double-stained for GFAP and BrdU using rabbit anti-GFAP and rat anti-BrdU, respectively. A counting grid was placed over the wells to count neuron and astrocyte numbers from eight identical fields for each condition. The average number of neurons and astrocytes (± SEM) was calculated for each condition. Each experiment was performed in duplicate and repeated three to four times.
Western blotting. Cells were lysed in 200 μl of ice-cold radioimmunoprecipitation assay (RIPA) buffer (1% Triton, 0.1% SDS, 0.5% deoxycholate, 20 mm Tris, pH 7.4, 150 mmNaCl, 10 mm NaF, 1 mmNa3VO4, 1 mmEDTA, 1 mm EGTA, and 0.2 mm PMSF), and insoluble material was removed by centrifugation at 10,000 ×g at 4°C for 10 min. Protein concentrations were quantitated by the method of Bradford (1976). Proteins were resolved by 7.5% SDS-PAGE and Western blotted with primary antibody [4G10 (1:2000), COX-2 (1:250), or ERK2 (1:5000)] overnight at 4°C. Antibody binding was detected via enhanced chemiluminescence (Pierce, Rockford, IL).
Monocyte differentiation. THP-1 monocytes were stimulated with 100 nm TPA in normal growth media for 48 hr to induce differentiation into an activated macrophage phenotype (Tsuchiya et al., 1982). Inhibition of monocyte differentiation was assayed by incubating the monocytes in the presence or absence of PPARγ agonists (10 μm 15d-PGJ2, 50 μm troglitazone, and 50 μm DHA) during the 48 hr stimulation with 100 nm TPA. Cells were fixed in 4% paraformaldehyde for 30 min at 37°C, and phase-contrast images were recorded.
Cyclooxygenase-2 expression. For Western analysis, THP-1 monocytes were incubated with TPA (100 nm) or fibrillar Aβ25–35 and Aβ1–40 (60 μm) for 18 hr in RPMI medium containing 5% FBS in the presence or absence of the various drugs, and COX-2 expression was evaluated by Western analysis. Cells were lysed in RIPA buffer, and aliquots of the cellular lysates were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed with an antibody recognizing COX-2. For immunocytochemistry, microglia (1.8 × 104 cells) were added to wells containing the bound peptides in Neurobasal media for 48 hr in the presence or absence of the indicated drugs. Cells were fixed and stained using an anti-COX-2 antibody.
TNFα and IL-6 reporter assays. Luciferase reporter constructs for the human IL-6 and TNFα genes were transfected into THP-1 cells using DEAE-dextran together with a β-galactosidase reporter construct to control for transfection efficiency (Yao et al., 1997). The cells were transfected and 48 hr later stimulated for 6 hr in serum-free RPMI media in the presence or absence of drugs and fibrillar Aβ25–35 (60 μm), Aβ1–40 (40 μm), or lipopolysaccharide (LPS; 5 μg/ml). The cells were lysed, and luciferase activity was measured and normalized to β-galactosidase activity. All conditions were performed in duplicate in three separate experiments.
Immunocytochemistry. For immunocytochemistry, cells were fixed in 4% paraformaldehyde for 30 min at 37°C. Neurons were stained with a mouse anti-MAP2 antibody (1:500). Astrocytes were double-stained with a rabbit anti-GFAP antibody (1:1000) and rat anti-BrdU (1:500). Microglial cells were stained with mouse anti-COX-2 (1:250) and rat anti-MAC-1 (1:20). Immunoreactivity was visualized using either 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories, Burlingame, CA) (MAP2, GFAP, and COX-2) or Vector VIP (Vector Laboratories) (BrdU) as the chromogens or indocarbocyanine (Cy3)-conjugated rabbit anti-mouse (1:200; The Jackson Laboratory, Bar Harbor, ME) (COX-2).
MTT reduction assay. Active mitochondrial dehydrogenases of living cells convert soluble MTT to a water-insoluble formazan. Formazan is soluble in isopropanol, and dissolved material is measured spectrophotometrically at a wavelength of 570 nm with background subtraction at 630 nm yielding absorbance as a function of concentration of reduced MTT. The MTT assay was performed according to the manufacturer's specifications (Sigma). Briefly, THP-1 cells were plated into Neurobasal media with B27 supplements (4.0 × 104/24-well tissue culture plate) in the presence of PPARγ agonists at the maximal concentrations used for promoting neuron survival for 48 hr. During the last 4 hr of incubation, MTT was added to the THP-1 cells at a final concentration of 0.1 μg/ml. The dye was dissolved in 0.04N HCl/isopropanol, and absorbances were read. Numbers of percent control viable cells were determined from absorbance values. Experiments were performed in triplicate in three independent experiments, and mean values (± SEM) were determined.
Statistical analysis. All experiments were performed in duplicate or triplicate a minimum of three to four times. Mean values (± SEM) for each experiment were determined, and values statistically different from controls were calculated using one-way ANOVA. The Tukey-Kramer multiple comparisons post test was used to determinep values.
RESULTS
PPARγ agonists prevent microglial- and monocyte-mediated neurotoxicity
Microglial activation is accompanied by the secretion of numerous acute-phase and proinflammatory products that typify macrophage responses in the periphery. Numerous studies have described the ability of microglial lineage cells to generate neurotoxic products in response to treatment with Aβ peptides (Banati et al., 1993; Giulian, 1993;Giulian et al., 1995, 1996). A variety of the microglial secretory products have been reported to be toxic to neurons including cytokines, chemokines, and reactive oxygen and nitrogen species as well as undefined neurotoxic components (Brown et al., 1996; Ii et al., 1996;Kretzschmar et al., 1997). The release of these neurotoxic products represents the outcome of a coordinated program of intracellular signaling events mediating a proinflammatory response. We have used a well characterized tissue culture model system in which highly purified populations of primary cortical neurons are cultured in conditioned media from THP-1 cells or primary microglia to evaluate and quantitate the elaboration of neurotoxic and proinflammatory products (Giulian et al., 1996; Combs et al., 1999). Conditioned medium was prepared by incubating THP-1 monocytes and primary mouse microglia in the absence or presence of Aβ fibrils for 48 hr. The conditioned medium was collected and applied to purified mouse cortical neuronal cultures, and neuronal viability was measured 72 hr later. Conditioned medium from untreated THP-1 cells and microglia exhibited little or no neurotoxicity. However, the conditioned medium from THP-1 cells and microglia exposed to fibrillar Aβ was highly neurotoxic, killing the majority of the neurons within 72 hr (Fig.1A,C). The cell death was not a consequence of dissociation of the bound Aβ fibrils from the plate into the media. We demonstrated previously in this system that no neuronal death occurs when medium is taken from wells containing only bound Aβ fibrils (and no THP-1 cells) and then applied to neurons (Combs et al., 1999). Furthermore, the neurotoxicity is specific to the fibrillar forms of the peptides because stimulation of THP-1 cells with a scrambled, nonfibillar form of the Aβ peptides did not result in neurotoxicity. This is consistent with our previous work demonstrating that only the fibrillar forms of the Aβ peptides are capable of stimulating an inflammatory response in microglia and monocytes (McDonald et al., 1997, 1998; Combs et al., 1999). If the THP-1 cells were exposed to Aβ in the presence of the NSAIDs and PPARγ agonists ibuprofen or indomethacin, the production of neurotoxins was inhibited. Similarly, the PPARγ agonists 15d-PGJ2 and DHA and the thiazolidinediones ciglitizone and troglitazone also arrested the production of neurotoxins. The specificity of the action of PPARγ ligands on microglia and monocytes to mediate neuroprotection was established by first stimulating THP-1 cells with bound Aβ fibrils in the absence of any drugs and then adding drugs directly to the neuronal cultures themselves together with the THP-1 cell-conditioned media (Fig. 1B), allowing the evaluation of the ability of PPARγ ligands to act on neuronal PPARγ to mediate neuron survival. No significant improvement in neuronal survival was achieved by direct application of PPARγ agonists to the neurons, demonstrating that the effects of these drugs were exerted via their action on cells of the microglial lineage and not on the neurons themselves (Fig. 1B). These data demonstrate that a variety of PPARγ agonists act to suppress the elaboration of proinflammatory neurotoxic products from activated microglia and macrophages.
Fig. 1.

PPARγ agonists prevent neuronal death induced by β-amyloid-stimulated microglia and monocytes. THP-1 monocytes (A, B) or microglia (C) were stimulated for 48 hr with β-amyloid by plating the cells into uncoated wells (black bars) or wells coated with fibrillar Aβ25–35 (light gray bars) or Aβ1–40 (speckled bars) or nonfibrillar, scrambled Aβ25–35 (scr; negative control; striped bar) (all used at 48 pmol/mm2). A, C, The cells were cultured for 48 hr in the absence or presence of DMSO vehicle (control), 15d-PGJ2 (10 μm), troglitazone (50 μm), ciglitizone (50 μm), DHA (50 μm), indomethacin (100 μm), ibuprofen (600 μm), or NS-398 (5 μm). The conditioned media (CM) were collected, added to purified cultures of mouse cortical neurons (E16; 4.0 × 104 neurons/well; 5 d in vitro), and incubated for 72 hr. The neuronal cultures were then fixed and stained for neuron-specific MAP2 protein, and surviving neurons were counted. B, In parallel experiments, the specificity of PPARγ ligand action for microglia and monocytes was demonstrated by plating THP-1 cells onto uncoated wells or wells coated with Aβ25–35 (light gray bars; 48 pmol/mm2) followed by incubation for 48 hr. The CM from the THP-1 cells stimulated with Aβ25–35 were collected and added to mouse cortical neurons (4.0 × 104 neurons/well; 5 d in vitro) along with PPARγ ligands (light gray bars): 15d-PGJ2 (5 μm), troglitazone (25 μm), ciglitizone (25 μm), DHA (50 μm), indomethacin (100 μm), and ibuprofen (600 μm). Media from unstimulated THP-1 cells were added to neurons along with DMSO vehicle (black bars; control). PPARγ agonists alone were added to neurons as well (stripedbars). The neuronal cultures were stained for neuron-specific MAP2 protein, and surviving neurons were counted. The mean values shown (± SEM) are representative of four independent experiments. Unpaired ANOVA was performed with Tukey-Kramer post comparison to evaluate statistical significance (* =p < 0.001).
Importantly, exposure of monocytes to Aβ in the presence of a specific inhibitor of COX-2 activity, NS-398, did not alter neurotoxicity of the conditioned media (Fig. 1A). Concentrations of NS-398 well above the reported IC50 for human COX-2 (0.1 μm) failed to promote neuron survival, providing direct evidence that cyclooxygenase products are not responsible for microglial-mediated neuronal toxicity (Ouellet and Percival, 1995). Additionally, others have shown recently using a similar experimental system that the neuroprotective effect of NSAIDs is independent of the ability of these drugs to inhibit cyclooxygenase activities (Klegeris et al., 1999).
Exposure of THP-1 monocytes or primary microglial cells to fibrillar forms of Aβ results in the stimulation of protein tyrosine phosphorylation as a consequence of the activation of the tyrosine kinases Lyn, Syk, FAK, and Pyk2 (McDonald et al., 1997, 1998; Combs et al., 1999). We tested whether PPARγ agonists could affect the activation of the kinases and elements of the signal transduction apparatus mediating the responses of these cells to Aβ. The PPARγ agonists 15d-PGJ2, ciglitizone, and troglitazone did not significantly alter the induction of protein tyrosine phosphorylation after Aβ exposure, demonstrating that PPARγ agonists do not interact with the principal catalytic components of the signal transduction cascades linked to the inflammatory responses in these cells (Fig.2A). Incubation of THP-1 cells with PPARγ ligands at the concentrations shown to provide maximal neuroprotection had no dramatic effects on the viability of the THP-1 cells (Fig. 2B). Although ibuprofen caused a slightly significant decrease in THP-1 cell survival during the 48 hr incubation, this could not account for the dramatic increase in neuronal survival resulting from the same treatment.
Fig. 2.

β-Amyloid fibrils activate a tyrosine kinase-dependent signaling cascade in microglial lineage cells that is unaffected by PPARγ agonists. A, The effect of PPARγ agonists on the activation of the Aβ-stimulated tyrosine kinase-signaling cascade was examined by incubating THP-1 cells for 24 hr with DMSO vehicle only or with 10 μm 15d-PGJ2, 50 μm ciglitizone, or 50 μmtroglitazone and then stimulating the cells with 60 μmfibrillar Aβ25–35 in suspension in serum-free media for 5 min. Cell lysates were examined by Western blot using the anti-phosphotyrosine antibody 4G10. B, The MTT reduction assay was performed on THP-1 cells stimulated with PPARγ agonists and NS-398 to evaluate the toxicity of these drugs. Cells were stimulated for 48 hr at 37°C with DMSO vehicle (control) or 10 μm 15d-PGJ2, 50 μm ciglitizone, 50 μm troglitazone, 50 μm DHA, 100 μm indomethacin, 600 μm ibuprofen, and 5 μm NS-398. MTT was added during the last 4 hr of stimulation. The percent control MTT reduction was calculated on the basis of the absorbance of the reduced MTT product formazan at 570 nm. Mean values are shown (± SEM) from an experiment performed in triplicate and independently repeated three times. Unpaired ANOVA was performed with Tukey-Kramer post comparison to evaluate statistical significance (* = p < 0.05).
Microglial-mediated astrocyte proliferation is blocked by PPARγ agonists
Astrogliosis is observed in a number of CNS diseases, including AD, and in response to both acute and chronic brain insults. A primary response of astrocytes in these settings is the elevated expression of the intermediate filament protein GFAP and the increased cell division that serve as prototypic markers of astrocyte activation. We tested whether Aβ-stimulated secretion of astrocytic mitogens by monocytes was inhibited by PPARγ agonist treatment of the monocytes. Exposure of primary mouse astrocytes to conditioned medium from untreated THP-1 monocytes did not lead to detectable differences in astrocytic DNA synthesis compared with that of control cultures (as measured by the incorporation of the nucleotide analog BrdU). However, conditioned medium from activated, Aβ-treated monocytes provoked a dramatic increase in the numbers of dividing astrocytes to levels ∼50% of that stimulated by 10% FBS (Fig.3). Importantly, conditioned media from THP-1 monocytes that were treated simultaneously with Aβ and the PPARγ agonist troglitazone did not provoke astrocyte cell division. Treatment of the monocytes with the PPARγ agonist reduced the number of BrdU-positive astrocytes by 85%. These observations provide evidence that the PPARγ agonists inhibit the production of monocytic and microglial secretory products that are responsible for astrocyte proliferation.
Fig. 3.

PPARγ agonists prevent astrocyte proliferation induced by β-amyloid-stimulated monocytes. Conditioned medium was collected from THP-1 monocytes that had been incubated alone or were stimulated with fibrillar Aβ for 48 hr by plating onto fixed, surface-bound fibrillar Aβ25–35 or Aβ1–40 or nonfibrillar, scrambled Aβ25–35 (negative control) (48 pmol/mm2) in the presence of DMSO vehicle or troglitazone (50 μm). The conditioned medium (CM) was collected from the THP-1 cultures and applied to purified mouse astrocyte cultures for 72 hr. Treatments added to astrocyte cultures were as follows: conditioned medium from THP-1 cells only with DMSO vehicle (control), conditioned medium from Aβ25–35-stimulated THP-1 cells (Aβ25–35 CM), conditioned medium from Aβ1–40-stimulated THP-1 cells (Aβ1–40 CM), conditioned medium from THP-1 cells stimulated with Aβ25–35 in the presence of 50 μm troglitazone (Aβ25–35/trog CM), medium alone with 50 μm troglitazone (trog), conditioned medium from Aβ25–35-stimulated THP-1 cells added along with direct addition of 50 μmtroglitazone to astrocytes (CM+trog), medium alone with 20 μm Aβ25–35 (Aβ25–35), conditioned medium from scrambled Aβ25–35-stimulated THP-1 cells (scrAβ25–35 CM), and medium alone with 10% heat-inactivated FBS (FBS). Addition of FBS served as a positive control for mitogen-stimulated astrocyte cell division. The control for the effect of direct Aβ peptide stimulation of astrocytes was performed by adding, in suspension, 20 μm Aβ25–35 directly to astrocytic cultures for 72 hr. The controls for the effects of PPARγ agonists on the astrocytes themselves were performed by adding 50 μm troglitazone directly to astrocytes or adding 50 μm troglitazone directly to astrocyte cultures together with conditioned media from Aβ-stimulated THP-1 cells. The astrocytes were fixed and double-labeled for GFAP and BrdU. The numbers of BrdU-positive astrocytes were counted and graphed as a percentage of control. Shown are mean values (± SEM) from one of three representative experiments performed in duplicate. Unpaired ANOVA was performed with Tukey-Kramer post comparison to evaluate statistical significance (* = p < 0.001).
Importantly, control studies established that direct Aβ peptide stimulation of astrocytes was not sufficient to induce astrocyte cell division, confirming the importance of monocyte- and microglial-secreted proinflammatory products in this response. Nonfibrillar, scrambled Aβ peptides were not able to stimulate THP-1 cells to secrete astrogliotic factors, demonstrating the specificity of this response for fibrillar peptides. We evaluated the effect of PPARγ agonists on astrocyte proliferation by treating astrocytes with Aβ-stimulated THP-1-conditioned media and then adding troglitazone. Troglitazone was able to reduce modestly the number of astrocytes stimulated to divide by conditioned media from Aβ-stimulated THP-1 cells (Fig. 3). This suggests that PPARγ agonists also act on astrocytes and suppress their response to proinflammatory stimuli.
PPARγ agonists prevent the differentiation of THP-1 monocytes into macrophages
Monocytes undergo a morphological and biochemical differentiation into a macrophage phenotype after exposure to phorbol ester or other activating stimuli (Tsuchiya et al., 1982). The phenotypic conversion of THP-1 monocytes into macrophages was stimulated by a 48 hr exposure of the cells to TPA (100 nm; Fig.4). Concomitant exposure of the cells to TPA and the PPARγ agonists 15d-PGJ2, DHA, or troglitazone blocked the differentiation of the cells. These data provide direct evidence that PPARγ agonists act to inhibit a broad range of cellular activities that participate in the differentiation of these cells. Moreover, these findings are consistent with a role for these agents acting as anti-inflammatory agents via their capacity to block the generation of a reactive phenotype in cells of this lineage.
Fig. 4.
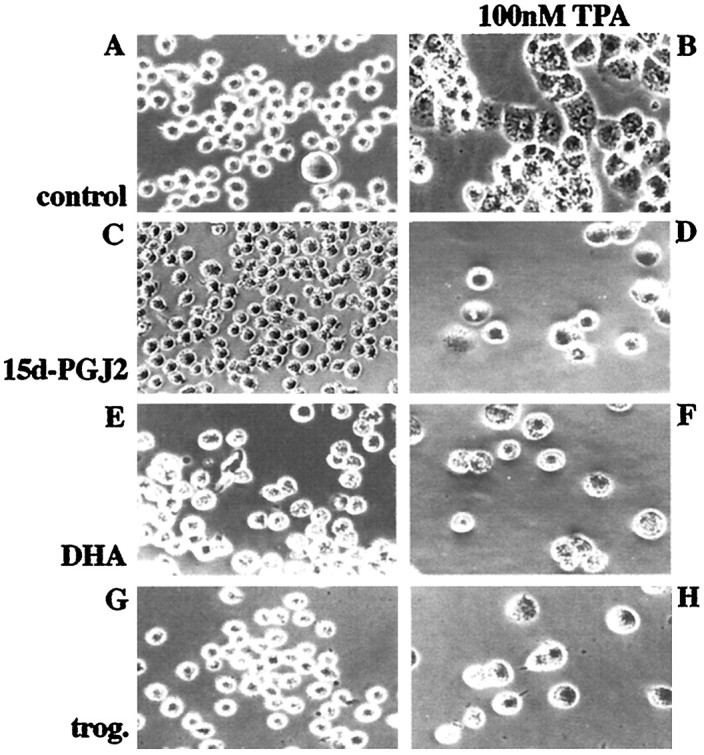
PPARγ agonists prevent differentiation of THP-1 monocytes into macrophages. THP-1 monocytes were incubated with vehicle only (control; DMSO and ethanol; A) or induced to differentiate into macrophages by treatment with 100 nm TPA for 48 hr (B, D, F, H) in the absence (A, B) or presence of 10 μm 15d-PGJ2 (C, D), 50 μmDHA (E, F), or 50 μm troglitazone (trog.; G, H).
PPARγ agonists prevent acquisition of an activated phenotype by microglia
Microglia are derived from a monocytic lineage and act functionally as macrophages in the brain (Banati et al., 1993). Microglia inducibly express a number of cell surface receptors when activated. Increased expression of the β2-integrin CD11b/CD18 (MAC-1) by plaque-associated microglia in the AD brain (McGeer and McGeer, 1995) as well as its transgenic mouse models (Sturchler-Pierrat et al., 1997; Frautschy et al., 1998) has been reported. Aβ treatment of primary microglial cultures resulted in morphological changes as well as dramatically increased expression of MAC-1 immunoreactivity. The induction of MAC-1 expression was inhibited by coincubating cultures with the PPARγ agonist troglitazone (Fig.5). These data demonstrate that PPARγ agonists act to inhibit those cellular activities responsible for activation and phenotypic conversion of microglial cells.
Fig. 5.

Aβ-stimulated MAC-1 expression is inhibited by PPARγ agonists. Primary mouse microglia were plated onto uncoated culture wells (A, C) or surface-bound fibrillar Aβ1–40 (48 pmol/mm2; B, D) for 48 hr in the presence of vehicle (DMSO; A, B) or 10 μm troglitazone (trog.; C, D). Cells were fixed and stained for MAC-1. Immunoreactivity was visualized using 3,3′-diaminobenzidine tetrahydrochloride as the chromogen.
IL-6 and TNFα expression is inhibited by PPARγ agonists
One consequence of microglial activation by Aβ or other immune stimuli is the stimulation of cytokine production. We tested whether PPARγ agonists would affect the Aβ-stimulated expression of the IL-6 and TNFα genes. We used a luciferase reporter linked to the promoter elements of the human genes. As predicted, stimulation of THP-1 cells with LPS resulted in increased activity of both the IL-6 and TNFα promoters and served simply as a positive control. Aβ treatment of THP-1 cells resulted in the stimulation of both promoter activities as well (Fig. 6), consistent with the previously reported effects of these peptides on cytokine production (Del Bo et al., 1995; Meda et al., 1996; Klegeris et al., 1997a; Yao et al., 1997; Fiala et al., 1998). Incubation of THP-1 cells with the natural PPARγ agonists 15d-PGJ2 and DHA resulted in inhibition of promoter activity. Similarly, the thiazolidinediones troglitazone and ciglitizone as well as ibuprofen and indomethacin also blocked expression of the reporter. These data demonstrate that a diverse range of PPARγ agonists efficiently suppressed expression of the IL-6 and TNFα genes.
Fig. 6.

PPARγ agonists inhibit IL-6 and TNFα gene expression. THP-1 cells were transiently transfected with TNFα–luciferase reporter (A, B) or IL-6–luciferase reporter (C, D) constructs and assayed for promoter activity 48 hr later. The cells were cotransfected with a β-galactosidase–reporter construct to control for transfection efficiency. During the last 4 hr the cells were incubated alone (blackbars), with fibrillar Aβ1–40 (40 μm;stripedbars;B, D) or Aβ25–35 (60 μm;speckledbars; A, C), or with 5 μg/ml LPS (positive control; open bars) in the presence or absence of troglitazone (20 μm), ciglitizone (50 uM), DHA (100 μm), 15d-PGJ2 (50 μm), ibuprofen (3 mm), or indomethacin (200 μm). The data shown represent the average (± SEM) of three independent experiments. Unpaired ANOVA was performed with Tukey-Kramer post comparison to evaluate statistical significance (* =p < 0.001).
Cyclooxygenase-2 expression is blocked by PPARγ agonists
COX-2 is inducibly expressed in response to a variety of immune stimuli. Aβ and TPA treatment of THP-1 monocytes resulted in the induction of COX-2 expression (Fig. 7). However, coincubation of the cells with the PPARγ agonist 15d-PGJ2 (Fig. 7B) inhibited the Aβ-mediated increase in COX-2 expression. Primary microglial cells stimulated with Aβ also showed a dramatic increase in COX-2 immunoreactivity (Fig.8) that was inhibited by coincubation of microglia with the PPARγ agonist troglitazone. Levels of ERK2 are not acutely regulated and thus serve as a control for protein loading. These observations are of particular significance because they potentially provide a novel therapeutic approach for suppression of COX-2 action in AD and other inflammatory disorders.
Fig. 7.

Aβ-stimulated COX-2 expression in THP-1 cells is inhibited by PPARγ agonists. A, THP-1 monocytes were incubated alone (c), with fibrillar Aβ25–35 (60 μm) or Aβ1–40 (60 μm) in suspension, or with 100 nm TPA for 18 hr in serum-free RPMI, and COX-2 expression was monitored by Western analysis of cellular lysates using an anti-COX-2-specific antibody. The blots were reprobed using an anti-ERK2 antibody as a control for protein loading. B,THP-1 cells were incubated in vehicle alone (c; DMSO) or stimulated with fibrillar Aβ25–35 (60 μm) or TPA (100 nm) in the presence or absence of the PPARγ agonist 15d-PGJ2 (50 μm).
Fig. 8.

Aβ-stimulated COX-2 expression in primary microglia is inhibited by PPARγ agonists. Primary mouse microglia were plated onto underivatized culture wells (A, C) or surface-bound Aβ1–40 (48 pmol/mm2; B, D) for 48 hr in the presence of vehicle (DMSO; A, B) or 10 μm troglitazone (trog.;C, D). Cells were fixed and stained for COX-2. Immunoreactivity was visualized using Cy3-conjugated rabbit anti-mouse antibody.
DISCUSSION
Deposition of β-amyloid into insoluble plaques represents the hallmark pathology of Alzheimer's disease brains. Genetic evidence of autosomal dominant linkage of AD to mutations in the gene coding for the precursor protein of Aβ peptides, amyloid precursor protein (APP), and mutations in the presenilin genes mediating proteolytic processing of APP argues that fibrillar peptide formation and deposition are key events in the pathophysiology of the disease. This study focuses on the secondary, inflammatory events that occur in the brain as a response to the extracellular deposition of Aβ fibrils. The primary immune effector cell responsible for this inflammatory component of the disease is the microglial cell. Microglia associated with the senile plaque exhibit enhanced expression of a variety of cell surface proteins and possess a ramified morphology, features that typify a reactive phenotype. It is clear that the sustained, intimate contact of microglia with the Aβ-containing plaques results in the prolonged activation of these cells and acquisition of a reactive phenotype (McGeer and McGeer, 1998, 1999; Kalaria, 1999). The consequence of microglial activation is a fulminating process resulting in the astrogliosis and neuronal cell death that characterize the pathology of the AD brain. Thus, the microglia respond to the deposited amyloid fibrils and are responsible for the generation of a local inflammatory response centered on the senile plaque.
We demonstrated previously that exposure of microglia to β-amyloid fibrils provokes the activation of complex intracellular tyrosine kinase-based signal transduction pathways leading to production of reactive oxygen species and neurotoxins (McDonald et al., 1997, 1998;Combs et al., 1999). Significantly, the intracellular mechanisms via which microglia respond to Aβ are similar to those used by these cells in response to other immune stimuli, resulting in the elaboration of an extraordinary range of bioactive molecules (McGeer and Rogers, 1992; McGeer et al., 1993, 1996). The primary downstream targets of these intracellular signaling pathways are transcription factors that positively regulate expression of cytokines and other proinflammatory products.
The involvement of an inflammatory component in the pathophysiology of AD is supported by a large body of data that has documented the detection of elevated levels of inflammatory cytokines in the AD brain and the presence of a number of acute-phase products (McGeer and Rogers, 1992; McGeer et al., 1993; McGeer and McGeer, 1996, 1997). Therapeutic approaches that have targeted inflammatory processes have proven to be an effective strategy for slowing disease progression and dramatically decreasing incidence and risk (Rogers et al., 1993; Rich et al., 1995; McGeer and McGeer, 1996; Stewart et al., 1997) and reducing the numbers of plaque-associated microglia (Mackenzie and Munoz, 1998). Thus, drugs are currently available that clearly offer a therapeutic option for the treatment of AD. Unfortunately, prolonged treatment with existing NSAIDs results in severe side effects, most prominently in the gastrointestinal tract, that occur primarily as a consequence of the inhibition of COX-1 activity.
Previously, the anti-inflammatory actions of NSAIDs and their therapeutic benefit in treating AD have been attributed to the ability of these drugs to inhibit the cyclooxygenases and resultant PGE2 production (Smith et al., 1996; Kaufmann et al., 1997). This view of the mechanism of NSAID action has led to the recent initiation of clinical trials using COX-2-specific inhibitors for the treatment of AD. However, the relative significance of COX-2 and PGE2 in the pathophysiology of AD is unknown. There is a surprisingly modest literature on the role of PGE2 in the brain and its potential involvement in AD pathophysiology (Kaufmann et al., 1997). Our data demonstrate that PPARγ ligands act to prevent the increase in Aβ-stimulated COX-2 expression in microglia and monocytes. However, we determined that the neuroprotective effect of both the NSAIDs and PPARγ ligands in our in vitro assays was not attributable to a reduction in cyclooxygenase activity because the COX-2-specific inhibitor NS-398 failed to promote neuron survival. We conclude that although microglial COX-2 expression increases in response to Aβ stimulation, its activity and subsequent prostaglandin production are not necessary components in the neuronal death process we observed. It is important to note that therapeutically efficacious doses of NSAIDs are achieved at concentrations substantially greater than those required to inhibit cyclooxygenase activity (Brooks and Day, 1991;Meade et al., 1993; Lehmann et al., 1997; Jiang et al., 1998). Moreover, extended use of aspirin, a potent COX inhibitor, is not associated with a reduction in the risk of AD (Stewart et al., 1997). These data suggest that NSAIDs must also act via other mechanisms. We argue, particularly in the case of AD, that the beneficial aspects of NSAID therapy are attributable principally to the actions of these drugs acting as agonists for the transcription factor PPARγ, rather than via their ability to inhibit cyclooxygenase activity. We suggest that PPARγ agonists could provide an alternative to traditional NSAIDs as an efficacious anti-inflammatory therapy for AD.
The present work documents that the Aβ-stimulated proinflammatory response in microglia and monocytes can be broadly inhibited by agonists of PPARγ, particularly by members of the thiazolidinedione class of drugs, attenuating the activation of both microglia and astrocytes and the consequent elaboration of neurotoxins. Specifically, we demonstrate that PPARγ agonists block Aβ-stimulated microglial activation as evidenced by inhibition of MAC-1 expression as well as the arrest of macrophage differentiation. We have also demonstrated that PPARγ agonists dramatically inhibit Aβ-stimulated cytokine and COX-2 expression and secretion of neurotoxic products by microglia and macrophages. It is presently unclear which microglial products, alone or combinatorially, mediate the neurotoxic response (Haga et al., 1993;Giulian et al., 1995, 1996; Goodwin et al., 1995; Meda et al., 1995,1996; Ii et al., 1996; Lorton et al., 1996; Klegeris and McGeer, 1997;Klegeris et al., 1997a,b; McDonald et al., 1997; Weldon et al., 1998). The identification of the neurotoxic factor(s) secreted by microglia and monocytes in response to Aβ stimulation remains elusive and controversial. Our data suggest that PPARγ agonists act to suppress production of a wide range of proinflammatory products. These findings are consistent with reports demonstrating that NSAIDs act via PPARγ to prevent microglial and macrophage activation as evidenced by their ability to suppress the transcription of proinflammatory cytokines, matrix metalloproteinase, scavenger receptor A, and inducible nitric oxide synthase (Lehmann et al., 1997; Jiang et al., 1998; Ricote et al., 1998; Petrova et al., 1999). Interestingly, it has recently been reported that levels of PPARγ increase in AD compared with controls (Kitamura et al., 1999).
The present data provide evidence that the primary mechanism by which NSAIDs intervene in disease progression is via their capacity to act as agonists for PPARγ and alter, at the transcriptional level, microglial production of proinflammatory products. It is of some significance that many PPARγ agonists exhibit substantial bioavailability after oral administration and have little or no toxicity associated with their use (Jha, 1999; Plosker and Faulds, 1999; Subramaniam, 1999). Several new PPARγ agonists of the thiazolidinedione class have been developed recently for application in the treatment of diabetes mellitus; however, their anti-inflammatory properties have not yet been explored. We suggest that nervous system diseases involving a microglial- or macrophage-mediated inflammatory cascade will be susceptible to PPARγ regulation. We conclude that PPARγ agonists may be of therapeutic use for the treatment of AD as well as other indications having an inflammatory component including stroke, peripheral neuropathies, and traumatic injury of the nervous system.
Footnotes
This work was supported by the National Institutes of Health Grant AG08012 to G.E.L. and by the generous support of the Blachett Hooker Rockefeller Foundation. C.K.C. was supported by the National Institutes of Health Training Grant HD0710422. IL-6 and TNFα constructs were a generous gift from Dr. Andre Nel. Troglitazone was a gift from Dr. Charles Burant. We thank Drs. Patrick McGeer and Andis Klegeris for helpful discussion and comments.
Correspondence should be addressed to Dr. Gary Landreth, Alzheimer Research Laboratory, E504, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:gel2@po.cwru.edu.
REFERENCES
- 1.Aisen PS. Inflammation and Alzheimer's disease: mechanisms and therapeutic strategies. Gerontology. 1997;43:143–149. doi: 10.1159/000213842. [DOI] [PubMed] [Google Scholar]
- 2.Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 3.Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkman DJ. The effect of aspirin and nonsteroidal anti-inflammatory drugs on prostaglandins. Am J Med. 1998;105:8S–12S. doi: 10.1016/s0002-9343(98)00069-2. [DOI] [PubMed] [Google Scholar]
- 5.Bolten WW. Scientific rationale for specific inhibition of COX-2. J Rheumatol Suppl. 1998;51:2–7. [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer's disease. J Neural Transm Suppl. 1998;53:127–140. doi: 10.1007/978-3-7091-6467-9_11. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E, Bohl J, Bratzke H. Evolution of Alzheimer's disease related cortical lesions. J Neural Transm Suppl. 1998;54:97–106. doi: 10.1007/978-3-7091-7508-8_9. [DOI] [PubMed] [Google Scholar]
- 9.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Brooks PM, Day RO. Nonsteroidal antiinflammatory drugs—differences and similarities. N Engl J Med. 1991;324:1716–1725. doi: 10.1056/NEJM199106133242407. [DOI] [PubMed] [Google Scholar]
- 11.Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 12.Combs CK, Johnson DJ, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Bo R, Angeretti N, Lucca E, De Simoni MG, Forloni G. Reciprocal control of inflammatory cytokines, IL-1 and IL-6 and β amyloid production in cultures. Neurosci Lett. 1995;188:70–74. doi: 10.1016/0304-3940(95)11384-9. [DOI] [PubMed] [Google Scholar]
- 14.Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- 15.Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- 16.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 17.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 18.Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia. 1993;7:102–110. doi: 10.1002/glia.440070116. [DOI] [PubMed] [Google Scholar]
- 19.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 20.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo Y-M, Roher AE. Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodwin JL, Uemura E, Cunnick JE. Microglia release of nitric oxide by the synergistic action of β-amyloid and IFN-γ. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- 22.Haga S, Ikeda K, Sato M, Ishii T. Synthetic Alzheimer amyloid β/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res. 1993;601:88–94. doi: 10.1016/0006-8993(93)91698-r. [DOI] [PubMed] [Google Scholar]
- 23.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. β-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 24.Jha RJ. Thiazolidinediones—the new insulin enhancers. Clin Exp Hypertens. 1999;21:157–166. doi: 10.3109/10641969909068658. [DOI] [PubMed] [Google Scholar]
- 25.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 26.Kalaria RN. Microglia and Alzheimer's disease. Curr Opin Hematol. 1999;6:15–24. doi: 10.1097/00062752-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun. 1999;254:582–586. doi: 10.1006/bbrc.1998.9981. [DOI] [PubMed] [Google Scholar]
- 29.Klegeris A, McGeer P. β-Amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 30.Klegeris A, Walker DG, McGeer PL. Interaction of Alzheimer beta-amyloid peptide with the human monocytic cell line THP-1 results in a protein kinase C-dependent secretion of tumor necrosis factor-alpha. Brain Res. 1997a;747:114–121. doi: 10.1016/s0006-8993(96)01229-2. [DOI] [PubMed] [Google Scholar]
- 31.Klegeris A, Walker DG, McGeer PL. Regulation of glutamate in cultures of human monocytic THP-1 and astrocytoma U-373 MG cells. J Neuroimmunol. 1997b;78:152–161. doi: 10.1016/s0165-5728(97)00094-5. [DOI] [PubMed] [Google Scholar]
- 32.Klegeris A, Walker DG, McGeer PL. Toxicity of human THP-1 cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs. Neuropharmacology. 1999;38:1017–1025. doi: 10.1016/s0028-3908(99)00014-3. [DOI] [PubMed] [Google Scholar]
- 33.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 34.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 35.Kretzschmar HA, Giese A, Brown DR, Herms J, Keller B, Schmidt B, Groschup M. Cell death in prion disease. J Neural Transm. 1997;50:191–210. doi: 10.1007/978-3-7091-6842-4_19. [DOI] [PubMed] [Google Scholar]
- 36.Lagenaur C, Lemmon V. An L1-like molecule, the 8D9 antigen, is a potent substrate for neurite extension. Proc Natl Acad Sci USA. 1987;84:7753–7757. doi: 10.1073/pnas.84.21.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 38.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 39.Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 40.Lorton D, Kocsis J-M, King L, Madden K, Brunden KR. β-Amyloid induces increased release of interleukin 1β from lipopolysaccharide-activated human monocytes. J Neuroimmunol. 1996;67:21–29. doi: 10.1016/0165-5728(96)00030-6. [DOI] [PubMed] [Google Scholar]
- 41.Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- 42.McCormack K. Roles of COX-1 and COX-2. J Rheumatol. 1998;25:2279–2281. [PubMed] [Google Scholar]
- 43.McDonald D, Bamberger M, Combs C, Landreth G. β-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP-1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGeer EG, McGeer PL. The role of immune system in neurodegenerative disorders. Mov Disord. 1997;12:855–888. doi: 10.1002/mds.870120604. [DOI] [PubMed] [Google Scholar]
- 46.McGeer EG, McGeer PL. The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol. 1998;33:371–378. doi: 10.1016/s0531-5565(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 47.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 48.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer's disease. Ann NY Acad Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 49.McGeer PL, McGeer EG. Inflammation of the brain in Alzheimer's disease: implications for therapy. J Leukoc Biol. 1999;65:409–415. doi: 10.1002/jlb.65.4.409. [DOI] [PubMed] [Google Scholar]
- 50.McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer's disease. Neurology. 1992;42:447–449. doi: 10.1212/wnl.42.2.447. [DOI] [PubMed] [Google Scholar]
- 51.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 52.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 53.Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isoenzymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- 54.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 55.Meda L, Bernasconi S, Bonaiuto C, Sozzani S, Zhou D, Otvos L, Jr, Mantovani A, Rossi F, Cassatella MA. β-Amyloid (25–35) peptide and IFN-γ synergistically induce the production of the chemotactic cytokine MCP-1/JE in monocytes and microglial cells. J Immunol. 1996;157:1213–1218. [PubMed] [Google Scholar]
- 56.Ouellet M, Percival MD. Effect of inhibitor time-dependency on selectivity towards cyclooxygenase isoforms. Biochem J. 1995;306:247–251. doi: 10.1042/bj3060247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pairet M, Van Ryn J. Experimental models used to investigate the differential inhibition of cyclooxygenase-1 and cyclooxygenase-2 by non-steroidal anti-inflammatory drugs. Inflamm Res. 1998;47:S93–S101. doi: 10.1007/s000110050289. [DOI] [PubMed] [Google Scholar]
- 58.Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-delta12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plosker GL, Faulds D. Troglitazone: a review of its use in the management of type 2 diabetes mellitus. Drugs. 1999;57:409–438. doi: 10.2165/00003495-199957030-00014. [DOI] [PubMed] [Google Scholar]
- 60.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 61.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 62.Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, Kogan F. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 63.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 64.Spiegelman B. PPARg in monocytes: less pain, any gain? Cell. 1998;93:153–155. doi: 10.1016/s0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- 65.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 66.Streit WJ, Kincaid-Colton CA. The brain's immune system. Sci Am. 1995;273:38–43. doi: 10.1038/scientificamerican1195-54. [DOI] [PubMed] [Google Scholar]
- 67.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subramaniam S. The emerging role of thiazolidinediones in the treatment of diabetes-mellitus and related disorders. Clin Exp Hypertens. 1999;21:121–136. doi: 10.3109/10641969909068655. [DOI] [PubMed] [Google Scholar]
- 69.Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, Tada K. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982;42:1530–1536. [PubMed] [Google Scholar]
- 70.Vane JR, Botting RM. Anti-inflammatory drugs and their mechanisms of action. Inflamm Res. 1998;47:S78–S87. doi: 10.1007/s000110050284. [DOI] [PubMed] [Google Scholar]
- 71.Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18:2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao J, Mackman N, Edgington TS, Fan S-T. Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 73.Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]