

Abstract
Albumin D-binding protein (DBP) is a PAR leucine zipper transcription factor that is expressed according to a robust circadian rhythm in the suprachiasmatic nuclei, harboring the circadian master clock, and in most peripheral tissues. Mice lacking DBP display a shorter circadian period in locomotor activity and are less active. Thus, although DBP is not essential for circadian rhythm generation, it does modulate important clock outputs. We studied the role of DBP in the circadian and homeostatic aspects of sleep regulation by comparing DBP deficient mice (dbp−/−) with their isogenic controls (dbp+/+) under light–dark (LD) and constant-dark (DD) baseline conditions, as well as after sleep loss. Whereas total sleep duration was similar in both genotypes, the amplitude of the circadian modulation of sleep time, as well as the consolidation of sleep episodes, was reduced in dbp−/− under both LD and DD conditions. Quantitative EEG analysis demonstrated a marked reduction in the amplitude of the sleep–wake-dependent changes in slow-wave sleep delta power and an increase in hippocampal theta peak frequency in dbp−/− mice. The sleep deprivation-induced compensatory rebound of EEG delta power was similar in both genotypes. In contrast, the rebound in paradoxical sleep was significant in dbp+/+ mice only. It is concluded that the transcriptional regulatory protein DBP modulates circadian and homeostatic aspects of sleep regulation.
Keywords: sleep homeostasis, non-REM sleep intensity, knock-out mice, clock-genes, EEG and circadian oscillations, simulation
The cells of the suprachiasmatic nuclei (SCN) constitute the circadian “clock” that drives the daily fluctuations in many behaviors and physiological parameters and keeps them entrained to the 24 hr light/dark cycle (Miller et al., 1996). Recently, enormous progress has been made in the understanding of the molecular basis of circadian rhythm generation in mice. Thus, several clock elements have been identified (for review, see Dunlap, 1999) of which CLOCK, mPER2, mCRY1, and mCRY2 have been shown to be essential for rhythm generation in mice (Antoch et al., 1997; van der Horst et al., 1999; Zheng et al., 1999). The mechanisms by which the molecular signals of the pacemaker are translated into overt rhythms, however, are less well studied. In Drosophila, several genes mediating clock output have been proposed (Hall, 1995) and, in the mouse, the CLOCK:BMAL1 heterodimer can activate vasopressin transcription (Jin et al., 1999). Another recent example of a murine circadian output gene is the transcription factor albumin D-binding protein (DBP) (Lopez-Molina et al., 1997). In addition to the presence of a strong circadian rhythmicity in both DBP protein and mRNA in a variety of peripheral tissues (Falvey et al., 1995; Fonjallaz et al., 1996), DBP mRNA also displays a strong circadian rhythm in the SCN, and the lack of DBP results in a shortening of the period of the circadian free-running rhythm and in a reduction of locomotor activity (Lopez-Molina et al., 1997).
The distribution and consolidation of sleep are directly controlled by the SCN (Dijk and Czeisler, 1995), and lesioning this structure abolishes the circadian sleep–wake rhythm, particularly the consolidation of sleep (Edgar et al., 1993). Of equal importance is the homeostatic control of sleep that, in concert with the circadian process, regulates the expression of sleep (Borbély, 1982). The homeostatic process reflects the need or propensity for sleep, which builds up during wakefulness and dissipates during sleep. For slow-wave sleep (SWS), EEG delta power is considered a marker of this process (Daan et al., 1984) because it exhibits a predictive quantitative relationship with the duration of previous wakefulness (Tobler and Borbély, 1986; Dijk et al., 1987). For paradoxical sleep (PS), it is the duration that is homeostatically regulated because a loss of PS is primarily compensated by an increase in time spent in PS (Kitahama and Valatx, 1980; Parmeggiani et al., 1980; Franken et al., 1991a,1993, 1999; Amici et al., 1998). Certain aspects of the regulation of sleep and the sleep EEG have a strong genetic component (Beijsterveldt and Boomsma, 1994; Franken et al., 1998, 1999; Tafti et al., 1999). More specifically, the regulation of PS in mice is determined by a few genes only (Valatx and Bugat, 1974; Kitahama and Valatx, 1980). Recently, we confirmed these findings by providing the first quantitative trait loci (QTL) for PS (Tafti et al., 1997), which corresponds to a genomic region that contains important candidate genes, including dbp.
Given the circadian expression of dbp in the SCN, its effect on circadian period, and the association of this gene with the above mentioned QTL, we assessed the effects of DBP on the expression and regulation of sleep under several conditions. We compared sleep and the EEG of mice lacking DBP with their wild-type controls under light–dark and constant-dark (DD) conditions, and after a 6 hr sleep deprivation (SD). Because both strains were otherwise isogenic, differences in sleep can be attributed to the presence or absence of DBP alone and not to differences in genetic background (Gerlai, 1996).
MATERIALS AND METHODS
The generation of 129/Ola mice carrying a null allele for thedbp gene (Lopez-Molina et al., 1997) and the methods concerning the recording and analysis of the EEG in mice (Franken et al., 1998, 1999) have been described in detail previously. The experimental protocols were approved by the local veterinary office (Office Vétérinaire Cantonal de Genève) and the ethical committee of the University of Geneva.
Eight male 129/Ola mice from a sixth generation intercross of progeny homozygous for the null allele (dbp−/−) were included in the present experiment. Eight male 129/Ola wild-type mice constituted their isogenic controls (dbp+/+). Genotypes did not differ in weight or age (dbp+/+ vs dbp−/−; age, 134 ± 8 vs 139 ± 5 d; p > 0.6; weight, 29 ± 1 vs 31 ± 2 gm; p > 0.3;t tests), and no differences were noted in gross morphology or in brain histology. All mice were individually housed in an experimental room under a 12 hr light/dark cycle. EEG and electromyogram (EMG) electrodes were implanted under pentobarbital anesthesia. Mice were allowed 10–14 d of recovery from surgery and habituation before the experiments.
In a first experiment, EEG and EMG signals were recorded continuously for four consecutive 24 hr periods, starting at lights-on (8:00 A.M.). Days 1 and 2 were considered baseline (BSL1 and BSL2). On day 3, starting at lights-on, mice were sleep-deprived for 6 hr by gentle handling (Franken et al., 1991a, 1999). The remaining 18 hr of days 3 and 4 were considered recovery (REC1 and REC2). The effect of SD on sleep and the sleep EEG was assessed by comparing the first 6 hr of REC1 with the first 6 hr of BSL2. The remaining three 12 hr periods of recovery were compared with the corresponding 12 hr periods of BSL2. In a second experiment, the contribution of light on the genotype-specific differences observed in the first experiment was studied. Four mice of each genotype were recorded for 88 hr or 3.7 d, starting 2 hr before lights-on. Ten days passed between the SD in the first experiment and the first day of the second experiment. Day 1, under the usual light/dark cycle, served as BSL. The remaining 2.6 d (DD1–DD3) were recorded under DD conditions. The protocols of the two experiments are illustrated in Figures 1 and 3.
Fig. 1.
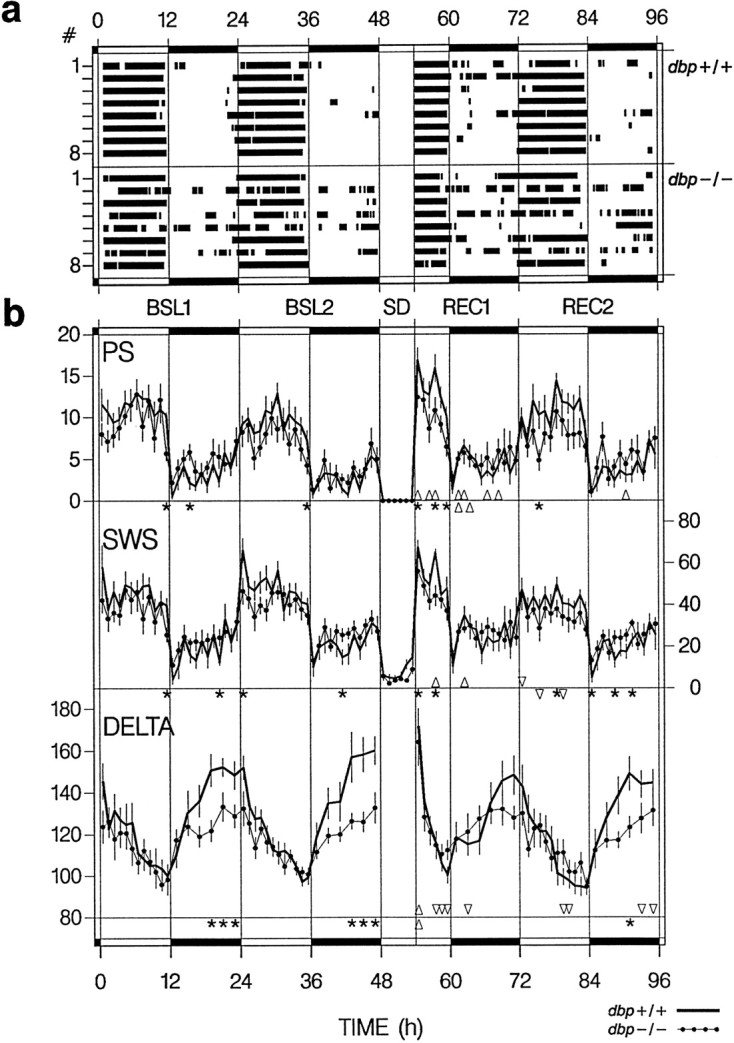
Distribution of sleep and SWS delta power.A, Individual distributions of rest episodes in BSL1 and BSL2, the 6 hr SD, and recovery days REC1 and REC2. Black bars indicate 15 min for which the 2 hr mean percentage sleep was larger than the baseline mean (see Materials and Methods). Individuals are indexed 1–8 for dbp+/+ (top 8 bars) and dbp−/− (bottom8 bars). Vertical lines andhorizontal black bars in the top andbottom mark the dark periods. B, Time course of PS, SWS, and SWS delta power (DELTA). Mean ± SEM hourly (or 2 hr for DELTA in the dark periods) values (n = 8 per genotype). The variables were affected by genotype (*p < 0.05 indicates intervals with significant genotype differences;t tests) and SD (Δ > BSL2 and ▿ < BSL2 indicate intervals in which recovery values differed from BSL2;p < 0.05; paired t tests; fordbp+/+ depicted above the reference line below each set of curves, for dbp−/− beneath). Three-way ANOVA;REC1: factor SD: PS, p < 0.0001 and SWS, p < 0.02; factor time: p< 0.0001; genotype × SD: DELTA, p < 0.04; genotype × time: PS, DELTA, p < 0.04 and SWS, p < 0.002; SD × time: PS, DELTA,p < 0.0001. REC2: factor SD: PS, SWS, p < 0.0005; factor time:p < 0.0001; genotype × time: PS, SWS,p < 0.01 and DELTA, p < 0.005; SD × time: PS, p < 0.05.
Fig. 3.

Time course of TS and SWS EEG delta power (DELTA) for a BSL under 12 hr light/dark, followed by 2.6 d under constant darkness (DD1–DD3). Top curvein each panel represents dbp+/+ (leftscaling), and the bottom curve representsdbp−/− (right scaling). Thin lines are repetitions of BSL. Δ > dbp+/+ and ▿ < dbp+/+ indicate intervals in which values differed between genotypes (p < 0.05;t tests; n = 4 per genotype).Black horizontal bars indicate dark periods, andshaded bars indicate the subjective light periods under DD. a, Hourly TS values as percentage recording time. Horizontal lines represent the mean TS in BSL, DD1–DD3. b, Hourly or 2 hr delta values for the (subjective) light or dark periods, respectively, as a percentage of the value in the last SWS hour in the BSL light period. Thick horizontal lines represent the mean level of DELTA in BSL, DD1–DD3. Peak and trough values are indicated by open squares (±SEM for time and magnitude). Only peak values and amplitude (peak − trough) varied with genotype [two-way ANOVA; factor genotype (amplitude, peak), p < 0.03; factor day,p > 0.6]. The difference between peak or trough times of consecutive days did not deviate from 24 hr (pairedt tests).
The analyses presented here are based on 2240 hr of EEG and EMG recording. Both signals were amplified, filtered, and analog-to-digital converted. The EEG signal was subjected to a Fast-Fourier Transform yielding power spectra between 0.125 and 25.125 Hz, with a 0.25 Hz frequency resolution per window of 4 sec. The behavior in each of these 2,016,000 4-sec epochs was classified as PS, SWS, or wakefulness (W) by visual inspection of the EEG and EMG signals. The amount and distribution of the behavioral states were analyzed by expressing them as a percentage of recording time. As an amplitude measure of the changes in sleep and wakefulness across the 24 hr day, the difference between the mean amount of sleep in the (subjective) light and dark periods was taken. To further evaluate the distribution of sleep over the day (i.e., circadian sleep consolidation), episodes in which both SWS and PS prevailed (i.e., “rest” episodes) were identified according to previously published criteria (Franken et al., 1999). In short, PS and SWS time were determined over 2 hr intervals that were offset by 15 min to produce a running average. Both variables were then expressed as a fraction of their mean amount in baseline (BSL1 and BSL2), and the two fractions were averaged, yielding one value per 15 min to which both PS and SWS contributed equally. Fifteen minute intervals in which this combined value was >1 were counted as rest. Consolidation of sleep was also assessed at the level of individual sleep episodes. The distribution of SWS and PS episode duration was analyzed according to previously published criteria (Tobler et al., 1997; Franken et al., 1999). According to their length, episodes were allotted to one of nine bins of logarithmically increasing size (4, 8–12, 16–28, 32–60, 64–124, 128–252, 256–508, 512–1020, and >1024 sec). The frequency in each bin was corrected for the total amount of each state by expressing it per hour of SWS or PS.
Mean EEG spectra were obtained by averaging the spectra of all 4 sec epochs scored as either PS or SWS in baseline (BSL1 and BSL2). The EEG spectrum during exploratory behavior was analyzed during the first 5 min after a cage change, 3 hr after lights-on on day 3, i.e., the middle of SD. During this period, all animals were engaged in exploratory behavior. EEG peak frequency was determined by calculating the distribution of the frequencies in which EEG power was highest within each of the 4 sec epochs selected for that state. The prevailing frequency was determined by fitting a normal distribution to the frequency distribution (Franken et al., 1998).
As a marker for SWS propensity, EEG delta power was calculated as the mean power over the frequency range between 1 and 4 Hz (Borbély, 1982; Daan et al., 1984). All SWS delta power measures were first expressed as a percentage of the individual mean delta power over the last 900 SWS epochs in the baseline light period to correct for individual differences in the absolute power. Previous observations in mice demonstrated that delta power steeply declines in the presence of SWS (Franken et al., 1999). Therefore, to obtain a better estimate of its initial level, delta power was averaged over the initial 15 min of SWS after SD and after light onset in baseline. In addition, for the 2 baseline days of the first experiment and the 3 d of the second experiment, daily peak and trough values were determined for SWS delta power by selecting the maximum and minimum delta power values calculated over 15 min or 225 consecutive (but not necessarily uninterrupted) 4 sec epochs scored as SWS, with a lag of 5 min (or 75 4-sec epoch scored as SWS). The peak trough difference was taken as the amplitude of the changes in SWS delta power across the 24 hr day.
All main effects of factors “genotype” (dbp−/− vsdbp+/+), “day” (baseline vs recovery or DD), “time-of-day” (1, 2, 6, or 12 hr values), and “bin” (bins 1–9 of episode duration distribution) were analyzed by ANOVA for repeated measures within genotype. Only those factors and interactions between factors are listed that reached significance levels (p < 0.05). When main effects were present,post hoc, two-sided t tests were performed to further evaluate differences between genotypes and paired ttests for differences within genotype. When more then two levels per factor were compared, Tukey's studentized range tests were performed to control the experimentwise error rate.
RESULTS
Sleep consolidation
Although the daily amounts of sleep and wakefulness did not differ between dbp+/+ and dbp−/− mice (Table1), several observations demonstrated differences in the distribution of sleep over the 24 hr day.dbp−/− mice displayed less sleep during the light or rest period and tended to have more sleep in the dark or active period (Table 1, Fig. 1b). Of the three behavioral states, the distribution of PS was generally affected the most by genotype. During the light periods of both baseline and recovery, dbp−/− mice displayed significantly less PS. In addition, during the entire recording period, the expression of PS was consistently more variable in dbp−/− mice (i.e., larger SEM, see Tables 1, 3).
Table 1.
Behavioral states in baseline
| W | SWS | PS | PS/SWS | ||
|---|---|---|---|---|---|
| 48 hr | dbp+/+ | 59.8 (2.7) | 33.5 (2.6) | 6.7 (0.2) | 21.0 (1.8) |
| dbp−/− | 61.9 (1.8) | 31.8 (1.7) | 6.2 (0.4) | 20.2 (1.5) | |
| p | 0.52 | 0.60 | 0.38 | 0.73 | |
| 24 hr L | dbp+/+ | 42.6 (3.1) | 47.1 (3.1) | 10.3 (0.3) | 22.9 (1.7) |
| dbp−/− | 52.5 (3.6) | 39.1 (3.1) | 8.5 (0.7) | 22.2 (1.5) | |
| p | 0.06 | 0.09 | 0.027 | 0.75 | |
| 24 hr D | dbp+/+ | 77.1 (3.2) | 19.9 (3.0) | 3.0 (0.3) | 17.6 (2.8) |
| dbp−/− | 71.5 (2.0) | 24.5 (1.6) | 4.0 (0.6) | 16.4 (2.0) | |
| p | 0.15 | 0.20 | 0.18 | 0.73 | |
| 6 hr L | dbp+/+ | 38.6 (3.0) | 51.9 (3.0) | 9.5 (0.4) | 18.8 (1.3) |
| dbp−/− | 51.1 (4.9) | 41.1 (4.2) | 7.8 (1.1) | 18.8 (2.4) | |
| p | 0.049 | 0.06 | 0.18 | 1.0 |
Mean ± SEM (n = 8 per genotype) recording time for W, SWS, PS, and percentage PS/SWS in the 2 baseline days. The entire 48 hr period (48 hr), the two 12 hr light (24 hr L) and dark (24 hr D) periods, and the first 6 hr of the second baseline (6 hr L) are shown. The 6 hr L values serve to contrast the values obtained in the first 6 hr of recovery. p indicates the probability of thet tests comparing the genotypes.
Table 3.
Behavioral states in recovery
| W | SWS | PS | PS/SWS | |||
|---|---|---|---|---|---|---|
| REC1 | 6 hr L | dbp+/+ | 32.6 (3.4)a | 54.0 (3.0) | 13.3 (0.7)a | 25.0 (1.6)a |
| dbp−/− | 45.4 (3.1) | 44.8 (2.8) | 9.8 (0.9) | 22.4 (2.5) | ||
| p | 0.015 | 0.042 | 0.008 | 0.37 | ||
| 12 hr D | dbp+/+ | 70.8 (4.2)b | 24.9 (4.0)b | 4.2 (0.5)a | 19.7 (3.1)a | |
| dbp−/− | 69.1 (1.0) | 26.1 (0.7) | 4.8 (0.7)b | 18.5 (2.6)b | ||
| p | 0.69 | 0.78 | 0.50 | 0.78 | ||
| REC2 | 12 hr L | dbp+/+ | 47.8 (2.3) | 41.5 (2.7) | 10.7 (0.7) | 27.0 (3.2) |
| dbp−/− | 56.1 (4.3)b | 34.3 (4.1)a | 7.7 (1.2) | 22.9 (3.7)b | ||
| p | 0.11 | 0.17 | 0.044 | 0.41 | ||
| 12 hr D | dbp+/+ | 76.5 (2.6) | 19.8 (2.5) | 3.6 (0.2)b | 19.9 (2.2)b | |
| dbp−/− | 71.4 (1.1) | 23.7 (1.1) | 4.9 (0.7)a | 21.1 (3.3)a | ||
| p | 0.09 | 0.17 | 0.09 | 0.77 |
Mean ± SEM percentage (n = 8 per genotype) recording time for W, SWS, and PS and the percentage PS/SWS for the light (L) and dark (D) periods for REC1 and REC2. Variables were affected by genotype and/or sleep deprivation (two-way ANOVA; REC1 light∶factor genotype: W, SWS, PS, p < 0.05; factor SD: W, PS, PS/SWS, p < 0.005 and SWS,p < 0.05; interaction: PS, p < 0.07; REC1 dark: factor SD: PS, PS/SWS, p < 0.001 and W,p < 0.05; REC2 light: factor SD: W, SWS, PS/SWS,p < 0.01; REC2 dark:factor SD:PS, PS/SWS,p < 0.005). p indicates the probability of the t tests comparing the genotypes.
ap < 0.005, bp < 0.05, significant difference from the second baseline day (Table 1); pairedt test.
The altered distribution resulted in a significantly smaller light–dark amplitude for all three behavioral states (Fig.2a). The lack of DBP affected the expression of SWS and PS to the same extent, leaving the PS/SWS ratio unchanged (Table1, Fig. 2a). These differences were a consequence of the presence in several of the dbp−/− mice of extended (>2 hr) rest episodes in the dark or active periods (and extended active episodes in the light or rest periods), whereas indbp+/+ mice, rest episodes were restricted to the light period (Fig. 1a).
Fig. 2.

Daily amplitude in sleep time. a,Filled (dbp+/+) and open(dbp−/−) bars indicate the mean ± SEM difference (n = 8 per genotype) in percentage recording time, between values in the light and dark periods in baseline days 1 and 2 for SWS, PS, and total sleep (TS) (SWS+PS) and the PS/SWS percentage. The light–dark difference for TS, SWS, and PS was smaller for dbp−/− (* p < 0.05; t test). Note that, for TS and SWS, the scaling is indicated on the left, and for PS and PS/SWS, the scaling is on the right.b, Mean ± SEM difference (n = 4 per genotype) between percentage TS in the (subjective) light and dark periods of BSL and days 1 and 2 under constant darkness (DD1 and DD2). For DD, it was assumed that circadian timing did not deviate from baseline (see Fig. 3b). The amplitude was smaller indbp−/− (* p < 0.02;t test) and was reduced under DD. Two-way ANOVA; factor genotype, p < 0.004; factor day (BSL, DD1, DD2),p < 0.003.
Under constant-dark conditions, the distribution of sleep remained remarkably stable in dbp+/+ mice (Fig.3a). Especially, the timing of the end of the major rest period did not differ from baseline and varied little between individuals. In dbp−/− mice, the absence of light resulted in an even more disrupted distribution of sleep than under light–dark conditions (Fig. 3a), which was accompanied by a further reduction in the amplitude of sleep time (Fig.2b).
A reduction in sleep consolidation was not only evident at a circadian level but also at the level of individual sleep episodes. Significantly more SWS episodes <2 min (and significantly less >2 min) were counted in dbp−/− (Fig.4a). Similar, but less robust, differences were observed for PS (Fig. 4b).
Fig. 4.

Frequency distribution of episode duration in baseline for SWS (a) and PS (b) over nine consecutive time bins (only lower bin limits are indicated). Filled(dbp+/+) and open(dbp−/−) vertical bars indicate mean ± SEM number of episodes per bin expressed per hour of SWS or PS (n = 8 per genotype). Frequency differed with genotype for both behavioral states (two-way ANOVA; factor genotype,p < 0.05; factor bin, p < 0.0001; interaction, p < 0.005; *p < 0.05 indicates bins with significant genotype differences; t tests).
EEG delta power in SWS
The SWS EEG is characterized by thalamocortical oscillations in the delta (1–4 Hz) frequency range (Steriade et al., 1993). Spectral analysis quantifies the contribution of specific frequencies to the EEG signal in terms of power (in V2 units). Power in the delta range varies according to the distribution of sleep and wakefulness in that it decreases in the presence of SWS and increases in the absence of SWS (Borbély, 1982; Daan et al., 1984). In accordance with this, SWS delta power decreased in the course of the light periods, i.e., the main rest period for mice, and increased in the dark periods (Fig. 1b). Fordbp+/+ mice, these changes were highly significant within both periods (one-way ANOVA factor time, p < 0.0001). For dbp−/−, the amplitude of the daily changes in delta power was strongly reduced. Calculated as the difference between peak and trough delta power in baseline, the overall amplitude was two-thirds of that observed in dbp+/+ (dbp+/+ vsdbp−/−, 88 ± 9 vs 59 ± 6%; p< 0.02; t test). With this reduction in amplitude, several other delta power measures differed accordingly between genotypes. Thus, the increase in delta power during the dark period was not significant in dbp−/− (one-way ANOVA factor time,p > 0.5 vs p < 0.0001 indbp+/+); the mean value reached in the last 2 hr of the dark periods was almost half of that for dbp+/+ (Fig.1b); the initial level at light onset (i.e., in the first 15 min of SWS) was lower (dbp+/+ vs dbp−/−, 150 ± 6 vs 128 ± 7%; p < 0.03;t test); the decrease in the course of the light period was only marginally significant (p < 0.05 vsp < 0.0001 in dbp+/+; one-way ANOVA); and finally, the overall mean baseline level of SWS delta power was lower (mean over 48 1 hr intervals per mouse; dbp+/+ vsdbp−/−, 145 ± 7 vs 124 ± 4%;p < 0.02; t test).
The genotype differences in amplitude of the daily changes in SWS delta power persisted in the absence of light and were smaller fordbp−/− because of lower peak values reached at the end of the subjective dark periods (Fig. 3b). The analysis of peak and trough delta power further revealed that, for both genotypes, neither the magnitude nor timing changed under constant darkness, despite the profound effect of this condition on the sleep–wake distribution in dbp−/− (Fig.3a).
The lower delta power values reached for dbp−/− in the last 6 hr of the dark periods of baseline did not result from an overall suppression of the amplitude of the EEG signal but rather from specific changes in the 1.0–9.5 Hz range (Fig.5a). This frequency range coincided with that in which EEG power in SWS decreased in the course of the baseline light period (Fig. 5b). These changes in the SWS EEG were paralleled by highly frequency-specific changes in the waking EEG. In the last 6 hr of the dark period, power in the 1.25–5.0, 6.5–9.0, and 17.0–19.25 Hz ranges of the waking EEG was significantly higher in dbp+/+ (Fig. 5c). Between 2.75 and 3.25 Hz, this increase was highly significant (p < 0.001; t test). Similar observations were made within genotypes when the waking EEG spectrum of the last 6 hr of the dark period was compared with that of the remaining 18 hr of baseline (analyses not shown). However, these changes were significant only for dbp+/+ mice, whereas the waking EEG spectra in these 18 hr did not differ between genotypes.
Fig. 5.
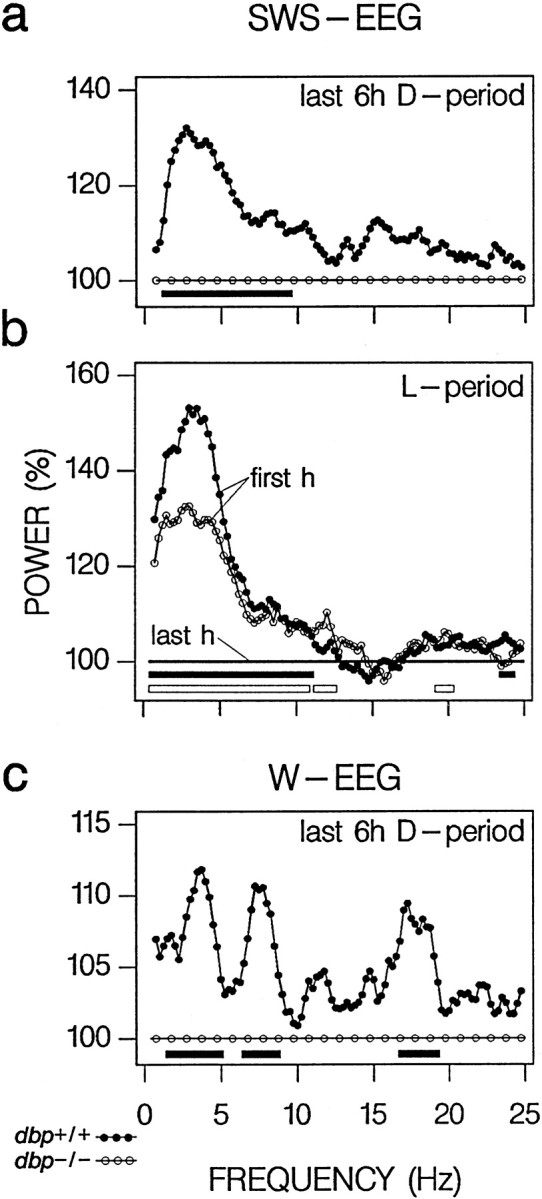
EEG spectra in the last 6 hr of the baseline dark (D) period. a, Difference in SWS EEG spectra between dbp+/+ and dbp−/− (n = 8 per genotype). Individual spectra were first expressed relative to spectral values in the last hour of SWS in the baseline light (L) period. Subsequently, the spectral profile for dbp+/+ (dots) was expressed as a percentage of that of dbp−/− mice (circles). Frequency bins with significant genotype differences in EEG power are indicated by filled horizontal bars below the curves (p < 0.05;t test). b, For comparison, the EEG changes over the light period are depicted as the difference between the SWS EEG spectra in the first recording hour and the last hour of SWS (100%) in the baseline light period fordbp+/+ (dots) and dbp−/− (circles). Significant differences are indicated byfilled (dbp+/+) and open(dbp−/−) horizontal bars(p < 0.05; paired t tests).c, Genotype differences in the waking (W) EEG in the last 6 hr of the baseline dark period. Individual spectra were expressed first relative to the values in the remaining 18 hr of baseline. Symbols as ina.
Theta peak frequency
Hippocampal theta oscillations (5–10 Hz) mark the EEG of both PS and exploratory behavior and are associated with learning, memory consolidation, and long-term potentiation (Vinogradova, 1995; Shors and Matzel, 1997; Vertes and Kocsis, 1997). The analysis of the EEG spectral profiles revealed that the prevailing theta frequency in both PS and exploratory behavior was ∼0.25 Hz higher fordbp−/− (Table 2). A nonsignificant higher and more narrow theta peak in dbp+/+ added to the changes observed between the EEG profiles of these two behavioral states, suggesting a slower but more regular theta rhythm for dbp+/+. The EEG of SWS displayed (nonsignificant) changes in peak frequency in the same direction for both theta (Table2) and delta (data not shown). Theta peak frequency during PS varied with time-of-day (two-way ANOVA with repeated measures: light or dark period, p < 0.0001; genotype, p < 0.005; interaction, p > 0.7), but the dark to light difference did not vary with genotype (dbp+/+, +0.30 Hz,p < 0.001; dbp−/−, +0.33 Hz,p < 0.0005; paired t tests), and thedbp−/− to dbp+/+ difference did not vary with time-of-day (light period, +0.24 Hz, p < 0.005; dark period, +0.26 Hz, p < 0.02; t tests).
Table 2.
Theta peak frequency in baseline
| Expl. behav. | PS | SWS | |
|---|---|---|---|
| dbp+/+ | 8.62 (0.10) | 7.31 (0.05) | 5.83 (0.10) |
| dbp−/− | 8.90 (0.08) | 7.54 (0.05) | 6.38 (0.25) |
| p | 0.042 | 0.005 | 0.08 |
Mean ± SEM EEG theta peak frequency (in Hertz;n = 8 per genotype) in exploratory behavior (Expl. behav.), PS, and SWS. Theta peak frequency varied with genotype and behavioral state (two-way ANOVA; factor genotype, p < 0.02; factor state, p < 0.0001; exploratory behavior > PS > SWS, p < 0.0001; Tukey's multiple range test). p indicates the probability of thet tests comparing the genotypes.
Compensatory rebound after sleep loss
Although especially toward the end of SD brief SWS episodes could not be avoided (dbp+/+ vs dbp−/−, 7.5 ± 1.8 vs 4.3 ± 1.1%; p > 0.1; t test) (Fig. 1b), the SD did result in a substantial reduction in sleep time relative to the corresponding 6 hr of BSL2 (88 vs 91% fordbp+/+ and dbp−/−, respectively) (Table 1).
After the SD, as in baseline, dbp−/− mice slept less in the light periods and somewhat more in the dark periods of recovery (Table 3, Fig. 1b). However, in the light period of recovery day 1, the differences observed in the baseline light-period were amplified by the SD, and significant differences between genotype were now reached for all three behavioral states (Table 3). In the light period of recovery day 2, the genotype differences were again limited to PS.
Compared with the initial 6 hr of BSL2, in the initial 6 hr of recovery day 1, a robust 140% increase in PS was observed in dbp+/+ mice at the cost of the amount of wakefulness (Table 3, Fig.1b). For dbp−/−, none of the behavioral states were significantly affected in this period, and the relative PS rebound tended to be larger for dbp+/+ (p < 0.07; t test). In the dark period of recovery day 1 also, SWS time was above baseline for dbp+/+, but the relative increase in PS was still more important judged by the continued increase in the PS/SWS ratio (Table 3). For dbp−/−, only for PS a modest increase was present. Interestingly, whereas fordbp+/+ values no longer deviated from baseline in the light period of recovery day 2, for dbp−/−, SWS time was now significantly below baseline. This was illustrated by the distribution of the rest episodes (Fig. 1a).
Immediately after the SD, high values of SWS delta power were reached for both genotypes (Fig. 1b), but in contrast to the findings in baseline, delta power in the initial 15 min of SWS did not differ between genotypes (dbp+/+ vs dbp−/−, 193 ± 11 vs 175 ± 12%; p > 0.3;t test). In addition, the highly significant increase in SWS delta power in these 15 min (as a percentage of delta power in the initial 15 min of SWS in baseline; dbp+/+, 129 ± 8%,p = 0.008; dbp−/−, 136 ± 6%,p = 0.0002; paired t tests) did not vary with genotype (p > 0.5; t test). Delta power rapidly decreased in the presence of SWS, which was illustrated by its short-lasting positive rebound (only significantly larger than baseline in the first 15 and 25 min of SWS fordbp+/+ and dbp−/−, respectively) and its steep decline during the first 6 hr of recovery. For dbp+/+, even an undershoot below baseline (“negative rebound”) (Franken et al., 1991a; Feinberg and Campbell, 1993) was observed after 3 hr, and delta power tended to remain below baseline thereafter (Fig. 1b). As a consequence, the large genotype difference in delta power observed in the second half of the baseline dark periods was absent in the first recovery day and still reduced in the second recovery day (Fig.1b). Previously, it has been demonstrated that a negative rebound in SWS delta power is a consequence of the increased SWS time after SD (Franken et al., 1991a,b). The absence of a negative rebound in dbp−/− (Fig. 1b) can thus result from the significantly smaller amount of SWS in the first 6 hr of recovery day 1 compared with dbp+/+ (Table 3).
DISCUSSION
DBP affects circadian sleep consolidation
The most conspicuous difference between dbp wild-type and mutant mice is the decreased circadian amplitude of the distribution of sleep observed in the latter. This attenuated amplitude in dbp−/− mice was observed under both light–dark and constant-dark conditions and can be attributed mainly to the occurrence in dbp−/− of important rest episodes in the active periods and of active episodes in the rest periods. Similar to what has been observed in several other inbred strains (Franken et al., 1999), indbp+/+ mice, rest and active episodes were confined to the light and dark periods, respectively. The reduction in sleep consolidation was not only evident at the circadian level but also at the level of individual sleep episodes. These findings suggests that DBP, in addition to changing the period of the circadian clock (Lopez-Molina et al., 1997), modifies the strength of the SCN output signal, which governs the distribution and consolidation of sleep and wakefulness over the day (Edgar et al., 1993; Dijk and Czeisler, 1995).
DBP affects the time course of SWS delta power
SWS delta power is thought to reflect a homeostatic process underlying the regulation of SWS propensity (Borbély, 1982; Daan et al., 1984) because it decreases in the course of the major rest period and increases in the major active period in a variety of mammals, including mice and men (Dijk et al., 1987; Franken et al., 1999). Furthermore, sleep deprivation results in an increase in SWS delta power that is proportional to its duration (Tobler and Borbély, 1986). Thus, the lower level of SWS delta power reached in the second half of the dark period suggests that, indbp−/− mice, SWS propensity was lower. This notion was supported by the frequency-specific differences between genotypes in the EEG of both SWS and wakefulness in this period. The genotype differences in the SWS EEG in the second half of the dark period strongly resembled the EEG changes that typically accompany the decrease in SWS propensity during the major rest period (Fig.5b) (Franken et al., 1991a; Dijk et al., 1997). Sleep propensity can also be monitored in the waking EEG (Cajochen et al., 1999, and references therein). In the rat, EEG power in wakefulness gradually increases as time awake progresses, and these changes are most pronounced in the delta (3–5 Hz) and theta (6–10 Hz) frequency range, and in the 16–19 Hz range (Franken et al., 1991a, 1993). Only in these three frequency bands, the waking EEG of dbp+/+ mice was increased compared with dbp−/− in the last 6 hr of the baseline dark period.
The daily variations in SWS delta power are thought to be primarily “driven” by the distribution of sleep and wakefulness and are not directly under circadian control (Dijk and Czeisler, 1995; Dijk et al., 1997). Therefore, the genotype differences have to be interpreted first as a secondary effect of the reduced circadian amplitude of SWS.dbp−/− mice did display 33 min more SWS in the dark period, although only in one 1 hour interval was this difference significant. Can this small increase in SWS time explain the large decrease in the level of SWS delta power, or does a lack ofdbp decelerate the build-up of SWS propensity during wakefulness? The changes in SWS delta power after the sleep deprivation demonstrated that delta power decreases rapidly in the presence of SWS; within 25 min of SWS, the positive rebound was dissipated. Using computer simulations, the relationship between the build-up of delta power in the absence of SWS and its exponential decrease in its presence have been quantified in the rat (Franken et al., 1991a, 1993). According to similar simulations in dbp+/+ anddbp−/− mice, we estimated the time constant of the increase at 5.5 hr and that of the decrease at 2.1 hr and found no differences between genotypes (analyses not shown). With this decrease rate, it can be calculated that, within 22 min of (continuous) SWS, delta power can be reduced by 50%. These analyses suggest that the accumulation of SWS propensity in the course of the dark period can, to a large extent, be discharged by the extra SWS time present indbp−/− and that the rate of accumulation does not differ between genotypes. The latter statement was further supported by the fact that, after 6 hr of enforced wakefulness, the initial level of SWS delta power did not differ between genotypes. The importance of the small amount of SWS present in the active period to preclude a large build-up of SWS propensity was also demonstrated in the rat (Franken et al., 1993). Depriving rats of the 2 hr of SWS normally present during the 12 hr active period resulted in a large increase in SWS delta power, comparable with that observed after a 24 hr sleep deprivation (Franken et al., 1991a).
The reduced amplitude in SWS delta power for dbp−/− resulted in a lower level of delta power across baseline. This suggests that, in dbp−/− mice, SWS propensity is generally lower, which is supported by the observation that their SWS is more fragmented. Negative correlations between the level of SWS delta power and SWS fragmentation have been reported previously in the rat (Franken et al., 1991a, 1993) and the mouse (Tobler et al., 1997; Franken et al., 1999).
DBP may act through its effect on locomotor activity
It has been reported previously that dbp−/− mice display less spontaneous locomotor activity, resulting mainly from a profound reduction in the last part of the dark period in whichdbp+/+ mice display maximum activity levels (Lopez-Molina et al., 1997) (Fig. 6). In mice, a reduction of locomotor activity can result in a reduced amplitude in the circadian distribution of sleep and in a more fragmented sleep (Welsh et al., 1988; Edgar et al., 1991a), which corresponds with the observations made in the present study. Furthermore, the distribution of spontaneous motor activity across the active period affects the free-running period of the circadian clock (Edgar et al., 1991b). Given the different distribution of locomotor activity in the two genotypes (Fig. 6), the shortening of tau observed in dbp−/− (Lopez-Molina et al., 1997) could be explained by this “activity feedback” to the circadian pacemaker (Edgar et al., 1991b). In addition, results from several studies suggest a causal relationship between the level of activity during wakefulness and the amount of SWS with high delta power during subsequent sleep (Horne and Moore, 1985;Mistlberger et al., 1987). In the present study, the levels of both SWS delta power and locomotor activity in dbp−/− started deviating in parallel from those in dbp+/+ (Fig. 6). This suggests that, in dbp−/−, the reduced activity might have contributed to the difference in the level of SWS delta power.
Fig. 6.

Time course of wakefulness (%W), SWS delta power (DELTA), and spontaneous locomotor activity (ACTIVITY) in baseline. Hourly values of time course of wakefulness and delta power from the present experiment (mean over 2 baselines) are aligned with hourly activity values [number of infrared beam breaks · hr−1 · d−1; mean over 10 d; dbp+/+, n = 33;dbp−/−, n = 25; from Lopez-Molina et al. (1997) with permission]. Whereas time course of wakefulness did not significantly differ between genotypes, activity during waking was reduced for most of the dark period in dbp−/− mice. This reduction paralleled the decrease in delta power indbp−/−. The relationship between activity during waking and delta power during SWS was underscored by a highly significant correlation (linear regression,r2 = 0.59; p< 0.0001; 24 1 hr values per genotype) that further improved when delta power was correlated with activity in the preceding hr (r2 = 0.76).
DBP may act through its effect on target genes
Because DBP is a transcriptional factor, its effects are likely to be mediated through its target genes. Thus far, genes whose expression is influenced by DBP have been identified only in the liver (Lavery et al., 1999, and references therein). The role of these, if any, in the regulation of sleep is unknown. Possibly, the cytochrome testosterone 15α-hydroxylase may be of relevance because it could affect the level of testosterone, which has pronounced effects on locomotor activity and the period of circadian rhythms in mice (Daan et al., 1975). Database screening for DBP-binding sites (5′ RTTATGTAAY) (Falvey et al., 1996) in promoters of other sleep-related genes revealed a 90% nucleotide match for such a sequence in the gene encoding tryptophan hydroxylase (TPH). TPH is the rate-limiting enzyme in the synthesis of the neurotransmitter serotonin, which has been implicated in sleep, especially in the regulation of PS (Jouvet, 1984; Boutrel et al., 1999). This is of special interest because the lack of DBP affected the expression and regulation of PS the most, as evidenced by the significant reduction in PS in the light periods, the higher variability in PS time, and the lack of a significant PS rebound after sleep loss. Serotonin has been further associated in the activity feedback signal that can modify circadian period (Mistlberger et al., 1998) and in locomotor activity per se (Reuter et al., 1997). Moreover, reduced serotonergic output from the raphe nuclei can increase the frequency of hippocampal theta (Vinogradova, 1995). Whether or not DBP can act on various sleep parameters via the modulation of TPH gene expression remains to be examined.
Based on various observations, Lopez-Molina et al. (1997) have concluded that dbp is a clock output gene rather than an essential clock gene. However, recent evidence suggests that circadiandbp transcription is controlled by the same molecular components that establish self-sustained oscillations in the expression of essential clock genes (J. A. Ripperger, L. P. Shearman, S. M. Reppert, and U. Schibler, unpublished observations). In the present study, we have demonstrated that DBP mostly affects those aspects of sleep that are known to be under direct circadian control but leaves the homeostatic, circadian-independent regulation of SWS unaffected. Hence, dbp links the molecular clockwork generating self-sustained circadian oscillations to complex circadian outputs, such as locomotor activity and sleep. The precise mechanisms by which DBP operates are still unknown, and their dissection will require the identification of relevant target genes.
Footnotes
This study was supported by the Swiss National Science Foundation Grants 31.45751.95 to M.T. and 31.47314.96 to U.S.
Correspondence should be addressed to Dr. Paul Franken, HUG Belle-Idée, Biochemistry and Neurophysiology Unit, Department of Psychiatry, University of Geneva, Chemin du Petit-Bel-Air 2, CH-1225 Chêne-Bourg, Switzerland. E-mail: paul.franken@hcuge.ch.
Dr. Lopez-Molina's present address: Laboratory of Plant Molecular Biology, Rockefeller University, New York, NY 10021.
REFERENCES
- 1.Amici R, Zamboni G, Perez E, Jones CA, Parmeggiani PL. The influence of a heavy thermal load on REM sleep in the rat. Brain Res. 1998;781:252–258. doi: 10.1016/s0006-8993(97)01242-0. [DOI] [PubMed] [Google Scholar]
- 2.Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, Sangoram AM, King DP, Pinto LH, Takahashi JS. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89:655–667. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beijsterveldt CEM, Boomsma DI. Genetics of the human electroencephalogram (EEG) and event-related brain potentials (ERPs): a review. Hum Genet. 1994;94:319–330. doi: 10.1007/BF00201587. [DOI] [PubMed] [Google Scholar]
- 4.Borbély AA. Sleep regulation: circadian rhythm and homeostasis. In: Ganten D, Pfaff D, editors. Sleep. Clinical and experimental aspects. Current topics in neuroendocrinology. Vol. 1. Springer; Berlin: 1982. pp. 83–103. [Google Scholar]
- 5.Boutrel B, Franc B, Hen R, Hamon M, Adrien J. Key role of 5-HT1B receptors in the regulation of paradoxical sleep as evidenced in 5-HT1B knock-out mice. J Neurosci. 1999;19:3204–3212. doi: 10.1523/JNEUROSCI.19-08-03204.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cajochen C, Khalsa SBS, Wyatt JK, Czeisler CA, Dijk DJ. EEG and ocular correlates of circadian melatonin phase and human performance decrements during sleep loss. Am J Physiol. 1999;277:R640–R649. doi: 10.1152/ajpregu.1999.277.3.r640. [DOI] [PubMed] [Google Scholar]
- 7.Daan S, Damassa D, Pittendrigh CS, Smith ER. An effect of castration and testosterone replacement on a circadian pacemaker in mice (Mus musculus). Proc Natl Acad Sci USA. 1975;72:3744–3747. doi: 10.1073/pnas.72.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–R183. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 9.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–3538. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dijk DJ, Beersma DG, Daan S. EEG power density during nap sleep: reflection of an hourglass measuring the duration of prior wakefulness. J Biol Rhythms. 1987;2:207–219. doi: 10.1177/074873048700200304. [DOI] [PubMed] [Google Scholar]
- 11.Dijk DJ, Shanahan TL, Duffy JF, Ronda JM, Czeisler CA. Variation of electroencephalographic activity during non-rapid eye movement and rapid eye movement sleep with phase of circadian melatonin rhythm in humans. J Physiol (Lond) 1997;505:851–858. doi: 10.1111/j.1469-7793.1997.851ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 13.Edgar DM, Kilduff TS, Martin CE, Dement WC. Influence of running wheel activity on free-running sleep/wake and drinking circadian rhythms in mice. Physiol Behav. 1991a;50:373–378. doi: 10.1016/0031-9384(91)90080-8. [DOI] [PubMed] [Google Scholar]
- 14.Edgar DM, Martin CE, Dement WC. Activity feedback to the mammalian circadian pacemaker: influence on observed measures of rhythm period length. J Biol Rhythms. 1991b;6:185–199. doi: 10.1177/074873049100600301. [DOI] [PubMed] [Google Scholar]
- 15.Edgar DM, Dement WC, Fuller CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep–wake regulation. J Neurosci. 1993;13:1065–1079. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falvey E, Fleury-Olela F, Schibler U. The rat hepatic leukemia factor (HLF) gene encodes two transcriptional activators with distinct circadian rhythms, tissue distributions and target preferences. EMBO J. 1995;14:4307–4317. doi: 10.1002/j.1460-2075.1995.tb00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falvey E, Marcacci L, Schibler U. DNA-binding specificity of PAR and C/EBP leucine zipper proteins: a single amino acid substitution in the C/EBP DNA-binding domain confers PAR-like specificity to C/EBP. Biol Chem. 1996;377:797–809. [PubMed] [Google Scholar]
- 18.Feinberg I, Campbell IG. Total sleep deprivation in the rat transiently abolishes the delta amplitude response to darkness: implications for the mechanism of the “negative delta rebound”. J Neurophysiol. 1993;70:2695–2699. doi: 10.1152/jn.1993.70.6.2695. [DOI] [PubMed] [Google Scholar]
- 19.Fonjallaz P, Ossipow V, Wanner G, Schibler U. The two PAR leucine zipper proteins, TEF and DBP, display similar circadian and tissue-specific expression, but have different target promoter preferences. EMBO J. 1996;15:351–362. [PMC free article] [PubMed] [Google Scholar]
- 20.Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in the rat: effects on electroencephalogram power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991a;261:R198–R208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 21.Franken P, Tobler I, Borbély AA. Sleep homeostasis in the rat: simulation of the time course of EEG slow-wave activity. Neurosci Lett. 1991b;130:141–144. doi: 10.1016/0304-3940(91)90382-4. [DOI] [PubMed] [Google Scholar]
- 22.Franken P, Tobler I, Borbély AA. Effects of 12-h sleep deprivation and of 12-h cold exposure on sleep regulation and cortical temperature in the rat. Physiol Behav. 1993;54:885–894. doi: 10.1016/0031-9384(93)90297-s. [DOI] [PubMed] [Google Scholar]
- 23.Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–R1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- 24.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- 25.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–189. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 26.Hall JC. Tripping along the trail to the molecular mechanisms of biological clocks. Trends Neurosci. 1995;18:230–240. doi: 10.1016/0166-2236(95)93908-g. [DOI] [PubMed] [Google Scholar]
- 27.Horne JA, Moore VJ. Sleep EEG effects of exercise with and without additional body cooling. Electroencephalogr Clin Neurophysiol. 1985;60:33–38. doi: 10.1016/0013-4694(85)90948-4. [DOI] [PubMed] [Google Scholar]
- 28.Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating output from the suprachiasmatic circadian clock. Cell. 1999;96:57–68. doi: 10.1016/s0092-8674(00)80959-9. [DOI] [PubMed] [Google Scholar]
- 29.Jouvet M. Neuromediators and hypnogenic factors. Rev Neurol (Paris) 1984;140:389–400. [PubMed] [Google Scholar]
- 30.Kitahama K, Valatx JL. Instrumental and pharmacological paradoxical sleep deprivation in mice: strain differences. Neuropharmacology. 1980;19:529–535. doi: 10.1016/0028-3908(80)90022-2. [DOI] [PubMed] [Google Scholar]
- 31.Lavery DJ, Lopez-Molina L, Margueron R, Fleury-Olela F, Conquet F, Schibler U, Bonfils C. Circadian expression of the steroid 15a-hydroxylase and coumarin 7-hydroxylase genes in mouse liver is regulated by the PAR leucine zipper transcription factor DBP. Mol Cell Biol. 1999;19:6488–6499. doi: 10.1128/mcb.19.10.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Molina L, Conquet F, Dubois-Dauphin M, Schibler U. The DBP gene is expressed according to a circadian rhythm in the SCN and influences circadian behavior. EMBO J. 1997;16:6762–6771. doi: 10.1093/emboj/16.22.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JD, Morin LP, Schwartz WJ, Moore RY. New insights into the mammalian circadian clock. Sleep. 1996;19:641–667. doi: 10.1093/sleep/19.8.641. [DOI] [PubMed] [Google Scholar]
- 34.Mistlberger R, Bergmann B, Rechtschaffen A. Period-amplitude analysis of rat electroencephalogram: effects of sleep deprivation and exercise. Sleep. 1987;10:508–522. [PubMed] [Google Scholar]
- 35.Mistlberger RE, Bossert JM, Holmes MM, Marchant EG. Serotonin and feedback effects of behavioral activity on circadian rhythms in mice. Behav Brain Res. 1998;96:93–99. doi: 10.1016/s0166-4328(98)00007-2. [DOI] [PubMed] [Google Scholar]
- 36.Parmeggiani PL, Cianci T, Calasso M, Zamboni G, Perez E. Quantitative analysis of short term deprivation and recovery of desynchronized sleep in cats. Electroencephalogr Clin Neurophysiol. 1980;50:293–302. doi: 10.1016/0013-4694(80)90157-1. [DOI] [PubMed] [Google Scholar]
- 37.Rueter LE, Fornal CA, Jacobs BL. A critical review of 5-HT brain microdialysis and behavior. Rev Neurosci. 1997;8:117–137. doi: 10.1515/revneuro.1997.8.2.117. [DOI] [PubMed] [Google Scholar]
- 38.Shors TJ, Matzel LD. Long-term potentiation: what's learning got to do with it? Behav Brain Sci. 1997;20:597–655. doi: 10.1017/s0140525x97001593. [DOI] [PubMed] [Google Scholar]
- 39.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 40.Tafti M, Franken P, Kitahama K, Malafosse A, Jouvet M, Valatx JL. Localization of candidate genomic regions influencing paradoxical sleep in mice. NeuroReport. 1997;8:3755–3758. doi: 10.1097/00001756-199712010-00019. [DOI] [PubMed] [Google Scholar]
- 41.Tafti M, Chollet D, Valatx JL, Franken P. Quantitative trait loci approach to the genetics of sleep in recombinant inbred mice. J Sleep Res [Suppl 1] 1999;8:37–43. doi: 10.1046/j.1365-2869.1999.00007.x. [DOI] [PubMed] [Google Scholar]
- 42.Tobler I, Borbély AA. Sleep EEG in the rat as a function of prior waking. Electroencephalogr Clin Neurophysiol. 1986;64:74–76. doi: 10.1016/0013-4694(86)90044-1. [DOI] [PubMed] [Google Scholar]
- 43.Tobler I, Deboer T, Fisher M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valatx JL, Bugat R. Facteurs génétiques dans le determinisme du cycle veille-sommeil chez la souris. Brain Res. 1974;69:315–330. doi: 10.1016/0006-8993(74)90009-2. [DOI] [PubMed] [Google Scholar]
- 45.van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JH, Yasui A. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 46.Vertes RP, Kocsis B. Brainstem-diencephalo-septohippocampal systems controlling the theta rhythm of the hippocampus. Neuroscience. 1997;81:893–926. doi: 10.1016/s0306-4522(97)00239-x. [DOI] [PubMed] [Google Scholar]
- 47.Vinogradova OS. Expression, control, and probable functional significance of the neuronal theta-rhythm. Prog Neurobiol. 1995;445:523–583. doi: 10.1016/0301-0082(94)00051-i. [DOI] [PubMed] [Google Scholar]
- 48.Welsh D, Richardson GS, Dement WC. Effect of running wheel availability on circadian patterns of sleep and wakefulness in mice. Physiol Behav. 1988;43:771–777. doi: 10.1016/0031-9384(88)90375-7. [DOI] [PubMed] [Google Scholar]
- 49.Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature. 1999;400:169–173. doi: 10.1038/22118. [DOI] [PubMed] [Google Scholar]