

Abstract
Brain-derived neurotrophic factor (BDNF) has trophic effects on serotonergic (5-HT) neurons in the adult brain and can prevent the severe loss of cortical 5-HT axons caused by the neurotoxinp-chloroamphetamine (PCA). However, it has not been determined whether BDNF promotes the survival of 5-HT axons during PCA-insult or facilitates their regenerative sprouting after injury. We show here that BDNF fails to protect most 5-HT axons from PCA-induced degeneration. Instead, chronic BDNF infusions markedly stimulate the sprouting of both intact and PCA-lesioned 5-HT axons, leading to a hyperinnervation at the neocortical infusion site. BDNF treatment promoted the regrowth of 5-HT axons when initiated up to a month after PCA administration. The sprouted axons persisted in cortex for at least 5 weeks after terminating exogenous BDNF delivery. BDNF also encouraged the regrowth of the 5-HT plexus in the hippocampus, but only in those lamina where 5-HT axons normally ramify. In addition, intracortical BDNF infusions induced a sustained local activation of the TrkB receptor. The dose–response profiles for BDNF to stimulate 5-HT sprouting and Trk signaling were remarkably similar, suggesting a physiological link between the two events; both responses were maximal at intermediate doses of BDNF but declined at higher doses (“inverted-U-shaped” dose–response curves). Underlying the downregulation of the Trk signal with excessive BDNF was a decline in full-length TrkB protein, but not truncated TrkB protein or TrkB mRNA levels. Thus, BDNF–TrkB signaling does not protect 5-HT neurons from axonal injury, but has a fundamental role in promoting the structural plasticity of these neurons in the adult brain.
Keywords: neurotrophin, BDNF, TrkB, serotonin, structural plasticity, sprouting, cerebral cortex, neurotoxicity, amphetamines, p-chloroamphetamine
The neurotrophin brain-derived neurotrophic factor (BDNF) has trophic effects on serotonergic neurons and thus is hypothesized to have a role in the treatment and/or pathophysiology of certain psychiatric disorders (Duman et al., 1997;Altar, 1999). A dysfunction of the 5-HT system has been implicated in a number of psychiatric syndromes, including severe depression, anxiety, impulsive/aggressive behaviors, and suicide (Baumgarten and Grozdanovic, 1995; Hen, 1996; Mann, 1998). Conventional treatments for many of these disorders augment 5-HT neurotransmission (Owens and Nemeroff, 1994; Fuller, 1996) while also increasing the endogenous expression of BDNF and TrkB (the signal-transducing receptor for BDNF) in the brain (Nibuya et al., 1995; Smith et al., 1997).
BDNF influences the phenotype, structural plasticity, and perhaps survival of 5-HT neurons (Eaton et al., 1995; Mamounas et al., 1995;Siuciak et al., 1996). For example, BDNF administration augments 5-HT metabolism (Altar et al., 1994; Siuciak et al., 1996, 1998) and stimulates serotonergic axonal growth in neocortex and spinal cord (Mamounas et al., 1995; Xu et al., 1995; Bregman et al., 1997). The enhancement of 5-HT neurotransmission by BDNF potentiates several behaviors regulated by serotonin (Siuciak et al., 1994; Pelleymounter et al., 1995) and produces antidepressant effects in animal models of depression (Siuciak et al., 1997). While novel neurotrophin-based pharmacotherapies are being proposed to treat depression and other mental illnesses, little is known still about the nature or mechanism of BDNF effects on 5-HT neurons in the adult brain.
We previously reported that chronic BDNF infusion in neocortex of adult rats prevents the loss of 5-HT axons caused by the neurotoxin p-chloroamphetamine (PCA); however, the mechanism for this effect was not clarified (Mamounas et al., 1995). Serotonergic neurons were exposed to BDNF for a considerable duration both before and after PCA administration, yielding a dense plexus of 5-HT axons at the cortical BDNF infusion site. Systemically administered PCA causes a rapid degeneration of most 5-HT axons in the forebrain (Axt et al., 1994;Mamounas et al., 1995) by a mechanism proposed to involve toxic free-radical formation within the nerve terminal (Gibb et al., 1994;Colado et al., 1997). Because BDNF can increase the activity of protective antioxidant enzymes in other free-radical injury models (Spina et al., 1992; Mattson et al., 1993, 1995), one likely interpretation of our initial findings is that BDNF protects 5-HT axons from degeneration by PCA. On the other hand, a unique feature of PCA neurotoxicity is that the brainstem 5-HT soma and many proximal axons are spared by this toxin, allowing a limited amount of axonal regrowth to occur slowly over several months (Mamounas et al., 1992; Axt et al., 1994). Thus, BDNF may not have prevented the PCA-induced degeneration of 5-HT terminals, but instead may have markedly stimulated the regenerative sprouting from the lesioned axons. In support of this prospect, BDNF has been shown to promote dendritic and axonal growth in the CNS (Cohen-Cory and Fraser, 1995; Cabelli et al., 1995; McAllister et al., 1995). Here, we exploited some of the unique features of the PCA-lesion model to determine whether BDNF protects 5-HT neurons from axonal injury by PCA or augments their regenerative sprouting after injury.
Our previous study suggested that the effects of BDNF on 5-HT neurons are mediated by TrkB (Mamounas et al., 1995). The relative potencies of the different neurotrophins to promote 5-HT axonal survival/sprouting in the PCA-lesion model (BDNF ≫ neurotrophin-3 > NGF = vehicle) paralleled their ability to activate TrkB in vitro(Bothwell, 1995). To evaluate further the relationship between BDNF–TrkB signal transduction and 5-HT neuronal plasticity, we examined the in vivo regulation of Trk tyrosine autophosphorylation (reflecting Trk activation; Kaplan and Miller, 1997) by local BDNF administration in cortex.
MATERIALS AND METHODS
Animals. Adult male Sprague Dawley rats (225–250 gm at the start of the experiment; n = 4–14/treatment group) were group-housed with ad libitum access to food and water and treated in accordance with National Institutes of Health guidelines for animal care and use.
Experimental paradigm. As outlined in Figure1, the temporal parameters of BDNF and PCA delivery were manipulated to determine whether BDNF prevents the PCA-induced degeneration of 5-HT axons or potentiates their regenerative sprouting after injury. Recombinant human BDNF (0.1–36 μg/d; 0.25–0.5 μl/hr; Amgen-Regeneron partnership) or sterile PBS (vehicle) were chronically infused into the right frontoparietal cortex or dorsal hippocampus for 1–21 d, as described below. To lesion 5-HT axons in the forebrain, rats were administered one subcutaneous injection of d-l,PCA (10 mg/kg for animals with BDNF infusions in the neocortex, 20 mg/kg for hippocampus; expressed as the free base; Sigma, St. Louis, MO). A higher dose of PCA was used to lesion 5-HT axons in the hippocampus because a subset of the hippocampal 5-HT fibers are more resistant to the neurotoxic effects of PCA (cf., Mamounas et al., 1991).
Fig. 1.
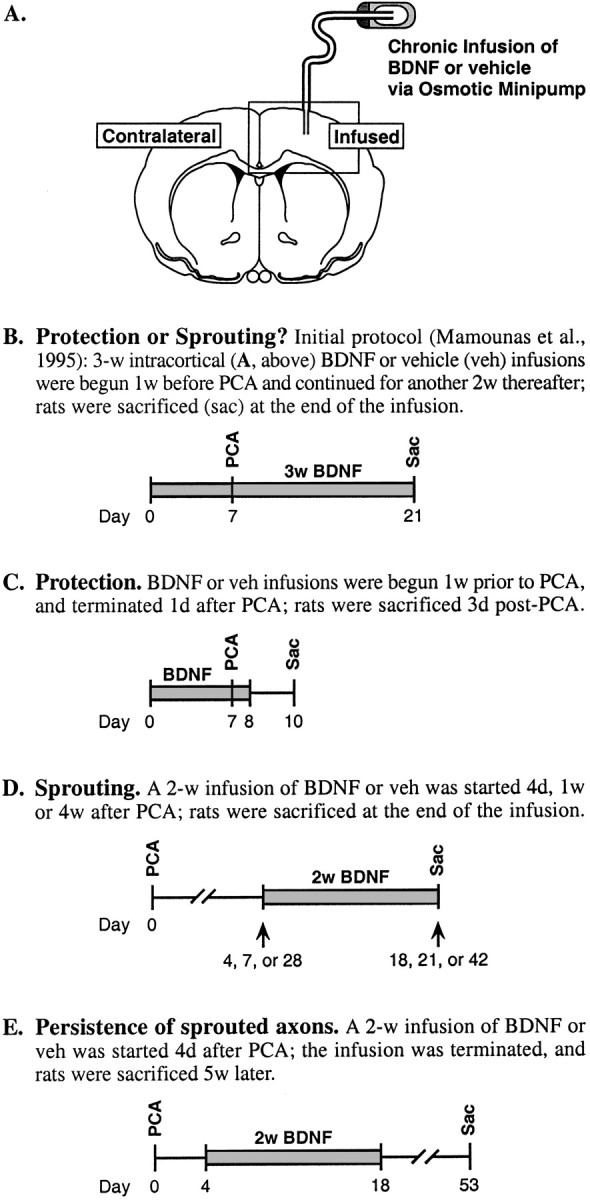
Method of BDNF infusion and experimental paradigms used to characterize the neurotrophic effects of BDNF on 5-HT axons in rat cortex. A, Cannula placement in frontoparietal cortex; rectangle overlaying the infused cortex depicts the location and orientation of subsequent photomicrographs.B–E, Treatment paradigms; the temporal parameters of BDNF (0.1–36 μg/d) and PCA (10 mg/kg, s.c., one time) delivery were manipulated to determine whether BDNF protects 5-HT neurons from axonal injury by PCA or augments their regenerative sprouting after injury.
For experiments analyzing the effects of the duration of the BDNF infusion on 5-HT axon density (see Fig. 6), treatment paradigms were modified slightly from that shown in Figure 1D. To control for the potential influence of “post-PCA survival time” on the experimental outcome, 3 d BDNF (or vehicle) infusions were started at 4 or 11 d after PCA administration (5-HT innervation evaluated at 7 or 14 d after PCA, respectively); 7 d BDNF/vehicle infusions were started at 7 d after PCA (5-HT innervation evaluated at 14 d after PCA); and 14 d BDNF/vehicle infusions were started at 4 or 7 d after PCA (5-HT innervation evaluated at 18 or 21 d post-PCA, respectively). Within either the 3 d or 14 d BDNF infusion experiments, varying the post-PCA survival time before starting the BDNF infusion did not lead to significant differences in 5-HT axon density (when expressed relative to the contralateral cortex; ANOVA,p > 0.05); therefore, these measures were collapsed for analysis in each infusion-duration experiment. Likewise, vehicle measures were pooled because the outcomes did not differ significantly when varying the infusion duration and/or post-PCA survival time.
Fig. 6.
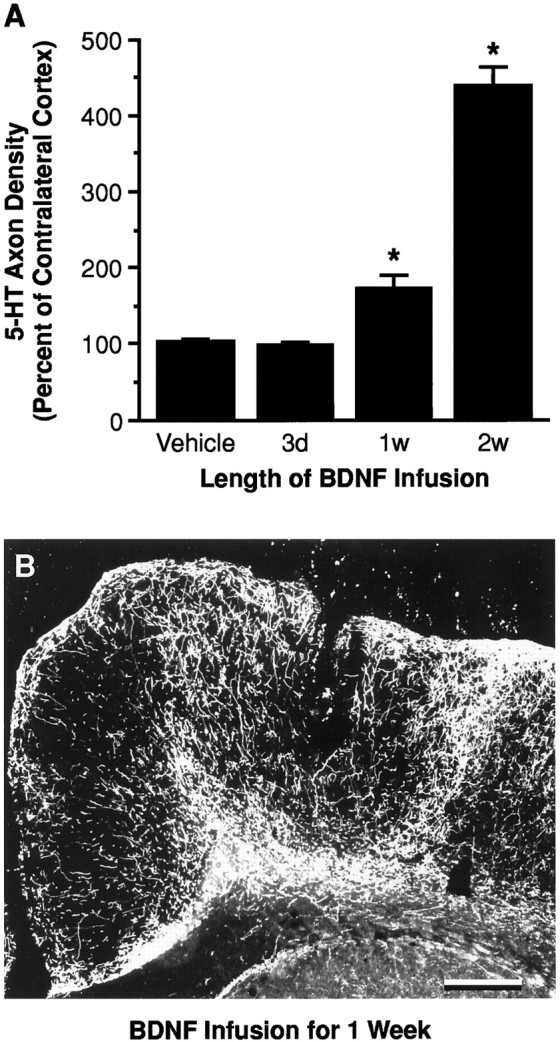
The serotonergic innervation after infusion of vehicle or BDNF (4 μg/d) for 3, 7, or 14 d in cortex of previously PCA-lesioned animals. A, SERT axon density was measured at the BDNF infusion site and expressed as a percentage of the density in the contralateral cortex (noninfused but PCA-lesioned). Treatment paradigms are described in Materials and Methods (Experimental paradigm). *p < 0.05, relative to the vehicle-infused and contralateral cortex (ANOVA followed by the Newman–Keuls multiple range test). B, Dark-field photomicrograph of 5-HT-immunoreactive axons after a 1 week BDNF infusion (4 μg/d; initiated at 7 d after PCA administration) in cortex. The effects of vehicle and 2 week BDNF (4 μg/d) infusions on 5-HT axons (using a similar treatment protocol) are shown in Figures2F and 4E, respectively. Scale bar, 0.5 mm.
BDNF administration. Under sterile conditions 4–18 hr before surgery, the Alzet (Alza, Palo Alto, CA) pump/cannula ensemble was filled with BDNF or vehicle and stored in sterile PBS at 37°C until surgical implantation. Animals were deeply anesthetized with a ketamine (80 mg/kg, i.p.) and xylazine (7 mg/kg, i.p.) mixture or with chloral hydrate (400 mg/kg, i.p.) for subsequent surgical procedures. As described (Mamounas et al., 1995), a 28–30 gauge stainless steel cannula (Plastics One, Roanoke, VA) connected via PE50 tubing to an Alzet osmotic minipump (model 1007D, 2002, or 2004; Alza) was implanted into the right frontoparietal cortex or dorsal hippocampus under stereotaxic guidance (coordinates for neocortex: 0.5 mm anterior and 2.5 mm lateral to bregma and 1.6 mm below the skull surface; for hippocampus: 3.8 mm posterior and 2.7 mm lateral to bregma and 4.0 mm below the skull surface). In cases in which the BDNF infusion was terminated before sacrifice (protocols: Fig.1C,E), the PE50 tubing was cut and sealed close to the cannula hub attachment before removing the Alzet osmotic pump. For brief exposure times, BDNF was injected into the frontoparietal cortex via a Hamilton syringe with a 30 gauge blunt-tip needle (4–12 μg of BDNF in 1 μl of sterile PBS infused over a 10 min period), and animals were decapitated 3 hr later.
Evaluation of monoamine innervation and BDNF diffusion.Brain tissue was processed for immunocytochemistry as described (Mamounas et al., 1991, 1995). To detect serotonergic fibers, free-floating coronal sections (30-μm-thick) were incubated in antiserum directed against serotonin (5-HT; diluted 1:15,000; Incstar, Stillwater, MN), the serotonin transporter (SERT; 1:12,500; Incstar, or gift from Dr. Randy Blakely) or the 5-HT-synthesizing enzyme tryptophan hydroxylase (TPH; 1:5000; Sigma). The 5-HT transporter (SERT) is a large transmembrane protein localized to serotonergic axon terminals in the forebrain, and has been shown to be an excellent presynaptic, structural marker of 5-HT axons (Qian et al., 1995; Sur et al., 1996). The catecholamine innervation was evaluated using an antibody against tyrosine hydroxylase (TH; 1:1500; Eugene Tech, Allendale, NJ). Bound Ig was visualized with the avidin–biotin-peroxidase method (Vector Laboratories, Burlingame, CA), using diaminobenzidine tetrachloride as the substrate, and the reaction product was intensified with either osmium (for 5-HT and SERT immunostained fibers), nickel (TH), or silver/gold (TPH). Cholinergic fibers were detected by acetylcholinesterase (AChE) histochemistry as described (Hedreen et al., 1985). To delineate the area of BDNF diffusion, adjacent sections were immunostained with a turkey anti-recombinant human BDNF antibody (rhBDNF; 1:7500; Amgen).
Axon density analysis. Similar innervation patterns and densities in response to PCA administration and/or BDNF infusions were obtained in neocortex when serotonergic fibers were immunostained for either 5-HT, SERT, or TPH. Detailed analyses of the serotonin innervation used 5-HT- and SERT-immunostained material: for the quantitative analysis (see Figs. 3, 5, 6A, 8), SERT-immunostained sections were used because of the slightly lower nonspecific background staining found with the SERT antibody; the dark-field photomicrographs (Figs. 2, 4, 6B,7) depict 5-HT-immunostained material.
Fig. 3.

The density of serotonin axons (SERT-immunoreactive) in frontoparietal cortex of PCA-lesioned animals and expressed as a percentage of the normal density found in the homologous cortex of intact animals. A, The normally slow reinnervation of cortex by 5-HT axons after PCA administration, as determined in the contralateral cortex of vehicle- and BDNF-infused animals (the two measures were not significantly different at any time point, and were thus collapsed for presentation). B,Effects of vehicle or BDNF infusions (12 μg/d) on PCA-lesioned 5-HT axons (measured locally at the infusion site), as investigated in the following treatment paradigms: Protection (protocol, Fig.1C; leftmost set of histograms); Sprouting (protocol, Fig. 1D) where 2 week BDNF infusions were initiated at 4 d or 4 weeks after PCA administration (center two sets of histograms, respectively); and Persistence of sprouted axons (protocol, Fig. 1E) at 5 weeks after terminating the BDNF infusion (rightmost set of histograms). Gray histogram bars, The contralateral cortex of vehicle- and BDNF-infused animals (the two values were not significantly different in any treatment paradigm, and were thus pooled); white bars,vehicle infusion in the ipsilateral (right) cortex; black bars, BDNF infusion. In all treatment paradigms, the 5-HT axon density was higher in the BDNF-infused cortex relative to the vehicle-infused and contralateral cortex (ANOVA followed by the Newman–Keuls multiple range test, p < 0.05), whereas the control conditions did not differ significantly (p > 0.05).
Fig. 5.

The density of serotonin axons (SERT-immunoreactive) after intracortical infusion of vehicle or 0.1, 0.4, 1, 4, 12, or 36 μg/d of BDNF (protocol, Fig.1D, 2 week infusions were started 4 d after PCA); measured in the area of BDNF-positive immunoreactivity (determined in adjacent sections) and expressed as a percentage of the normal density in intact animals. Closed squares,Vehicle or BDNF infusions in the ipsilateral (right) cortex;open squares, the contralateral (noninfused) side of cortex. ANOVA followed by the Bonferonni post hoc test revealed that 5-HT axon density was significantly higher after 0.4–12 μg/d of BDNF relative to vehicle; the 12 and 36 μg/d doses of BDNF resulted in a lower density than the 4 μg/d dose (F(6,24) = 15; p < 0.0001).
Fig. 8.

The densities of serotonergic (SERT), catecholaminergic (TH), and cholinergic (AChE) axons after intracortical infusion of vehicle or BDNF (4 μg/d) for 18 d in intact (non-PCA-lesioned) rats. Values were measured within the area of BDNF-positive immunoreactivity in cortex and expressed as a percentage of the normal density found in the contralateral cortex. White histogram bars,vehicle infusions; black bars, BDNF infusions. *p < 0.05, relative to the vehicle-infused and contralateral cortex (ANOVA followed by the Newman–Keuls multiple range test).
Fig. 2.
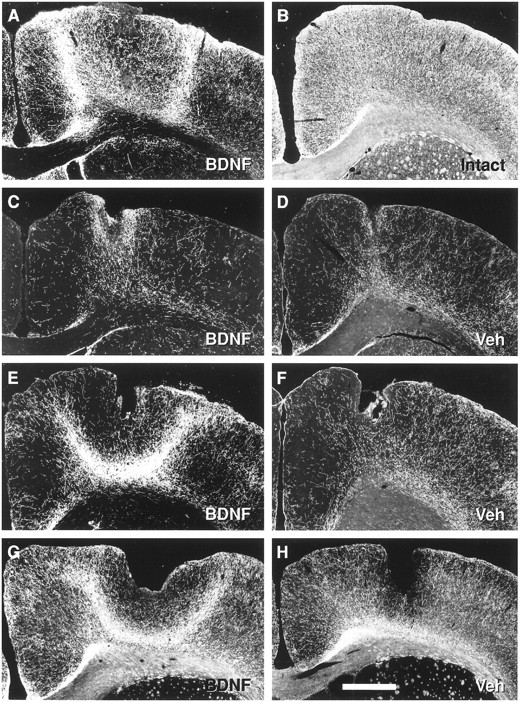
Effects of vehicle or BDNF (12 μg/d) infusions on serotonergic axons in cortex of PCA-lesioned rats. Dark-field photomicrographs depict bright 5-HT-immunoreactive axons on a dark background (coronal sections). A, Protection or sprouting? (protocol, Fig. 1B); 5-HT neurons were exposed to BDNF for a considerable duration both before and after PCA administration, yielding a dense plexus of 5-HT axons at the BDNF infusion site in cortex (cannula tract in center).B, Intact; the normal 5-HT innervation in cortex of non-PCA-lesioned, non-infused rats. C, D,Protection (protocol, Fig. 1C); few 5-HT axons are spared when BDNF (C) infusions are limited to the 1 week period before PCA administration, and animals are killed 3 d after PCA (D, vehicle). E,F, Sprouting (protocol, Fig. 1D); robust sprouting of prelesioned 5-HT axons is found when 2 week BDNF (E) but not vehicle (F) infusions are started 4 d after PCA. G,H, Persistence of sprouted axons (protocol, Fig.1E); once eliciting the 5-HT sprouting response with BDNF (G), the sprouted fibers persist for at least 5 weeks after terminating the BDNF infusion (H, vehicle). Scale bar, 1 mm.
Fig. 4.
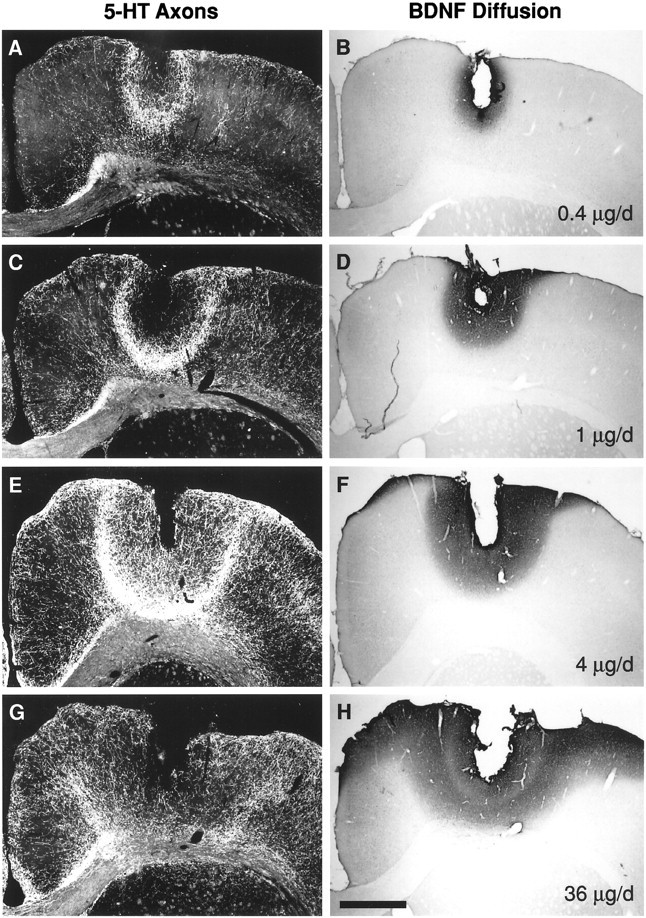
The dose–response profile for BDNF (0.4–36 μg/d) to stimulate 5-HT axon sprouting in cortex (protocol, Fig.1D, 2 week BDNF infusions were started 4 d after PCA). A, C, E, G, 5-HT-immunoreactive axons in cortex (dark-field photomicrographs). B, D, F, H, BDNF diffusion as determined by BDNF immunocytochemistry in adjacent sections (bright-field photomicrographs). A, B, BDNF, 0.4 μg/d; C, D, BDNF, 1 μg/d; E, F,BDNF, 4 μg/d; G, H, 36 μg/d of BDNF. The effects of vehicle infusion and the 12 μg/d dose of BDNF on 5-HT axons (using this same treatment protocol) are shown in Figure 2, Fand E, respectively. Scale bar, 1 mm.
Fig. 7.
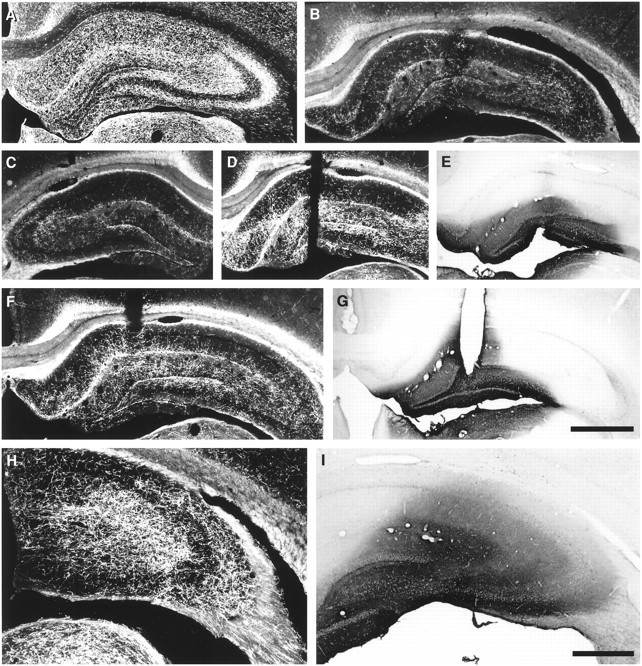
The serotonergic innervation in dorsal hippocampus after local infusion of vehicle or BDNF (4 μg/d) in PCA-lesioned animals (protocol, Fig. 1D, 2 week infusions were started 1 week after PCA). A–D, F, H,5-HT-immunoreactive axons in hippocampus (dark-field photomicrographs; coronal sections); E, G, I, area of BDNF diffusion as determined by BDNF immunocytochemistry in adjacent sections (bright-field photomicrographs). A, The normal 5-HT innervation in hippocampus of intact rats. B, Vehicle infusion in the PCA-lesioned hippocampus (note cannula tract at center). C, The extent of 5-HT denervation normally seen 3 weeks after PCA administration in the contralateral hippocampus.D, E, Sprouting of 5-HT axons in the dentate gyrus (D) after local infusion of BDNF (E). F, G, Sprouting of 5-HT axons in CA1 and the dentate gyrus (F) in response to BDNF infusion (G). H, I, Sprouting of 5-HT axons in CA3 (H) after infusion of BDNF (I); note the higher magnification inH and I than in A–G. Scale bars: A–G (shown in G), 1 mm;H, I (shown in I), 0.5 mm.
Fiber density was quantified using a Zeiss microscope/KS400 image analysis system. Axons were visualized using dark-field illumination at a magnification of 450× (20× objective), yielding excellent resolution of bright, well-delineated axons on a dark background. The microscrope field of interest was captured by a Sony CCD black and white video camera, and the image was contrast-enhanced by histogram stretching, followed by thresholding to convert to a binary image. A grid consisting of single pixels spaced 5 × 5 μm apart (in the x- by y-plane) was overlaid on the binary axon image. Axons intercepting grid points were counted and expressed per unit area of the region analyzed. For each animal, measurements were taken from three coronal sections (instrument thickness setting, 30 μm; postprocessing thickness, 21 ± 0.7 μm) spaced 360 μm apart, with the center section at the level of the BDNF cannula tract. In each section, six different microscope fields (470 × 620 μm each) were analyzed within the area of BDNF diffusion in cortex, excluding the cannula tract and any damaged tissue immediate (within ∼ 0.1–0.2 mm) to the cannula. Adjacent sections immunostained for BDNF were used to delineate the area of BDNF diffusion (see Fig. 4). At higher magnification, a gradient of BDNF diffusion could be observed: an extremely dense central core of BDNF immunoreactivity extended ∼1.0–1.3 mm radially from the infusion cannula (at the 4–12 μg/d BDNF doses), with a much paler rim of staining extending another 0.2–0.3 mm (Mamounas et al., 1995: see Fig.5). For vehicle infusions, selected fields were located within a 2–3 mm diameter area centered at the cannula tract (approximate area of diffusion for the 4–12 μg/d BDNF doses). For each animal, the same analysis was conducted in the contralateral cortex, with comparison fields “mirrored” across the midline. To assess the normal innervation density, 5-HT axon density was measured in the homologous cortex of intact (non-PCA-lesioned, non-infused) animals.
Microdissection of BDNF-infused cortex for Trk activation assays. At the end of the BDNF infusion in cortex, rats were decapitated, and the brains were rapidly removed and chilled for 5 min in a semifrozen “slushy” of PBS. Rapidly and under ice-cold conditions, brains were placed in a calibrated brain blocker/slicer (Aster Industries, Pittsburgh, PA), and a 3-mm-thick coronal slab (centered around the BDNF infusion site in the rostral–caudal direction) was cut and placed flat on an ice-chilled metal block. A chilled 3.0 mm diameter (ID) stainless steel tube (sample corer: Fine Science Tools, Foster City, CA) was centered over the BDNF cannula tract and lowered through the cortex. The cylinder of cortical tissue (∼21 mm3) was gently expelled, immediately frozen on dry ice, and stored at−80°C until used.
Trk protein analysis. Trk tyrosine autophosphorylation was assayed as described (Kaplan et al., 1991a,b, 1993). Briefly, lysates were prepared from fresh-frozen cortical tissue and then assayed for total protein (BCA reagent; Pierce, Rockford, IL). Samples were normalized to 1.0 mg total protein in a 1.25 ml final volume. An aliquot (18 μg of total protein) was removed to measure BDNF levels by Western blotting using a rabbit anti-BDNF N-20 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) as described (Smith et al., 1997). Trk family proteins were immunoprecipitated with rabbit anti-panTrk 203 antibody which reacts with the catalytic isoforms of TrkA, TrkB, and TrkC, and then electrophoresed on 7.5% SDS-PAGE minigels. Protein blots were probed with mouse monoclonal anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology, Lake Placid, NY), and analyzed using an ECL chemiluminescence system (Amersham, Arlington Heights, IL).
To assay the total levels of catalytic TrkB protein (full-length; gp145 TrkB), the blots were stripped and reprobed with a rabbit anti-TrkB antibody (TrkB-out), and analyzed with ECL. The anti-TrkB antibody was generated to amino acids 45–60 in the extracellular domain of rat TrkB. Levels of the truncated isoform of TrkB (gp95 TrkB) were measured by re-precipitating the lysates with wheat germ lectin-agarose (WGA; Pharmacia, Piscataway, NJ). Because WGA binds the extracellular glycosylated residues of Trk, the residual Trk isoforms (e.g., truncated) in the lysates are collected; lysates were Western blotted and probed with the TrkB-out antibody as described above.
To quantify Trk proteins, the samples to be compared (e.g., vehicle and different doses of BDNF; see Figs. 9, 10) were run on adjacent lanes on the same gel. During ECL, sheets of x-ray film were exposed to each blot for varying lengths of time. Films were scanned (UVP Image Store 7500), and optical densities within a band were quantified using NIH Image software, only using values within the linear range of the film. Using this approach, relative comparisons between the vehicle and BDNF were made, and the data are expressed as a percentage of the vehicle value.
Fig. 9.

The dose–response profile for BDNF infusions to stimulate Trk activity (as reflected by Trk tyrosine autophosphorylation) in cortex. BDNF (0.1–12 μg/d) or vehicle was chronically infused into cortex for 24 hr before assaying Trk proteins at the BDNF infusion site. A, Immunoblots of Trk tyrosine autophosphorylation (Trk P-tyr) and total full-length (gp145) TrkB protein after intracortical BDNF infusion in intact (left) or PCA-lesioned (right; 10 mg/kg, s.c., administered 1 week before starting the intracortical infusion) animals. Top set of immunoblots, To assay the levels of Trk P-tyr, Trk family proteins were immunoprecipitated (IP) with anti-panTrk (Trk) antibody, and Western-blotted (WB) with anti-phosphotyrosine (P-tyr) antibody (4G10; Upstate Biotechnology);middle, above immunoblots were reprobed with TrkB antibody to measure total levels of catalytic (full-length; gp145) TrkB protein; bottom, to assess the levels of exogenously delivered BDNF protein, an aliquot of the same lysate (used in each case above) was Western-blotted with anti-BDNF antibody.B, Quantitation of Trk P-tyr (top; expressed as -fold induction over vehicle infusion) and total full-length (gp145) TrkB protein (bottom; expressed as a percentage of vehicle). Open circles, Vehicle or BDNF infusions in cortex; closed circles, homologous cortex from naive (i.e., noninfused) animals.
Fig. 10.

Cortical Trk tyrosine autophosphorylation after local BDNF administration. A, Three hours after a single injection of vehicle or BDNF (4 or 12 μg). B, Chronic infusion of vehicle or BDNF (4 μg/d) for 7 d. Trk P-tyr, Tyrosine-phosphorylated Trk; gp145 TrkB, total full-length (catalytic) TrkB protein; gp95 TrkB, truncated isoform of the TrkB protein; BDNF, exogenously delivered BDNF. Levels are quantified in Table 1.
Ribonuclease protection analysis for TrkB mRNA levels. RNA was extracted using RNA STAT-60 (Tel-Test, Friendswood, TX) following the manufacturer's recommendations. RNase protection experiments, using ∼10 μg of total RNA, were performed as previously described (Tessarollo et al., 1992) using the RPA kit (Ambion, Austin, TX). A TrkB-specific cDNA probe that spans nucleotides 698–928 of the extracellular domain of the rat sequence was used to generate an antisense RNA probe which detects all TrkB isoforms in the RNase protection analysis. The GAPDH-specific probe (Ambion) was included in the same reaction mixture as a means of assessing relative levels of RNA present in each hybridization. Because GAPDH mRNA levels did not differ significantly across treatment groups in this study, the results are expressed as the ratio of TrkB to GAPDH mRNA levels.
Statistical analysis. The statistical significance of changes in 5-HT axon density or Trk biochemical measurements was assessed by a one- or two-factor ANOVA. The 5-HT axon density data (Fig. 3B) were analyzed with a two-factor (BDNF infusion × side of cortex) ANOVA with repeated measures on side of cortex. In the case of significant main effects or interactions, post hoc comparisons were performed using the Newman–Keuls multiple range test or by using the Bonferonni test for a priori comparisons of dose–response relationships (see Figs. 5,9B). Significance levels were accepted at p< 0.05.
RESULTS
BDNF treatment fails to protect most 5-HT axons from PCA-induced degeneration
Serotonin axons are normally found at high densities throughout the forebrain, including the neocortex (Fig. 2B). Subcutaneous administration of the amphetamine neurotoxin PCA (10 mg/kg, one time) caused a rapid (by 3 d) and severe loss of 5-HT axons throughout the forebrain, an effect that was seen when using several different markers of 5-HT axon viability including immunostaining for serotonin itself (Fig. 2D), TPH (the synthesizing enzyme for serotonin; data not shown) or the serotonin transporter (SERT; Fig. 3A). To determine whether BDNF could protect 5-HT axons from PCA-induced degeneration, chronic intracortical infusions of BDNF (12 μg/d) were started 1 week before PCA administration and terminated 1 d after PCA (protocol, Fig.1C). An evaluation of the 5-HT innervation 2 d later (3 d after PCA) revealed only minimal sparing of 5-HT-, TPH- or SERT-immunostained serotonergic axons in the cortical area immediately adjacent to the BDNF infusion cannula (25 ± 6% of the intact density; Fig. 3B, Protection). Although the density of 5-HT axons was statistically higher in the BDNF-infused cortex relative to the vehicle-infused or contralateral cortex (Fig.3B; ANOVA, F(1,7) = 16.9;p < 0.005), the spared 5-HT axons were highly localized to within a radius of 0.3–0.5 mm from the BDNF infusion cannula (Fig. 2C). Since the 12 μg/d dose of BDNF diffuses ∼1.5 mm radially from the infusion cannula (Mamounas et al., 1995), the neuroprotective effects of BDNF for 5-HT axons did not extend peripherally throughout the entire area of BDNF diffusion in cortex.
BDNF stimulates the sprouting of PCA-lesioned 5-HT axons in cortex
In the contralateral and vehicle-infused cortex (and other non-BDNF-infused cortical areas), we observed a slow, but incomplete sprouting response from the PCA-lesioned 5-HT axons, occurring over the 3–53 d after PCA administration (the range of post-PCA survival times used in this study; Fig. 3A). Although there was a partial recovery of the 5-HT axon density in the non-BDNF-infused cortex between 3 d (9 ± 2% of the intact control; see also, Fig.2D) and 53 d (45 ± 2% of intact; Fig.2H) after PCA administration, the normal 5-HT innervation density was not restored up to 1 year after treatment with PCA (data not shown; Mamounas et al., 1992; Axt et al., 1994).
To determine whether BDNF treatment could facilitate this slow, partial 5-HT sprouting response in cortex, 2 week BDNF infusions were initiated at various times after PCA administration (protocol, Fig.1D). BDNF infusions (0.4–12 μg/d) markedly stimulated the sprouting of the lesioned 5-HT axons when the infusions were started at 4 d (Figs. 2E, 3B,4, 5; ANOVA, F(1,15) = 26; p< 0.0001), 1 week (Fig.6A; ANOVA,F(3,14) = 101; p < 0.0001), or even 4 weeks (Fig. 3B; ANOVA,F(1,13) = 92; p < 0.0001) after treatment with PCA. The sprouted 5-HT axons were highly localized to the area of BDNF diffusion in cortex (Fig. 4); as assessed by qualitative examination, cortical areas further than ∼1–2 mm from the BDNF infusion site did not exhibit a higher density of 5-HT axons when compared to the contralateral cortex. Importantly, once having induced 5-HT axonal sprouting with BDNF (protocol, Fig.1E), the sprouted fibers persisted in cortex for at least 5 weeks after terminating the BDNF infusion (Figs. 2G,3B, Persistence 5w post BDNF; ANOVA,F(1,15) = 426; p < 0.0001).
At the higher BDNF doses (e.g., 4–12 μg/d), the sprouted 5-HT axons were concentrated along the perimeter of the area of BDNF diffusion in cortex (i.e., at the peripheral edge of the dense central core of BDNF immunoreactivity and extending into the paler outer zone of BDNF staining; see Materials and Methods); much lower densities were found closer to or further away from the infusion cannula (Figs.2E, 4). This zone of maximal sprouting may have represented an optimal concentration of BDNF for effects on 5-HT axons; thus, the higher BDNF concentrations found close to the infusion cannula (White and Schwartz, 1994) may have limited the sprouting response. To test this possibility, we examined the dose–response profile for BDNF (0.1–36 μg/d) to stimulate 5-HT axon sprouting in cortex (protocol, Fig. 1D; 2 week BDNF infusions were started 4 d after PCA). As predicted, the dose–response profile for BDNF effects on 5-HT axons displayed an inverted-U-shaped curve: BDNF infusions began to stimulate 5-HT axon sprouting at the 0.4 μg/d dose (Figs. 4A, 5), reaching a maximal response at the 4 μg/d dose (Figs. 4E, 5); there was a decline in the response to BDNF at the higher doses (12–36 μg/d; Figs. 4G, 5).
The duration of the BDNF infusion (4 μg/d for 3–14 d; Fig. 6) was also a significant factor in the magnitude of the 5-HT axonal sprouting response in neocortex. Three day infusions of BDNF failed to increase the density of 5-HT axons above control levels. One week BDNF infusions only partially stimulated the sprouting of the PCA-lesioned serotonergic axons, whereas more potent effects were seen with 2 week BDNF infusions.
BDNF-stimulated 5-HT axon sprouting in the hippocampus: tropic or trophic influences of BDNF on serotonergic neurons?
Because most areas of neocortex have a relatively uniform distribution of 5-HT axons across cortical layers (Fig.2B), it was difficult to evaluate from the neocortical experiments whether BDNF influences the direction of 5-HT axonal growth, perhaps redirecting the fibers to inappropriate target areas (tropic effect), or increasing axonal arborization and/or elongation within the appropriate termination zones (trophic influence). In contrast to the neocortex, the hippocampus has a more heterogeneous distribution of 5-HT fibers across its laminae (Fig.7A; Lidov et al., 1980;Mamounas et al., 1991), allowing a better analysis of the nature of BDNF influences on the serotonergic innervation in this structure. After lesioning most of the hippocampal 5-HT fibers with PCA (Fig.7C), BDNF infusions in hippocampus (4 μg/d for 2 weeks, initiated 1 week after PCA; protocol, Fig. 1D) caused robust sprouting of 5-HT axons locally in the area of BDNF diffusion (Fig. 7D,F,H); vehicle infusions had minimal effects on 5-HT axons in this structure (Fig. 7B). Within the BDNF-infused regions of hippocampus, the pattern of reinnervation by 5-HT fibers was reminiscent of the normal 5-HT innervation pattern found in the intact hippocampus (Fig. 7A). Thus, in the presence of BDNF, the sprouting axons ramified extensively in their normal termination zones –including stratum oriens and stratum radiatum of CA3 (Fig. 7H) and CA1 (Fig.7D,F), stratum lacunosum of CA1 (Fig.7D,F), and in the molecular layer and polymorphic zone of the dentate gyrus (Fig. 7D,F). In contrast, the presence of exogenous BDNF protein failed to entice the regrowing 5-HT fibers to enter into the stratum lucidum of CA3 (i.e., the mossy fiber termination zone; Fig. 7D–I), an area normally devoid of 5-HT axons in the intact hippocampus (Fig. 7A). In fact, there was often a sharp line of demarcation between the robust sprouting found in some terminal fields and the lack of sprouting within the neighboring stratum lucidum, despite the diffusion of BDNF to this layer (Fig. 7E,G,I).
Transmitter specificity of BDNF-stimulated sprouting in the intact cortex
Our previous study showed that BDNF infusions cause robust sprouting of intact (non PCA-lesioned) 5-HT axons in neocortex (Mamounas et al., 1995). To investigate whether BDNF can stimulate axon sprouting from other populations of uninjured subcortical neurons with axonal projections to cortex, BDNF was chronically infused (4 μg/d for 18 d) in frontoparietal cortex of intact rats, and the densities of serotonergic (SERT-immunoreactive), catecholaminergic (TH-immunoreactive) or cholinergic (by AChE histochemistry) fibers were measured in the BDNF-infused cortex. BDNF infusions increased (by 37 ± 4%) the density of 5-HT axons above the normal levels found in the vehicle-infused or contralateral cortex (Fig.8; ANOVA: F1,12 = 36, p < 0.0001), but failed to increase the densities of either catecholaminergic or cholinergic axons above normal (p > 0.05).
BDNF infusions stimulate Trk activation in cortex
To evaluate the relationship between the cortical plasticity described here and BDNF-mediated TrkB signaling events, the present study examined the in vivo regulation of Trk tyrosine autophosphorylation (reflecting Trk activation) by chronic BDNF infusion in neocortex. After BDNF administration, the cortical tissue surrounding the BDNF infusion cannula tip [∼(3 mm)3] was microdissected, and Trk activity was assayed as described in Materials and Methods. BDNF administration in cortex markedly stimulated Trk tyrosine phosphorylation (Trk P-tyr) above basal or vehicle levels when assayed 3 hr after a single injection of BDNF (4 or 12 μg; Fig.10A) or after chronic infusion of BDNF for 24 hr (0.4–12 μg/24 hr; Fig. 9) or 7 d (4 μg/d; Fig. 10B). Like the highly localized 5-HT sprouting response described above, Trk activity was maximally induced at the BDNF infusion site (within the 3 mm diameter cylinder of cortical tissue centered at the infusion cannula), with a much lower Trk P-tyr signal (and correspondingly lower exogenous BDNF protein levels) found in more distant cortical samples (cylinders located 2–5 mm from the BDNF infusion cannula; data not shown).
Dose–response profile for BDNF to stimulate Trk tyrosine phosphorylation
By 24 hr of continuous infusion, BDNF protein levels (by immunoblots) had reached steady-state levels in cortex, when compared to longer infusion times (Figs. 9, 10B). With 24 hr BDNF infusions into the cortex of intact animals, the dose–response profile for BDNF to stimulate Trk activity (Fig. 9) was remarkably similar to the dose–response profile shown above for stimulating cortical 5-HT axon sprouting (Figs. 4, 5). The induction of Trk P-tyr in cortex began at the 0.4 μg/d dose of BDNF, reached a maximal level at 4 μg/d, and declined at the 12 μg/d dose (Fig. 9; ANOVA,F(6,39) = 45; p < 0.0001, followed by the Bonferonni test for individual dose comparisons). Because of the nearly identical dose–response profiles for the infused BDNF to stimulate Trk P-tyr and 5-HT axon sprouting, we next examined whether the Trk P-tyr signal emanated primarily from the 5-HT axons in cortex. However, after lesioning most 5-HT axons in cortex with PCA, there was not a significant loss of Trk P-tyr at any dose of BDNF tested (0.4–12 μg infused over 24 hr, at 1 week after PCA administration) when compared to intact animals (p > 0.05; Fig. 9A). Thus, the major portion of the Trk P-tyr signal in response to BDNF arises from the target field (e.g., cortical neurons/glia, other innervating axons in cortex); even so, Trk signaling may occur in the 5-HT terminals as well, but below the level of detection in our assay (because the 5-HT axons comprise but a small fraction of the total neuropil in cortex).
Regulation of total TrkB protein by chronic BDNF infusions
To explore the mechanism for the downregulation of Trk activity at the higher BDNF doses (Fig. 9), we examined the regulation of total TrkB protein and mRNA by chronic BDNF infusion for 1 d (Fig. 9) or 7 d (Fig. 10B, Table1) in the neocortex. In parallel with a reduced Trk P-tyr signal, a 1 d infusion of BDNF at the 12 μg/d dose, but not at the lower doses (0.1–4 μg/d), produced a downregulation of full-length TrkB protein (gp145; catalytic) levels relative to vehicle or naïve controls (Fig. 9; ANOVA,F(6,42) = 3.9; p < 0.005, followed by the Bonferonni test for individual dose comparisons). An intermediate dose of BDNF when infused for a longer duration (4 μg/d for 7 d) also caused the concurrent downregulation of Trk P-tyr and full-length TrkB protein levels in the cortex. Thus, after a 7 d infusion of BDNF at 4 μg/d, the Trk P-tyr signal was still elevated relative to vehicle controls (Fig.10B, Table 1), but was lower than after a 1 d infusion of the same daily BDNF dose (Fig. 9). Accompanying this duration-dependent decline in Trk activity was a reduction in full-length TrkB protein levels occurring after a 7 d (Fig.10B, Table 1), but not 1 d (Fig. 9), BDNF infusion at the 4 μg/d dose. We observed a marginal reduction in truncated (gp95) TrkB protein levels with 7 d BDNF (4 μg/d) infusions (Fig. 10, Table 1; p = 0.08). The reduction in full-length TrkB protein levels caused by 7 d BDNF infusions was not associated with a reduction in TrkB mRNA levels (Table 1;p > 0.05).
Table 1.
Effects of chronic 7 d infusion of BDNF (4 μg/d) or vehicle in rat frontoparietal cortex on TrkB protein and mRNA expression
| TrkB Protein/mRNA Levels | Vehicle | BDNF | Student's t test (two-tailed) |
|---|---|---|---|
| Trk tyrosine phosphorylation | 1.0 ± 0.4 | 3.3 ± 0.2 | t12 = 4.8;p < 0.0005 |
| (Trk P-tyr; fold induction over vehicle) | |||
| Full-length TrkB protein | 100 ± 3 | 66 ± 7 | t12 = 4.5; p < 0.001 |
| (gp145 TrkB; percent vehicle) | |||
| Truncated TrkB protein | 100 ± 5 | 78 ± 9 | t6 = 2.1; p < 0.08, NS |
| (gp95 TrkB; percent vehicle) | |||
| TrkB/GAPDH mRNA | 100 ± 5 | 88 ± 6 | t8 = 1.6; p = 0.15, NS |
| (percent vehicle) |
At the end of a 7 d intracortical infusion of BDNF (4 μg/d) or vehicle in intact animals, the infused cortex was microdissected, and levels of Trk protein were assayed by immunoblotting (data shown in Fig. 10). TrkB mRNA was analyzed by RNAse protection assay (described in Materials and Methods) using a cDNA probe that recognizes all TrkB isoforms; a GAPDH probe was included in the same reaction mixture to assess relative levels of RNA present in each hybridization (data calculated as the ratio of TrkB to GAPDH). Values shown are means ± SEM for 4–7 rats per group. NS, Not significant at p < 0.05.
DISCUSSION
Our results indicate that chronic BDNF infusion (at doses as low as 0.4 μg/d for 1–2 weeks) in 5-HT terminal fields of frontoparietal neocortex or hippocampus markedly stimulates the regenerative sprouting of 5-HT axons previously lesioned by PCA. Unexpectedly, we found that BDNF treatment fails to protect most 5-HT axons from PCA-induced degeneration. Because BDNF can also elicit sprouting from uninjured (non-PCA-lesioned) 5-HT axons, but not catecholamine or cholinergic fibers, our findings suggest that BDNF has a trophic role in promoting serotonergic axonal growth and remodeling in the adult brain.
After ablating most 5-HT axons with PCA, BDNF infusions rapidly stimulate the sprouting of the lesioned or residual 5-HT axons, leading to a complete recovery of the innervation at the cortical BDNF infusion site. The ability of BDNF to elicit 5-HT sprouting is not restricted to a narrow “critical period” after axonal injury: BDNF treatment can markedly stimulate the regrowth of 5-HT axons whether initiated shortly after PCA administration (at 4–7 d) or up to a month after neurotoxin treatment. Importantly, once eliciting 5-HT sprouting with BDNF, the sprouted fibers persist in cortex for at least 5 weeks after terminating exogenous BDNF delivery. This contrasts with an NGF-induced expansion of the peripheral sympathetic innervation in the hippocampus or along brain vasculature that is completely reversed within 1–3 weeks after terminating exogenous NGF administration (Isaacson and Crutcher, 1995; Conner and Varon, 1996). Unlike the aberrant sympathetic sprouting in the hippocampus, the regenerating 5-HT axons reinnervate their normal target fields and thus may form stable local interactions (e.g., via synapses, endogenous trophic support mechanisms) that sustain the newly formed axons after removal of exogenous BDNF.
In contrast to the remarkable sprouting response elicited by BDNF, we found that BDNF treatment only minimally protects 5-HT axons from PCA-induced degeneration. Moreover, because 5-HT neurons were chronically exposed to BDNF for 1 week before PCA administration (protocol, Fig. 1C), it is possible that the few “surviving” 5-HT axons (found immediately adjacent to the BDNF infusion cannula) represent not “protection”, but an accelerated sprouting reaction from the injured axons during the 3 d after PCA. Although the direct application of BDNF to 5-HT axon terminals in cortex fails to promote their survival, neuroprotection in this experimental model may instead require elevated levels of BDNF within the 5-HT soma located in the midbrain raphe complex. However, the 1 week pretreatment with BDNF used in our protection paradigm (Fig.1C) likely allowed sufficient time for retrograde transport of the neurotrophin to raphe 5-HT cell bodies (Koliatsos and Price, 1993; Sobreviela et al., 1996) before PCA insult. Also, in pilot experiments, we found minimal protection of cortical 5-HT axons from PCA damage when BDNF was chronically infused near 5-HT cell bodies in the dorsal raphe (J. A. Siuciak, L. A. Mamounas, and C. A. Altar, unpublished observations).
BDNF is retrogradely transported from 5-HT axon terminals in cortex to cell bodies located in the midbrain raphe nuclei (Sobreviela et al., 1996), and TrkB is expressed in 5-HT neurons (Merlio et al., 1992). In this study, the time required for intracortical BDNF infusions to initiate local 5-HT sprouting responses (between 4 and 7 d) is consistent with a traditional retrograde signaling mechanism (Campenot, 1994; Segal and Greenberg, 1996) being responsible for the growth and remodeling of these axons. This model predicts that BDNF infused in cortex will initiate retrograde signaling from the 5-HT terminals to their somata followed by transcriptional changes and anterograde transport of newly synthesized proteins to the 5-HT terminal branches. However, the widespread anatomical organization of the serotonergic projection to cortex and the highly localized 5-HT sprouting response (in exact register with the location of exogenous BDNF) are incongruent with the notion that a retrograde mechanism is exclusivelyresponsible for the 5-HT axonal growth in cortex. Serotonergic axons arising from 5-HT neurons in the raphe nuclei do not project to their forebrain targets in a point-to-point, precisely topographic fashion, but instead are highly branching with individual 5-HT neurons having axonal terminations in many divergent cortical target fields (Molliver, 1987). Thus, the local application of BDNF to cortex should lead to a globalized and distributed 5-HT sprouting response in the ipsilateral cortex, if mediated entirely through retrograde means. Instead, the highly restricted pattern of 5-HT sprouting suggests that a local mode of action for BDNF is also involved in the sprouting response and may be an in vivo example of the local regulation of neurite growth observed by Campenot (1994) in compartmented sympathetic cultures. Although the molecular mechanisms for the local control of 5-HT axon growth by BDNF are not known, BDNF may induce local signaling events within the 5-HT axon terminals in cortex, thereby “tagging” (Frey and Morris, 1998) or “priming” (Gallo and Letourneau, 1998) these axons for the subsequent recruitment and reorganization of cytoskeletal proteins involved in the sprouting reaction. Or, a paracrine-like mechanism may be involved where BDNF induces the secretion of other instructive factors (e.g., cell adhesion molecules) from cortical neurons or glial cells at the BDNF infusion site which in turn interact with the regenerating 5-HT axons.
The importance of the target field in defining the spatial pattern of 5-HT axon sprouting in response to BDNF is also evident in the hippocampus. BDNF treatment encourages the regrowth of the 5-HT plexus only in those hippocampal lamina where 5-HT fibers ordinarily ramify, thereby restoring the normal pattern of 5-HT innervation in this structure. Thus, exogenous BDNF does not appear to be “chemotropic” for 5-HT neurons in the mature brain and cannot readily redirect the growth of 5-HT fibers into inappropriate termination zones. In the developing brain, BDNF administration increases the size of axonal arbors without disrupting the normal lamina-specific pattern of innervation (Cabelli et al., 1995; Inoue and Sanes, 1997). Likewise, NGF-induced sympathetic fiber sprouting in the hippocampus exhibits a striking laminar topography (Conner and Varon, 1995). Our data, as well, suggest that molecular cues (attractive/repulsive) in the target field, either constitutively expressed or induced by BDNF, are involved in sculpting the pattern of 5-HT axonal regrowth elicited by BDNF.
The highly localized 5-HT sprouting response is accompanied by the local activation of TrkB signaling (as reflected by Trk tyrosine autophosphorylation) with cortical BDNF infusions. The remarkably similar inverted-U-shaped dose–response profiles for BDNF to stimulate 5-HT sprouting and Trk-signaling responses suggest a physiological link between the two events. Also, a sustained elevation of the Trk signal with chronic BDNF infusion (for at least 1 week in our in vivo model) overlaps with the time required for BDNF to initiate 5-HT sprouting responses in cortex. The bulk of the Trk signal elicited by BDNF was found to originate from nonserotonergic elements at the cortical infusion site (local neurons/glia, the collective neuropil); thus, BDNF–TrkB signaling cascades occurring in the target field may create a favorable local environment for the regenerating fibers. Still, BDNF may also induce critical TrkB signaling events within the 5-HT terminals at the infusion site, although occurring below the level of detection in our experimental system. Further insight into potential BDNF–TrkB signaling events occurring in 5-HT neurons in this in vivo setting will require the development of highly sensitive in situ methods.
Interestingly, both the 5-HT sprouting and Trk-signaling responses are maximal at intermediate BDNF doses and are depressed at higher doses. In concert with the degradation of the Trk signal with excessive BDNF (higher doses, prolonged infusion), we found an underlying downregulation of full-length (catalytic) TrkB protein, but not truncated TrkB protein or TrkB mRNA levels in cortex. Local BDNF infusions also reduce full-length TrkB protein in the midbrain and olfactory bulb (Frank et al., 1997). As we found in neocortex, this downregulation of the protein is not associated with decreased TrkB mRNA expression, whether measuring TrkB message levels locally at the BDNF infusion site or in neuronal populations afferent to the infusion (Frank et al., 1997). Instead, excessive BDNF administration may lead to the increased degradation and turnover of TrkB protein with time, perhaps because of a shunting of the internalized BDNF–TrkB complex (Ehlers et al., 1995; Grimes et al., 1996) to endosomal-lysosomal pathways as occurs in vitro with NGF-TrkA (Grimes et al., 1996). Moreover, in parallel with our in vivo findings, high concentrations of NGF have been shown to inhibit neurite outgrowth from cultured sensory neurons (Levi-Montalcini and Angeletti, 1968; Conti et al., 1997). Taken together, these data have therapeutic implications with regard to administering pharmacological doses of BDNF during disease and demonstrate the importance of optimal BDNF levels for promoting both 5-HT regenerative sprouting and TrkB signal transduction in the brain.
Although the traditional view holds that the neurotrophins have a major role in promoting the survival of responsive neuronal populations during development or after injury in the mature nervous system (Lindsay et al., 1994; Koliatsos and Mocchetti, 1997), more recent evidence suggests a much broader role for the neurotrophins (particularly BDNF) in modifying neuronal phenotype, function, and plasticity (Lo, 1995; Thoenen, 1995; Altar and DiStefano, 1998), including the structural remodeling of dendrites and axons in the CNS (Cabelli et al., 1995; Cohen-Cory and Fraser, 1995; McAllister et al., 1995). With regard to the serotonergic system, BDNF administration augments 5-HT metabolism in the adult brain (Altar et al., 1994;Siuciak et al., 1996), presumably by increasing the expression of tryptophan hydroxylase (Siuciak et al., 1998). Chronic infusion of BDNF in the dorsal raphe nucleus alters the spontaneous firing pattern of 5-HT neurons in this nucleus (Celada et al., 1996). Also, heterozygous BDNF-deficient mutant mice develop functional disturbances in the brain serotonergic system in conjunction with 5-HT-sensitive behavioral abnormalities (Lyons et al., 1999). Our findings expand the role of BDNF as a regulator of 5-HT neuron function and include the demonstration of a remarkable structural plasticity of these neurons, whether intact or injured, in response to BDNF. We hypothesize that, in the adult brain, BDNF–TrkB signaling primarily serves to modulate the physiology and plasticity of 5-HT neurons, as opposed to being a major protective factor for these neurons during stress or insult.
Footnotes
This work was supported by National Institute of Mental Health Grant R29MH85433 (L.A.M.), a National Research Council National Institute on Aging/National Institutes of Health award (L.A.M.), and by the National Cancer Institute, Department of Health and Human Services, under contract with ABL (D.R.K, L.T., and W.E.L.). We thank Dr. Vassilis Koliatsos for helpful discussion and review of this manuscript, Dr. Randy Blakely for generously supplying the SERT antibody, Dr. Karen Axt for advice and assistance with SERT immunohistochemistry, and Dr. MaryAnn Wilson for advice and critical comments on axon density analysis.
Correspondence should be addressed to Dr. Laura A. Mamounas, Department of Pathology/Division of Neuropathology, The Johns Hopkins University School of Medicine, 558 Ross Building, 720 Rutland Avenue, Baltimore, MD 21205. E-mail: Mamounas@welchlink.welch.jhu.edu.
REFERENCES
- 1.Altar CA. Neurotrophins and depression. Trends Pharmacol Sci. 1999;20:59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- 2.Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21:433–437. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- 3.Altar CA, Boylan CB, Fritsche M, Jackson C, Hyman C, Lindsay RM. The neurotrophins NT-4/5 and BDNF augment serotonin, dopamine, and GABAergic systems during behaviorally effective infusions to the substantia nigra. Exp Neurol. 1994;130:31–40. doi: 10.1006/exnr.1994.1182. [DOI] [PubMed] [Google Scholar]
- 4.Axt KJ, Mamounas LA, Molliver ME. Structural features of amphetamine neurotoxicity in the brain. In: Cho AK, Segal DS, editors. Amphetamine and its analogs: psychopharmacology, toxicology, and abuse. Academic; San Diego: 1994. pp. 315–367. [Google Scholar]
- 5.Baumgarten HG, Grozdanovic Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry. 1995;28:73–79. doi: 10.1055/s-2007-979623. [DOI] [PubMed] [Google Scholar]
- 6.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 7.Bregman BS, McAtee M, Dai HN, Kuhn PL. Neurotrophic factors increase axonal growth after spinal cord injury and transplantation in the adult rat. Exp Neurol. 1997;148:475–494. doi: 10.1006/exnr.1997.6705. [DOI] [PubMed] [Google Scholar]
- 8.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 9.Campenot RB. NGF and the local control of nerve terminal growth. J Neurobiol. 1994;25:599–611. doi: 10.1002/neu.480250603. [DOI] [PubMed] [Google Scholar]
- 10.Celada P, Siuciak JA, Tran TM, Altar CA, Tepper JM. Local infusion of brain-derived neurotrophic factor modifies the firing pattern of dorsal raphé serotonergic neurons. Brain Res. 1996;712:293–298. doi: 10.1016/0006-8993(95)01469-1. [DOI] [PubMed] [Google Scholar]
- 11.Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- 12.Colado MI, O'Shea E, Granados R, Murray TK, Green AR. In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA (“ecstasy”) and p-chloroamphetamine but not the degeneration following fenfluramine. Br J Pharmacol. 1997;121:889–900. doi: 10.1038/sj.bjp.0701213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conner JM, Varon S. Effects of exogenous nerve growth factor upon sympathetic sprouting into the hippocampal formation. Exp Neurol. 1995;136:123–135. doi: 10.1006/exnr.1995.1089. [DOI] [PubMed] [Google Scholar]
- 14.Conner JM, Varon S. Maintenance of sympathetic innervation into the hippocampal formation requires a continuous local availability of nerve growth factor. Neuroscience. 1996;72:933–945. doi: 10.1016/0306-4522(95)00598-6. [DOI] [PubMed] [Google Scholar]
- 15.Conti AM, Fischer SJ, Windebank AJ. Inhibition of axonal growth from sensory neurons by excess nerve growth factor. Ann Neurol. 1997;42:838–846. doi: 10.1002/ana.410420604. [DOI] [PubMed] [Google Scholar]
- 16.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 17.Eaton MJ, Staley JK, Globus MYT, Whittemore SR. Developmental regulation of early serotonergic neuronal differentiation: the role of brain-derived neurotrophic factor and membrane depolarization. Dev Biol. 1995;170:169–182. doi: 10.1006/dbio.1995.1205. [DOI] [PubMed] [Google Scholar]
- 18.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank L, Wiegand SJ, Siuciak JA, Lindsay RM, Rudge JS. Effects of BDNF infusion on the regulation of TrkB protein and message in adult rat brain. Exp Neurol. 1997;145:62–70. doi: 10.1006/exnr.1997.6440. [DOI] [PubMed] [Google Scholar]
- 20.Frey U, Morris RGM. Synaptic tagging: Implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- 21.Fuller RW. The influence of fluoexetine on aggressive behavior. Neuropharmacology. 1996;14:77–81. doi: 10.1016/0893-133X(95)00110-Y. [DOI] [PubMed] [Google Scholar]
- 22.Gallo G, Letourneau PC. Localized sources of neurotrophins initiate axon collateral sprouting. J Neurosci. 1998;18:5403–5414. doi: 10.1523/JNEUROSCI.18-14-05403.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibb JW, Hanson GR, Johnson M. Neurochemical mechanisms of toxicity. In: Cho AK, Segal DS, editors. Amphetamine and it's analogs: psychopharmacology, toxicology, and abuse. Academic; San Diego: 1994. pp. 269–295. [Google Scholar]
- 24.Grimes ML, Zhou J, Beattie EC, Uyen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedreen JC, Bacon SJ, Price DL. A modified histochemical technique to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 26.Hen R. Mean genes. Neuron. 1996;16:17–21. doi: 10.1016/s0896-6273(00)80019-7. [DOI] [PubMed] [Google Scholar]
- 27.Inoue A, Sanes JR. Lamina-specific connectivity in the brain: regulation by N-cadherin, neurotrophins, and glucoconjugates. Science. 1997;276:1428–1431. doi: 10.1126/science.276.5317.1428. [DOI] [PubMed] [Google Scholar]
- 28.Isaacson LG, Crutcher KA. The duration of sprouted cerebrovascular axons following intracranial infusion of nerve growth factor. Exp Neurol. 1995;131:174–179. doi: 10.1016/0014-4886(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991a;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- 33.Koliatsos VE, Mocchetti I. Trophic factors as therapeutic agents for disease characterized by neuronal death. In: Koliatsos VE, Ratan RR, editors. Cell death and diseases of the nervous system. Humana; Totowa, NJ: 1997. [Google Scholar]
- 34.Koliatsos VE, Price DL. Retrograde axonal transport. Applications in trophic factor research. In: Boulton AA, Baker GB, Hefti F, editors. Animal models of neurological disorders. Humana; Clifton, NJ: 1993. pp. 247–290. [Google Scholar]
- 35.Levi-Montalcini R, Angeletti PU. Nerve growth factor. Physiol Rev. 1968;48:534–569. doi: 10.1152/physrev.1968.48.3.534. [DOI] [PubMed] [Google Scholar]
- 36.Lidov HGW, Grzanna R, Molliver ME. The serotonin innervation of the cerebral cortex in the rat–an immunocytochemical analysis. Neuroscience. 1980;5:207–227. doi: 10.1016/0306-4522(80)90099-8. [DOI] [PubMed] [Google Scholar]
- 37.Lindsay RM, Wiegand SJ, Altar CA, DiStefano PS. Neurotrophic factors: from molecule to man. Trends Neurosci. 1994;17:182–190. doi: 10.1016/0166-2236(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 38.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 39.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L (1999) BDNF-deficient mice develop aggressiveness and bulimia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA, in press. [DOI] [PMC free article] [PubMed]
- 40.Mamounas LA, Mullen CA, O'Hearn E, Molliver ME. Dual serotonergic projections to forebrain in the rat: morphologically distinct 5-HT axon terminals exhibit differential vulnerability to neurotoxic amphetamine derivatives. J Comp Neurol. 1991;314:558–586. doi: 10.1002/cne.903140312. [DOI] [PubMed] [Google Scholar]
- 41.Mamounas LA, Wilson MA, Axt KJ, Molliver ME. Morphological aspects of serotonergic innervation. Adv Biosci. 1992;85:97–118. [Google Scholar]
- 42.Mamounas LA, Blue ME, Siuciak JA, Altar CA. Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain. J Neurosci. 1995;15:7929–7939. doi: 10.1523/JNEUROSCI.15-12-07929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mann JJ. The neurobiology of suicide. Nat Med. 1998;4:25–30. doi: 10.1038/nm0198-025. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Cheng B, Smith-Swintosky VL. Mechanisms of neurotrophic factor protection against calcium- and free radical-mediated excitotoxic injury: Implications for treating neurodegenerative disorders. Exp Neurol. 1993;124:89–95. doi: 10.1006/exnr.1993.1178. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 46.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 47.Merlio J-P, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- 48.Molliver ME. Serontonergic neuronal systems: what their anatomic organization tells us about function. J Clin Psychopharmacol. 1987;7:3S–23S. [PubMed] [Google Scholar]
- 49.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem. 1994;40:288–295. [PubMed] [Google Scholar]
- 51.Pelleymounter MA, Cullen MJ, Wellman CL. Characteristics of BDNF-induced weight loss. Exp Neurol. 1995;131:229–238. doi: 10.1016/0014-4886(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 52.Qian Y, Melikian HE, Rye DB, Levey AI, Blakely RD. Identification and characterization of antidepressant-sensitive serotonin transporter proteins using site-specific antibodies. J Neurosci. 1995;15:1261–1274. doi: 10.1523/JNEUROSCI.15-02-01261.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 54.Siuciak JA, Altar CA, Wiegand SJ, Lindsay RM. Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res. 1994;633:326–330. doi: 10.1016/0006-8993(94)91556-3. [DOI] [PubMed] [Google Scholar]
- 55.Siuciak JA, Boylan C, Fritsche M, Altar CA, Lindsay RM. BDNF increases monoaminergic activity in rat brain following intracerebroventricular or intraparenchymal administration. Brain Res. 1996;710:11–20. doi: 10.1016/0006-8993(95)01289-3. [DOI] [PubMed] [Google Scholar]
- 56.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 57.Siuciak JA, Clark MS, Rind HB, Whittemore SR, Russo AF. BDNF induction of tryptophan hydroxylase mRNA levels in the rat brain. J Neurosci Res. 1998;52:149–158. doi: 10.1002/(SICI)1097-4547(19980415)52:2<149::AID-JNR3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 58.Smith MA, Zhang L-X, Lyons WE, Mamounas LA. Anterograde transport of endogenous brain-derived neurotrophic factor in hippocampal mossy fibers. NeuroReport. 1997;8:1829–1834. doi: 10.1097/00001756-199705260-00008. [DOI] [PubMed] [Google Scholar]
- 59.Sobreviela T, Pagcatipunan M, Kroin JS, Mufson EJ. Retrograde transport of brain-derived neurotrophic factor (BDNF) following infusion in neo- and limbic cortex in rat: relationship to BDNF mRNA expressing neurons. J Comp Neurol. 1996;375:417–444. doi: 10.1002/(SICI)1096-9861(19961118)375:3<417::AID-CNE6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 60.Spina MB, Squinto SP, Miller J, Lindsay RM, Hyman C. Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxydopamine and N-methyl-4-phenylpyridinium ion toxicity: involvement of the glutathione system. J Neurochem. 1992;59:99–106. doi: 10.1111/j.1471-4159.1992.tb08880.x. [DOI] [PubMed] [Google Scholar]
- 61.Sur C, Betz H, Schloss P. Immunocytochemical detection of the serotonin transporter in rat brain. Neuroscience. 1996;73:217–231. doi: 10.1016/0306-4522(96)00030-9. [DOI] [PubMed] [Google Scholar]
- 62.Tessarollo L, Nagarajan L, Parada LF. c-ros: the vertebrate homolog of the sevenless tyrosine kinase receptor is tightly regulated during organogenesis in mouse embryonic development. Development. 1992;115:11–20. doi: 10.1242/dev.115.1.11. [DOI] [PubMed] [Google Scholar]
- 63.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 64.White JD, Schwartz MW. Using osmotic minipumps for intracranial delivery of amino acids and peptides. In: Flanagan TR, Emerich DF, Winn SR, editors. Providing pharmacological access to the brain: alternate approaches. Academic; Boston: 1994. pp. 187–200. [Google Scholar]
- 65.Xu XM, Guénard V, Kleitman N, Aebischer P, Bunge MB. A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol. 1995;134:261–272. doi: 10.1006/exnr.1995.1056. [DOI] [PubMed] [Google Scholar]